

Abstract
Substituted cyclopropenes have recently attracted attention as stable “mini-tags” that are highly reactive dienophiles with the bioorthogonal tetrazine functional group. Despite this interest, the synthesis of stable cyclopropenes is not trivial and their reactivity patterns are poorly understood. Here, the synthesis and comparison of the reactivity of a series of 1-methyl-3-substituted cyclopropenes with different functional handles is described. The rates at which the various substituted cyclopropenes undergo Diels–Alder cycloadditions with 1,2,4,5-tetrazines were measured. Depending on the substituents, the rates of cycloadditions vary by over two orders of magnitude. The substituents also have a dramatic effect on aqueous stability. An outcome of these studies is the discovery of a novel 3-amidomethyl substituted methylcyclopropene tag that reacts twice as fast as the fastest previously disclosed 1-methyl-3-substituted cyclopropene while retaining excellent aqueous stability. Furthermore, this new cyclopropene is better suited for bioconjugation applications and this is demonstrated through using DNA templated tetrazine ligations. The effect of tetrazine structure on cyclopropene reaction rate was also studied. Surprisingly, 3-amidomethyl substituted methylcyclopropene reacts faster than trans-cyclooctenol with a sterically hindered and extremely stable tert-butyl substituted tetrazine. Density functional theory calculations and the distortion/interaction analysis of activation energies provide insights into the origins of these reactivity differences and a guide to the development of future tetrazine coupling partners. The newly disclosed cyclopropenes have kinetic and stability advantages compared to previously reported dienophiles and will be highly useful for applications in organic synthesis, bioorthogonal reactions, and materials science.
Keywords: bioconjugation, bioorthogonal cycloaddition, cyclopropene, density functional calculations, tetrazine
Introduction
Tetrazine inverse-electron-demand Diels–Alder cycloadditions are a recently introduced class of rapid reactions driven by the high electrophilicity of the tetrazine and high exothermicity of reaction with alkenes.[1] These reactions have found widespread application in bioorthogonal chemistry,[2] materials science,[3] and imaging research.[4] Initial efforts focused on the coupling of substituted 1,2,4,5-tetrazines with highly strained dienophiles, such as trans-cyclooctene, norbornene, and cyclooctyne.[5] These studies revealed that tetrazine cycloadditions could occur extremely rapidly with high yield in mild and aqueous conditions. The features of this reaction immediately drew several comparisons to the popular azide-alkyne cycloaddition, commonly referred to as “click” chemistry.[6] However, whereas azides and alkynes have the benefit of being low molecular weight “mini-tags” and are straightforward to synthesize, tetrazines and previously used strained dienophiles are both larger in size and harder to access synthetically. In an effort to overcome those drawbacks, our group recently developed a metal-catalyzed one-pot synthesis of unsymmetric 1,2,4,5-tetrazines.[7] However, this did not address the problem of the larger size of the coupling partners. For instance, a common application of bioorthogonal chemistry is to introduce a small bioorthogonal reactive handle into a biologically active molecule that is metabolically incorporated through promiscuous enzymatic pathways.[8] These pathways are often extremely specific to the size of the handle. For instance, elegant studies of the sialic acid salvage pathway have determined that N-acetyl mannosamine derivatives can be accepted below a threshold size limit and, based on these and other prior studies, it was clear that neither tetrazines nor high molecular weight strained dienophiles would be expected to be incorporated.[9] Furthermore, the solubility and size of the bioorthogonal dienophile tags influence the pharmacokinetics of the proteins and small molecules appended, which is an important consideration for in vivo applications.[4, 10] Finally, despite their proven applications, there have been stability concerns with highly reactive dienophiles, such as cyclooctynes and trans-cyclooctenes. The former are known to react with biologically relevant nucleophiles,[11] and the latter have the potential to isomerize to the very slow reacting cis-cyclooctene, especially in the presence of biological functional groups, such as thiols.[12]
A possible solution to the challenge of large dienophiles was to explore the use of smaller cyclopropenes as tetrazine reactive partners. In his pioneering studies of tetrazine reactivity with dienophiles, Sauer described and measured the rapid reaction of tetrazine with cyclopropene and 3-methylcyclopropene.[13] However, development of a practical cyclopropene mini-tag required balancing the rate of reaction, which depends heavily on substitution pattern, and the poor stability of unsubstituted cyclopropenes, which tend to react with each other through ene chemistry.[14] Considering these requirements, we introduced two methylcyclopropene mini-tags as fast and stable dienophiles for reaction with tetrazines.[12b] The tags are capable of rapidly reacting with tetrazines, elicit fluorogenic responses with quenched tetrazine-fluorophores, and were shown to be suitable for live-cell labeling of lipid analogues. Subsequent work utilized the small size and high reactivity of cyclopropenes as coupling agents. For instance, the Lin group developed cyclopropene tags as bioorthogonal chemical reporters that can be labeled via photoclick chemistry and introduced site-specifically on proteins.[15] The Prescher group elegantly utilized the 3-carbamoyl substituted methylcyclopropene tag to introduce modified neuramic acids on glycans and reveal them through tetrazine cycloaddition.[16] An important aspect of methylcyclopropenes is their potential to undergo tetrazine cycloaddition in parallel with traditional azide–alkyne cycloaddition chemistry. This feature was suggested based on theory[17] and has been verified experimentally.[16, 18] Furthermore, we recently demonstrated the ability of methylcyclopropene modified N-acetyl mannosamines to be used in metabolic imaging, an application that is highly sensitive to the steric bulk of appended bioorthogonal probes.[18] Thus, it appears that 1-methyl-3-substituted cyclopropenes are suitable as chemical mini-tags, readily and rapidly modified by inverse-electron-demand Diels–Alder cycloaddition with tetrazines and alternative coupling partners.
Given their potential utility in a wide range of applications, the development of novel methylcyclopropene handles would be of great interest, particularly if improvements could be made to their reaction kinetics with tetrazines and their stability in aqueous media. With these considerations in mind, we designed and synthesized several new methylcyclopropenes with functional handles substituted at the C3 position. These methylcyclopropenes are straightforward to synthesize from commercially available precursors and show varied reactivity and aqueous stability based on substitution pattern. An important outcome of these studies was the discovery of a novel 3-amidomethyl-1-methylcyclopropene that reacts significantly faster than previously reported methylcyclopropenes. Additionally, we have adapted the newly disclosed dienophile for bio-conjugation applications. Compared to previous probes, the novel amide-linked cyclopropene shows improved performance in bioconjugation applications. This feature was demonstrated by comparing cyclopropene probe stability and performance during the DNA templated detection of nucleic acid sequences. Tetrazine structure can greatly affect biological stability and reaction rate with dienophiles. As such, we compared the reactivity of the 3-amidomethyl-1-methylcyclopropene with several tetrazines to quantitate how tetrazine structure affects cyclopropene reaction rate. During this study, we surprisingly found that the 3-amidomethyl-1-methylcyclopropene reacts faster than a highly strained trans-cyclooctenol with a sterically hindered tert-butyl substituted tetrazine. This result is significant for future biological applications since tert-butyl substituted tetrazines are extremely resistant to biologically relevant nucleophiles, more so than other more commonly used methyl- and hydrogen-terminated tetrazines.
In an effort to better understand the reactivity patterns observed, we performed quantum mechanical simulations of the cyclopropene–tetrazine cycloaddition. We found that the distortion energy required for methylcyclopropene to achieve the transition-state geometry is dramatically smaller than that for the acyclic alkene, greatly accelerating the reaction. We selected several models of the synthesized methylcyclopropenes, and computed the transition states with 3-methyl-6-phenyl-1,2,4,5-tetrazine. The activation free energies were calculated for water, as well as the relative rate constants, which show good correspondence with the experimental data. These calculations reveal a correlation between the activation free energy and the cyclopropene HOMO energy. In addition, calculations support a previously overlooked advantage of 1-methyl-3-substituted cyclopropenes as mini-tags: their reactivities are not sensitive to the size of tetrazines. Our study sheds significant light on the reactivity of the methylcyclopropene mini-tag and will likely guide future work aiming on improving the rate of reaction with tetrazines, further promoting the use of methylcyclopropene and related mini-tags in chemical transformations, biological research, and materials science.
Results and Discussion
Synthesis of methylcyclopropenes
Although a wide variety of cyclopropenes are synthetically accessible, our primary concern was to create a series of cyclopropene mini-tags that were reactive with 1,2,4,5-tetrazines while possessing adequate aqueous stability.[19] Additionally, the appended tag should be of low molecular weight to minimize steric perturbation. In his previous studies, Sauer demonstrated that cyclopropene and 3-methylcyclopropene were both highly reactive with 1,2,4,5-tetrazine, but the 3,3-dimethylcyclopropene lowered the rate of reaction by more than three orders of magnitude.[20] This illustrated the importance of restricting substitution at the C3 position. Cyclopropenes without substituents at the C1 position showed rapid reactivity with tetrazine, but were not amenable to overnight storage due to rapid degradation. As a method to improve stability while conserving reactivity, we appended a methyl group to the alkene as a minimal steric perturbation. This successfully led to stable yet reactive cyclopropene mini-tags, and based on this previous experience, we limited our current study to 1-methyl-3-substituted cyclopropenes. In our initial studies, we noted that the substituents on the C3 position greatly affect reactivity in the Diels–Alder cycloaddition.[12b] For instance, the rate of cycloaddition of 3-carbamoyloxymethyl-1-methylcyclopropene 2 is approximately one-hundred times faster than 3-carbamoyl-1-methylcyclopropene 1 when reacting with tetrazine 3 in aqueous solution (Figure 1). This would be consistent with prior work, which demonstrated that electron-rich dienophiles react more rapidly in inverse-electron-demand Diels–Alder reactions.[21]
Figure 1.
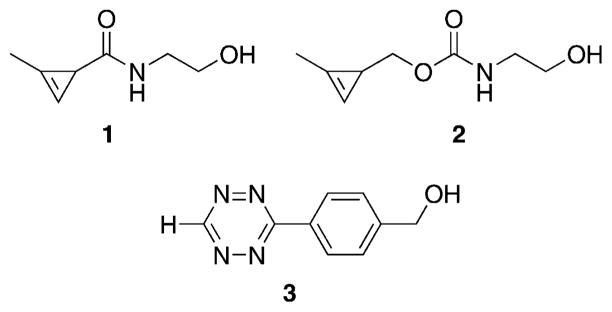
Previously studied methylcyclopropenes 1 and 2 react with tetrazine 3 in aqueous solution.
That the reaction rate could be modified so dramatically motivated us to explore how modifying the C3 position of methylcyclopropenes affects the kinetics and aqueous stability. Simultaneously, to easily introduce these groups to molecules of interest, we explored substituents possessing reactive handles that could be used for further modification. Herein, we report the synthesis of these new methylcyclopropene mini-tags.
The synthesis of minimally substituted cyclopropenes is challenging, as cyclopropenes can be volatile and prone to polymerization. Previously, we accessed cyclopropene alcohol 4 through rhodium-catalyzed cyclopropenation of trimethylsilylpropyne with ethyldiazoacetate followed by ester reduction (Scheme 1).[12b] Using the cyclopropene alcohol 4 as a starting material, we initially attempted to transform the alcohol into an amine by substitution chemistry. The inclusion of an amine was expected to increase the reaction rate while simultaneously providing a handle for later functionalization. However, conversion of the hydroxyl into a good leaving group leads to complex mixtures of degradation products.
Scheme 1.
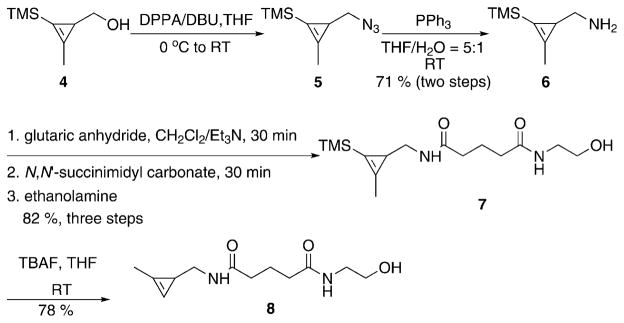
Synthesis of cyclopropene 8. DPPA=diphenylphosphoryl azide; DBU =1,8-diazabicyclo[5.4.0]undec-7-ene; TBAF =tetra-n-butylammonium fluoride; TMS =trimethylsilyl.
As an alternative approach, we attempted to form the amine by first converting the alcohol directly to an azide followed by reduction. To our delight, treatment with diphenylphosphoryl azide (DPPA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) can convert alcohol 4 into azide 5 in high yield. After simple purification, the azide is reduced with triphenylphosphine yielding amine 6. Both cyclopropene azide 5 and amine 6 are volatile and, although useful as synthetic intermediates, were not amenable to long-term storage. However, amine 6 could readily react to form stable amides or secondary amines. For instance, reaction with glutaric anhydride or methyl bromoacetate formed the corresponding amide 7 and secondary amine 9. Unlike 6, compounds 7 and 9 were amenable to long-term storage. The introduced carboxyl handle could also be further coupled with primary amines, such as ethanolamine, and the subsequent desilylation by TBAF afforded compounds 8 and 13 (Scheme 1 and Scheme 2).
Scheme 2.
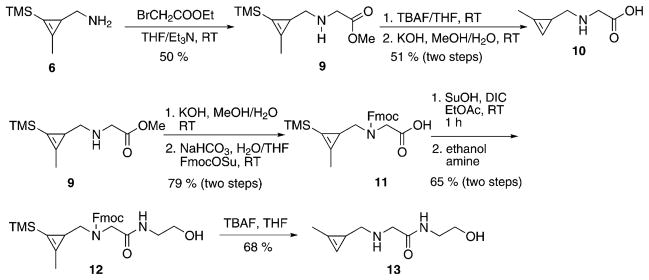
Synthesis of cyclopropenes 10 and 13. TBAF =tetra-n-butylammonium fluoride; TMS =trimethylsilyl; Su =succinimidyl; Fmoc =fluorenyl-methyloxycarbonyl.
We also explored whether cyclopropene alcohol 4 could be converted into an aldehyde by Dess–Martin oxidation. This reaction proved successful, although aldehyde 14 is volatile and unstable, slowly degrading over 48 h when stored at −20 °C. However, aldehyde 14 could be converted to α,β-unsaturated ester 15 through Horner–Wadsworth–Emmons reaction. Simultaneous hydrolysis of the ester and deprotection of the trimethylsilyl group with potassium hydroxide afforded carboxylic acid 16. This agent could react with amines forming stable amide 17 (Scheme 3).
Scheme 3.
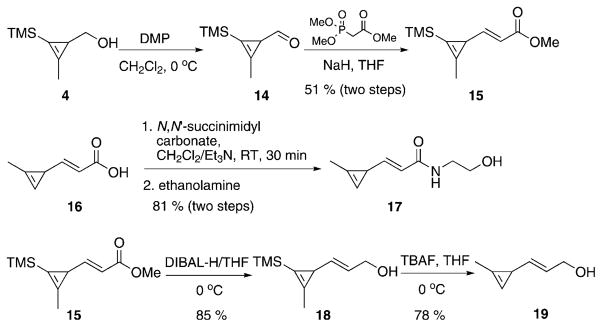
Synthesis of cyclopropenes 16, 17, and 19. DMP =Dess–Martin periodinane; TBAF =tetra-n-butylammonium fluoride; TMS =trimethylsilyl.
Previously, we have shown that eliminating an electron withdrawing carbonyl group can improve the rate of cyclopropene–tetrazine cycloaddition. To test the effect of the carbonyl group, we synthesized cyclopropene 19 through first reducing cyclopropene ester 15 followed by trimethylsilyl deprotection (Scheme 3). However, it should be noted that compound 19 was not stable to long-term storage at −20 °C, and was used immediately following purification.
Kinetic measurements and stability studies
After synthesizing the new cyclopropene mini-tags, we next sought to compare their reactivity with tetrazines. Additionally, we assayed their aqueous stability, given the ultimate goal of applications in bioorthogonal coupling reactions. In order to monitor kinetics, we conducted reactions of eight representative 1-methyl-3-substituted cyclopropenes with tetrazine 20 under pseudo-first-order conditions, and tracked the Diels–Alder reaction by monitoring the disappearance of the characteristic tetrazine absorption at 520 nm, similar to previous reports (Figure 2a).[5b, 22] Our choice of tetrazine 20 was motivated by previous work demonstrating that methyl-terminated tetrazines are more resistant to decomposition in biologically relevant media when compared to hydrogen-terminated tetrazines.[22] For cyclopropene stability experiments, we incubated methylcyclopropenes in aqueous deuterated solutions (D2O/[D6]DMSO, 4:1) as indicated, and monitored cyclopropene stability using NMR spectroscopy. The results of these experiments are summarized in Table 1, and the data are shown in Figure 2.
Figure 2.

Tetrazine–cyclopropene kinetics. a) Representative set of UV/Vis absorbance scans during the reaction of the fastest stable cyclopropene (entry 2, Table 1) with tetrazine 20 after background subtraction. Tetrazine absorbance peak at 520 nm decreases as a function of reaction progress. b) Data points and fitted lines for the reactions shown in Table 1. Tetrazine 20 was treated with the reported cyclopropene entries at 1 and 10 mM, respectively, in 50 mM MOPS, pH 7.5, 250 mM NaCl with specific conditions indicated in Table 1.
Table 1.
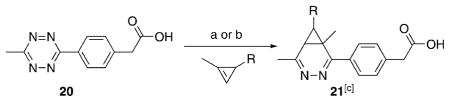
| ||||
|---|---|---|---|---|
| Entry | Cyclopropene | t1/2 [s] | k2 [M−1 s−1] | Stability |
| 1[a] |
|
135.1 | 0.74 ± 0.04 | unstable[d] |
| 2[a] |
|
153.8 | 0.65 ± 0.01 | stable[e, f] |
| 3[a] |
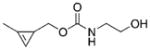
|
273.2 | 0.366 ± 0.005 | stable[e, f] |
| 4[a] |
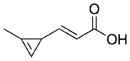
|
297.6 | 0.336 ± 0.006 | unstable[e] |
| 5[a] |
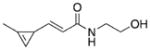
|
500.0 | 0.20 ± 0.01 | stable[e] |
| 6[b] |
|
2571 | 0.0389 ± 0.0007 | stable[e] |
| 7[b] |
|
3333 | 0.030 ± 0.002 | stable[e] |
| 8[b] |
|
21 277 | 0.0047 ± 0.0004 | stable[e] |
1.0 mM 20, 10.0 mM cyclopropene, 50 mM MOPS buffer, pH 7.5, 250 mM NaCl, RT;
1.0 mM 20, 10.0 mM cyclopropene, 25 % DMF in MOPS buffer, RT;
reaction of cyclopropene with tetrazine 20 leads to formation of diazanorcaradiene isomers (only one regioisomer is depicted);
D2O/[D6]DMSO = 4:1, RT;
D2O/[D6]DMSO = 4:1, 37 °C;
D2O/[D6]DMSO = 4:1, 37 °C, mixture with L-cysteine.
Of the stable cyclopropenes, the amide derived from 3-ami-nomethyl-1-methylcyclopropene (entry 2, Table 1) is the fastest with a second-order rate constant of 0.65 M−1 s−1. This rate is approximately twice as fast as that of the previously synthesized 3-carbamoyloxymethyl-1-methylcyclopropene (entry 3). The methylcyclopropenes derived from aldehyde precursors are slower, and the carboxylic acid (entry 4) proved to be unstable to incubation in aqueous solvent at 37 °C. However, conjugation with the primary amine to form an amide (entry 5) greatly improved stability, but significantly lowered the rate of reaction with tetrazine. As expected from prior studies, reduction of the ester to the alcohol greatly increased the reaction rate (entry 1). However, the resulting compound was unstable in aqueous solution, completely decomposing in 10.5 h when dissolved in D2O/[D6]DMSO = 4:1 at room temperature. The secondary amines resulting from 3-aminomethyl-1-methylcyclopropene (entries 6 and 7) showed a reduced rate of reaction with tetrazine in aqueous solvent, although they were found to be stable. Finally, 3-carbamoyl-1-methylcyclopropene (entry 8), was relatively sluggish with respect to tetrazine reactivity, but also proved to be highly stable. For comparison, the rate of tetrazine reaction with entry 8 was approximately 138-times slower than that using the fastest stable cyclopropene handle (entry 2).
One of the potential drawbacks of highly strained trans-cyclooctenes is their propensity for trans/cis isomerization in the presence of nucleophiles, such as thiols.[12] Recent work from several groups has demonstrated this property. Since the cycloaddition between tetrazine and cis-cyclooctene is expected to have a second-order reaction rate five orders of magnitude slower than the reaction with trans-cyclooctene, the cis form is expected to be significantly less active.[20] This could be a problem, particularly for long-term storage in complex media or for biological studies that require lengthy dienophile incubation. Our recent work with 1-methyl-3-substituted cyclopropenes indicated that these species are resistant to degradation when challenged with cysteine in aqueous conditions. As cyclopropene 8 is more reactive than cyclopropene 2 (as shown in the experiment above) as well as highly stable in aqueous deuterated solutions (entry 2, Table 1), we used this compound for incubation with L-cysteine in 4:1 D2O/[D6]DMSO at 37 °C, over-night. No decomposition could be observed by NMR spectroscopy. Given its rapid kinetics and excellent stability, we believe that 3-amidomethyl substituted methylcyclopropenes should be excellent mini-tags and superior to previously introduced methylcyclopropenes. However, it is noteworthy that the method of synthetic introduction should be taken into account when deciding on the use of a specific tag, as certain reactive handles will be easier to introduce onto molecular scaffolds given the available functional groups.
Biological application: improved stability and performance of cyclopropene amide modified DNA ligation probes
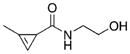
In addition to improved reaction rates, the amide linkage may have greater biological stability compared to the previously introduced carbamate linkage. Carbamates can be prone to decomposition in the proximity of biologically relevant nucleophiles and enzymes, such as esterases.[24] To determine if cyclopropene amide possessed improved stability relevant for bio-conjugation applications, we synthesized cyclopropene amide NHS ester 35, which can be conveniently conjugated to primary amine containing biomolecules, such as lipids, antibodies, proteins, or DNA. We conjugated the NHS ester 35 to a short oligonucleotide and used the resulting DNA–cyclopropene amide 37 to compare its stability to a previously described cyclopropene carbamate linkage 38 (Figure 3a).[23] Such 3′ dienophile-modified oligonucleotide probes are expected to undergo DNA-templated cycloaddition with quenched 5′ fluorescein–tetrazine oligonucleotide probes in the presence of a complementary DNA template (Figure 3b).[25] Reaction elicits a fluorogenic response as the tetrazine quencher is consumed. Although the tetrazine probe is highly stable over lengthy periods of time in buffer and biological media, methylcyclopropene probe 38 linked by a carbamate linkage is susceptible to degradation over extended periods and multiple freeze-thaw cycles (as determined by HPLC, Figure 3d). This prevents the long-term storage of such constructs and limits their biological application. However, attachment of the novel cyclopropene amide 35 to oligonucleotide probes (Figure 3c) dramatically reduced the decomposition rate and the probes are viable after long-term storage and multiple freeze-thaw cycles with minimal loss of reactivity. In a head-to-head comparison, we submitted samples to five freeze-thaw cycles over a one week period and compared the ability of amide-linked cyclopropene 37 and carbamate-linked cyclopropenes 38 to elicit a fluorogenic reaction from a quenched tetrazine probe in the presence of an appropriate DNA template (Figure 3e). After storage, the amide 37 remained viable and a strong fluorogenic response was elicited by the template. In contrast, the degraded carbamate 38 elicited a minimal turn-on response. We believe that the newly disclosed amide linked methylcyclopropenes will find broad application for their long-term stability when conjugated to biological molecules.
Figure 3.
Stability of cyclopropene–DNA oligonucleotide probes. a) Synthesis scheme of the modified DNA cyclopropene probes. b) Modified DNA probe reaction upon hybridization to a fully complimentary DNA template. Illustration depicts the placement of fluorescein (star) and tetrazine quencher that results in fluorescence turn-on (green star) after reaction with the cyclopropene probe. c) After five freeze-thaw cycles, 13 nucleotide 3′ cyclopropene amide probe 37 shows lower than 1 % degradation, based on a gradient HPLC trace of 260 nm absorbance peak containing expected probe product mass peaks. Initial and final HPLC traces are shown overlaid. d) Similarly, after five freeze-thaw cycles, 13 nucleotide 3′ cyclopropene carbamate probe 38 shows 95 % degradation, estimated from the HPLC trace peak areas. e) Cyclopropene–DNA probes (37 in green trace, 38 in blue) were treated with the neighboring tetrazine probes along a DNA template. Reaction progress was measured by the unquenching of fluorescein emission that is initially quenched by the unreacted tetrazine (trace in black). Final reacted fluorescein emission traces using the cyclopropene probes after freeze-thaw cycles are shown in green and blue, respectively. Inset: the reactions upon completion, visualized under a long range UV lamp. Stars correspond with measured fluorescence scan colors (cyclopropene 37 reactions in green, 38 in blue). Cyclopropene 38 reaction product was confirmed previously,[23] cyclopropene 37 reactant and the reaction product are confirmed in Figure S2 in the Supporting Information.
Dependence of methylcyclopropene reaction rate on tetrazine structure
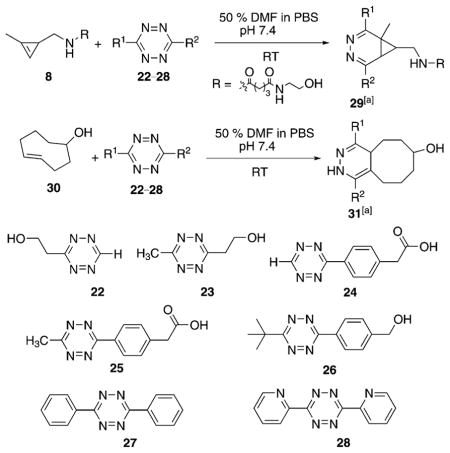
Having identified amides derived from 3-aminomethyl-1-methylcyclopropene as highly reactive and stable dienophiles for inverse-electron-demand Diels–Alder reaction with tetrazines, we next measured the reaction rates of the cyclopropene amide 8 with six different tetrazines to quantify how tetrazine structure affects the rate of cyclopropene–tetrazine cycloaddition. The functional group substitution at the C3 and C6 position of 1,2,4,5-tetrazines can dramatically affect the stability of tetrazines in biological media as well as their reactivity with dienophiles. Tetrazines with electron-rich substituents are highly stable while possessing slower reaction rates, whereas those with electron-poor substituents have lower stability in biological media but have faster reaction rates.[5a] We chose substituted tetrazines based on our previous experience of synthesizing aliphatic tetrazines.[7] Alkoxyltetrazines have also been shown to be easily accessible, highly stable, and useful for a variety of applications, particularly in materials science.[26] However, calculations indicated that the electron-rich substituents, such as methoxy and dimethylamino groups, would greatly decrease the tetrazine reactivity in the inverse-electron-demand Diels–Alder cycloaddition (for details, see Figure S4 in the Supporting Information).
As such, the reactivity of monoaryl tetrazine 24 is the greatest with methylcyclopropene 8, and additional alkyl or aryl substitution on the tetrazine decreases the rates (Table 2). We also tested the reactivity of a sterically hindered tert-butyl substituted tetrazine 26 (for the synthesis, see the Supporting Information). It was found that tetrazine 26 reacts only two-times slower than methyl substituted tetrazine 25 (entries 4 and 5, Table 2). We also compared the tetrazine cycloaddition reaction rates of cyclopropene 8 with trans-cycloctene 30 (Table 2). Previous work had indicated that for sterically less encumbered tetrazines, such as monosubstituted tetrazines, trans-cyclooctene reacts 2–3 orders of magnitude faster than methylcyclopropenes.[12b,22] Although we observed relative rates in line with this observation for most of the studied tetrazines, we surprisingly found that methylcyclopropene 8 reacts with tert-butyl modified tetrazine 26 faster than trans-cyclooctene 30. This result might be due to the larger size of the trans-cyclooctene dienophile, which may have limited accessibility to tetrazines that are flanked by sterically bulky substituents, such as the tert-butyl group. In contrast to trans-cycloctene, methylcyclopropenes are mini-tags of similar size to azides and alkynes. The low molecular weight of cyclopropenes has previously been used to enable metabolic labeling applications. The current work demonstrates that the small size of the methylcyclopropene dienophile is also important for enabling a rapid reaction rate with sterically hindered tetrazines, such as those substituted by tert-butyl groups.
Table 2.
| |||
|---|---|---|---|
| Entry | Tetrazine | k2,MCP [M−1 s−1] | k2,TCO [M−1 s−1] |
| 1[b] | 22 | 1.037 ± 0.007 | >100 |
| 2[b] | 23 | 0.0810 ± 0.0003 | 31.5 ± 0.3 |
| 3[b] | 24 | 3.34 ± 0.01 | >100 |
| 4[b] | 25 | 0.288 ± 0.002 | >100 |
| 5[b, c] | 26 | 0.131 ± 0.004 | 0.086 ± 0.002 |
| 6[d] | 27 | 0.1755 ± 0.0005 | 49 ± 42 |
| 7[d] | 28 | 2.29 ± 0.02 | >100 |
Reactions of cyclopropene 8 and trans-cyclooctene 30 with tetrazine lead to the formation of isomers (only one isomer is depicted);
1.0 mM tetrazine, 10.0 mM cyclopropene or (E)-cyclooct-4-enol, reaction conditions: 50 % DMF, 50 % PBS, pH 7.4, RT;
experiment done in triplicate;
1.0 mM tetrazine, 10.0 mM cyclopropene or (E)-cyclooct-4-enol, reaction conditions: 70 % DMF, 30 % PBS, pH 7.4, RT.
This result has important implications for future biological applications, as tert-butyl tetrazines are extremely resistant to degradation in biologically relevant media. We have found that sterically hindered tetrazines possessing a tert-butyl group are significantly more stable in comparison to hydrogen- and methyl-terminated tetrazines (Figure S1 in the Supporting Information). Using the characteristic visible absorption of tetrazines at approximately 520 nm to monitor stability, tert-butyl tetrazine 26 showed negligible degradation in the presence of buffer over 48 h whereas hydrogen- and methyl-terminated tetrazines degraded 3 and 10 %, respectively. Tetrazines have previously been shown to be susceptible to nucleophilic attack by thiols, with degradation rates varying depending on the substitution pattern.[5a] Remarkably, one equivalent of L-cysteine causes <1 % degradation of tert-butyl modified tetrazine 26 over 3 h. However, equimolar L-cysteine results in 4 % degradation of methyl-terminated tetrazine 25 and 20 % degradation of hydrogen-terminated tetrazine 24 over the same time-scale (for full experimental details, see the Supporting Information). Control reactions in the absence of nucleophiles (see the Supporting Information) showed no loss of tetrazine signal, indicating that the loss of absorption was not due to intrinsic tetrazine decomposition or light induced degradation.[27] Since methylcyclopropenes are also highly stable, even under conditions in which trans-cyclooctene is known to isomerize, the tert-butyl tetrazine and methylcyclopropene amide functional groups are a highly stable yet mutually reactive bioorthogonal coupling pair. In order to further characterize this reaction, we performed an LC-MS trace of tert-butyl tetrazine 26 reacting with methylcyclopropene amide 8 in 1:1 DMF/H2O (Figure 4). The reaction resulted in isomers of 3,4-diazanorcaradiene derivatives 39 in 97 % yield based on reacted started material through an electrocyclic ring-opening and closing process.[16, 28] The products appear to be stable to hydrolysis over the 18 h reaction in line with previous reports of diazanorcaradiene stability.[29] We are currently investigating the implementation of this reactive pair in bioconjugation applications.
Figure 4.

Reaction between cyclopropene amide 8 with tetrazine 26. HPLC trace of purified tetrazine 26 (blue) is shown overlaid with the trace of the products (red) of the reaction between 8 (1.1 mM) and 26 (1.0 mM) in 1:1 DMF and H2O after 18 h. Inset: the MS trace of the reaction with the selected ion monitoring at m/z 457.2 is shown in black (i.e., [M +H]+ peak). Only one isomer of 39 is depicted in the scheme above.
DFT calculations
Quantitative insights into the high rate of the cyclopropene–tetrazine cycloaddition and the effect of substituents on the reactions of 1-methyl-3-substituted cyclopropenes with 1,2,4,5-tetrazines were obtained with density functional theory (DFT) calculations.[30] M06-2X,[31] a density functional that gives relatively accurate energetics for cycloadditions,[32] was used in this computational study. First we compared the reactivities of methylcyclopropene and its unstrained counterpart 2-methyl-2-butene in the inverse-electron-demand Diels–Alder reactions with 3-methyl-6-phenyltetrazine (Figure 5). The energy of the highest occupied molecular orbital (HOMO) of 2-methyl-2-butene is −9.00 eV, higher than that of methylcyclopropene (−9.31 eV). Frontier molecular orbital (FMO) analysis would predict that the acyclic alkene is electronically more reactive. However, transition-state calculations showed that the activation free energy for the cycloaddition of methylcyclopropene with tetrazine via TS2 is 7.3 kcal mol−1 lower than that of 2-methyl-2-butene via TS1 (19.8 vs. 27.1 kcal mol−1, Figure 5). This corresponds to an increase of rate by over five orders of magnitude. Our distortion/interaction analysis[17, 33] reveals the origin of the high rate of the cyclopropene–tetrazine cycloaddition: the distortion energy required for methylcyclopropene to achieve the transition-state geometry is dramatically smaller than that for 2-methyl-2-butene (8.8 vs. 14.6 kcal mol−1, Figure 5). The highly strained three-membered ring of methyl-cyclopropene results in very easy distortion of the C–H and C–C bonds out of the C=C bond plane,[34] which is a prominent distortion in the transition state. Next, we located the Diels–Alder transition states for reactions of 3-methyl-6-phenyltetrazine with five 1-methyl-3-substituted cyclopropenes to model the variety of dienophiles studied experimentally (Figure 5). The activation free energies in water and relative rate constants at 298 K were determined and are shown below the transition structures in Figure 5. Compared to the activation barrier for methylcyclopropene (TS2), the experimentally tested substituents at the C3 position of methylcyclopropenes (TS3–6) increase the activation barriers for cycloadditions with tetrazine. The reactivity trends found experimentally are reproduced by these calculations. The calculations also reveal that the cycloaddition is always preferred on the face away from the 3-substituent of the cyclopropene.
Figure 5.

M06-2X/6-31G(d)-optimized transition-state structures for the Diels–Alder reactions of 3-methyl-6-phenyltetrazine with 2-methyl-2-butene and six methylcyclopropenes (distances in Å) and M06-2X/6-311+G(d,p)//6-31G(d)-computed activation free energies in water (Gwater, in kcal mol−1) and relative rate constants (krel).
We also analyzed the activation barriers of these reactions using the distortion/interaction model.[17, 33] The distortion energies are nearly identical, so the differences in reactivity arise from differences in interaction energies, which are related to the degree of charge transfer from the cyclopropene highest occupied molecular orbital (HOMO) to the tetrazine lowest unoccupied molecular orbital (LUMO).[35] There is a good linear correlation between the activation free energy and the cyclopropene HOMO energy (Figure 6). The experimentally employed substituents are all inductively electron-withdrawing as compared with hydrogen, and lower the HOMO energy, decreasing favorable charge-transfer interaction with the low-lying tetrazine LUMO. Calculations show that the methyl substituent can serve as an inductively electron-donating group, elevating the HOMO energy of cyclopropene. Therefore, it is predicted that the cyclopropene with a pure alkyl (methyl) 3-substituent (TS7, Figure 5) will have larger relative rate constant compared to the cyclopropenes studied here. Indeed, this is consistent with past observation that 3-methylcyclopropene reacts more rapidly with 1,2,4,5-tetrazines compared to cyclopropene.[20]
Figure 6.

Plot of activation free energy versus cyclopropene HOMO energy for various model 1-methyl-3-substituted cyclopropenes (Gwater = −5.72 EHOMO−33.4, r2 =0.95).
To better understand the effect of tetrazine substituents on the cyclopropene and trans-cyclooctene reaction rates, we investigated the Diels–Alder reactions of 3-phenyltetrazine, 3-methyl-6-phenyltetrazine, and 3-tert-butyl-6-phenyltetrazine with both 3-formamidomethyl-1-methylcyclopropene and trans-cyclooctene, using DFT calculations (Figure 7 and Figure 8). Computational results indicated that, for the small methylcyclopropene, the sterically bulky 3-tert-butyl-6-phenyltetrazine reacts only four-times slower than 3-phenyltetrazine. The distortion energies required for tetrazines are very close despite the obvious difference in size (Figure 7). This supports a significant advantage of 1-methyl-3-substituted cyclopropenes as mini-tags: their reactivities are not sensitive to the size of tetrazines and other coupling partners. By contrast, for the trans-cyclooctene–tetrazine cycloaddition, the change of one substituent of tetrazine from hydrogen to tert-butyl group leads to a decrease of rate by over four orders of magnitude (Figure 8). In the transition state TS12, to avoid the steric repulsions between the trans-cyclooctene and the bulky tert-butyl group, the dihedral angle of tetrazine plane decreases by 7.2° as compared with that in TS10 (142.8° vs. 150.0°, Figure 8). This needs more distortion energy of tetrazine (20.1 vs. 14.0 kcal mol−1), resulting in a higher activation barrier (20.8 vs. 15.2 kcal mol−1, Figure 8).
Figure 7.

M06-2X/6-31G(d)-optimized transition-state structures for the Diels–Alder reactions of 3-formamidomethyl-1-methylcyclopropene with three tetrazines (distances in Å, dihedral angles in deg) and M06-2X/6-311 +G(d,p)//6-31G(d)-computed activation free energies in water (Gwater, in kcal mol−1) and relative rate constants (krel).
Figure 8.

M06-2X/6-31G(d)-optimized transition-state structures for the Diels–Alder reactions of trans-cyclooctene with three tetrazines (distances in Å, dihedral angles in deg) and M06-2X/6-311 +G(d,p)//6-31G(d)-computed activation free energies in water (Gwater, in kcalmol−1) and relative rate constants (krel).
Conclusion
We have synthesized and characterized the reactivity of several novel 1-methyl-3-substituted cyclopropenes as “mini-tag” dienophiles for bioorthogonal reactions with electron poor tetrazines. These reactions are irreversible, do not require a catalyst, and can be performed in a mutually orthogonal manner with azide-dibenzocyclooctyne cycloadditions. We have developed synthetic protocols to access several stable 1-methyl-3-subsituted cyclopropenes that react with tetrazines at rates spanning over two orders of magnitude dependent on the substituent at the C3 position. Importantly, we have found that amidomethyl substitution on the C3 position of methylcyclopropene leads to a new class of cyclopropene mini-tags that react with tetrazines faster than previously reported cyclopropene handles while maintaining stability in aqueous solution in the presence of thiols. These new tags are significantly more stable in bioconjugation applications, and we have demonstrated this feature by using these probes for the fluorogenic detection of DNA templates. Additionally, we compared the reactivity of 3-amidomethyl-1-methylcyclopropene with several different substituted tetrazines. Unexpectedly, we discovered that amidomethyl-substituted cyclopropene reacts faster than a trans-cyclooctene with a sterically hindered yet extremely stable tert-butyl substituted tetrazine. This feature is significant for future bioconjugation applications that require reactive bioorthogonal probes that are highly stable to degradation. In order to better understand the origins of the reactivity differences, we performed DFT calculations and the distortion/interaction analysis of activation barriers. The high rate of the cyclopropene–tetrazine cycloaddition is because the distortion energy required for methylcyclopropene to achieve the transition-state geometry is dramatically smaller than that for the acyclic alkene. The computational results reproduced the experimentally observed reactivity trend of methylcyclopropenes with various C3 substituents and show that the activation barriers have a good correlation with the cyclopropene HOMO energies. Besides, calculations support a key advantage of cyclopropene mini-tags: their reactivities are not sensitive to the size of tetrazines and other coupling partners. The mechanistic information provides a guide to the future development of bioorthogonal cycloadditions. Methylcyclopropenes have size and stability advantages compared to previously reported tetrazine reactive dienophiles and, for these reasons, will likely find widespread use in bioorthogonal chemistry applications.
Experimental Section
Materials
All chemicals were obtained from commercial sources and used without further purification. Thin layer chromatography (TLC) was performed on silica gel. Chromatographic purifications were conducted using 40–63 μm silica gel. All mixtures of solvents are given in v/v ratio. 1H and 13C NMR spectroscopy was performed on a Varian NMR at 400 (1H) or 100 (13C) MHz and a Jeol NMR at 500 (1H) or 125 (13C) MHz. All 13C NMR spectra were proton decoupled. Phosphate-buffered saline (PBS) was used, diluted to 1 0 from the commercial 10 0 formulation, at the final 10 mM phosphate buffer, pH 7.4, 0.154 M NaCl (Sigma).
Kinetics determination
Tetrazine 20 (1 mM) was treated with a tenfold excess of methylcyclopropenes (Table 1) in 3-(N-morpholino)propanesulfonic acid (50 mM; MOPS) buffer, pH 7.5, and 250 mM NaCl (Figure 2 and Table 1). Characteristic tetrazine peak absorbance at 520 nm disappears upon reaction, and was tracked over time by absorbance scans using a UV/Vis spectrophotometer (Nanodrop 2000c, Thermo Scientific) in a quartz cuvette (Figure 2a). Tetrazine peak intensities were background-adjusted by subtracting an extrapolated straight line between the intensities preceding and following the peak. Reaction data were processed using GraphPad Prism 6.0 and fit to nonlinear regressions of one phase decay. Resulting observed pseudo-first-order kobs values were converted to the reported k2 second-order rate constants as k2 = kobs/[cyclopropene]0. Reaction halftimes were calculated as t1/2 = 1/(k2[cyclopropene]0) as applicable for the second-order reaction.
Supplementary Material
Acknowledgments
N.K.D. gratefully acknowledges discussions with Prof. Carlos Guerrero and support from the National Institutes of Health (K01EB010078) and the University of California, San Diego. K.N.H. gratefully acknowledges the National Science Foundation (CHE-1059084) for financial support of this research. Calculations were performed on the Hoffman2 Cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201304225.
Contributor Information
Prof. K. N. Houk, Email: houk@chem.ucla.edu.
Prof. Neal K. Devaraj, Email: ndevaraj@ucsd.edu.
References
- 1.Devaraj NK, Weissleder R. Acc Chem Res. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Hansell CF, Espeel P, Stamenovic MM, Barker IA, Dove AP, Du Prez FE, O’Reilly RK. J Am Chem Soc. 2011;133:13828–13831. doi: 10.1021/ja203957h. [DOI] [PubMed] [Google Scholar]; b) Zhou H, Johnson JA. Angew Chem. 2013;125:2291–2294. [Google Scholar]; Angew Chem Int Ed. 2013;52:2235–2238. doi: 10.1002/anie.201207966. [DOI] [PubMed] [Google Scholar]
- 4.Devaraj NK, Thurber GM, Keliher EJ, Marinelli B, Weissleder R. Proc Natl Acad Sci USA. 2012;109:4762–4767. doi: 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Kolb HC, Finn MG, Sharpless KB. Angew Chem. 2001;113:2056–2075. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Karver MR, Li W, Sahu S, Devaraj NK. Angew Chem. 2012;124:5312–5315. doi: 10.1002/anie.201201117. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:5222–5225. doi: 10.1002/anie.201201117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sletten EM, Bertozzi CR. Angew Chem. 2009;121:7108–7133. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, Bertozzi CR. J Am Chem Soc. 2002;124:14893–14902. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
- 10.Rossin R, Verkerk PR, van den Bosch SM, Vulders RC, Verel I, Lub J, Robillard MS. Angew Chem. 2010;122:3447–3450. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:3375–3378. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 11.van Geel R, Pruijn GJ, van Delft FL, Boelens WC. Bioconjugate Chem. 2012;23:392–398. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- 12.a) Taylor MT, Blackman ML, Dmitrenko O, Fox JM. J Am Chem Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang J, Seckute J, Cole CM, Devaraj NK. Angew Chem. 2012;124:7594–7597. [Google Scholar]; Angew Chem Int Ed. 2012;51:7476–7479. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rossin R, van den Bosch SM, Ten Hoeve W, Carvelli M, Versteegen RM, Lub J, Robillard MS. Bioconjugate Chem. 2013;24:1210–1217. doi: 10.1021/bc400153y. [DOI] [PubMed] [Google Scholar]
- 13.Sauer J, Heinrichs G. Tetrahedron Lett. 1966;7:4979–4984. [Google Scholar]
- 14.a) Dowd P, Gold A. Tetrahedron Lett. 1969;10:85–86. [Google Scholar]; b) Yan N, Liu X, Pallerla MK, Fox JM. J Org Chem. 2008;73:4283–4286. doi: 10.1021/jo800042w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Pan Y, Wang Z, Wang J, Lin Q. Angew Chem. 2012;124:10752–10756. [Google Scholar]; Angew Chem Int Ed. 2012;51:10600–10604. doi: 10.1002/anie.201205352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc. 2012;134:18638–18643. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 17.Liang Y, Mackey JL, Lopez SA, Liu F, Houk KN. J Am Chem Soc. 2012;134:17904–17907. doi: 10.1021/ja309241e. [DOI] [PubMed] [Google Scholar]
- 18.Cole CM, Yang J, Seckute J, Devaraj NK. Chem Bio Chem. 2013;14:205–208. doi: 10.1002/cbic.201200719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moerdyk JP, Bielawski CW. J Am Chem Soc. 2012;134:6116–6119. doi: 10.1021/ja3014105. [DOI] [PubMed] [Google Scholar]
- 20.Thalhammer F, Wallfahrer U, Sauer J. Tetrahedron Lett. 1990;31:6851–6854. [Google Scholar]
- 21.Sauer J, Heldmann DK, Hetzenegger J, Krauthan J, Sichert H, Schuster J. Eur J Org Chem. 1998:2885–2896. [Google Scholar]
- 22.Karver MR, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2011;22:2263–2270. doi: 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seckute J, Yang J, Devaraj NK. Nucleic Acids Res. 2013;41:e148. doi: 10.1093/nar/gkt540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Hutchins JE, Fife TH. J Am Chem Soc. 1973;95:3786–3790. doi: 10.1021/ja00792a051. [DOI] [PubMed] [Google Scholar]; b) Cha SW, Gu HK, Lee KP, Lee MH, Han SS, Jeong TC. Toxicol Lett. 2000;115:173–181. doi: 10.1016/s0378-4274(00)00176-4. [DOI] [PubMed] [Google Scholar]; c) Sogorb MA, Vilanova E. Toxicol Lett. 2002;128:215–228. doi: 10.1016/s0378-4274(01)00543-4. [DOI] [PubMed] [Google Scholar]
- 25.a) Silverman AP, Kool ET. Chem Rev. 2006;106:3775–3789. doi: 10.1021/cr050057+. [DOI] [PubMed] [Google Scholar]; b) Xu Y, Karalkar NB, Kool ET. Nat Biotechnol. 2001;19:148–152. doi: 10.1038/84414. [DOI] [PubMed] [Google Scholar]; c) Xu Y, Kool ET. Nucleic Acids Res. 1999;27:875–881. doi: 10.1093/nar/27.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Audebert P, Sadki S, Miomandre F, Clavier G. Electrochem Commun. 2004;6:144–147. [Google Scholar]; b) Audebert P, Sadki S, Miomandre F, Clavier G, Vernieres MC, Saoud M, Hapiot P. New J Chem. 2004;28:387–392. [Google Scholar]; c) Clavier G, Audebert P. Chem Rev. 2010;110:3299–3314. doi: 10.1021/cr900357e. [DOI] [PubMed] [Google Scholar]; d) Janowska I, Miomandre F, Clavier G, Audebert P, Zakrzewski J, Thi KH, Ledoux-Rak I. J Phys Chem A. 2006;110:12971–12975. doi: 10.1021/jp062186f. [DOI] [PubMed] [Google Scholar]
- 27.a) Burland DM, Carmona F, Pacansky J. Chem Phys Lett. 1978;56:221–226. [Google Scholar]; b) Chavez DE, Hanson SK, Veauthier JM, Parrish DA. Angew Chem. 2013;125:7014–7017. doi: 10.1002/anie.201302128. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:6876–6879. doi: 10.1002/anie.201302128. [DOI] [PubMed] [Google Scholar]; c) Chavez DE, Hiskey MA, Gilardi RD. Angew Chem. 2000;112:1861–1863. doi: 10.1002/(sici)1521-3773(20000515)39:10<1791::aid-anie1791>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:1791–1793. doi: 10.1002/(sici)1521-3773(20000515)39:10<1791::aid-anie1791>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; d) Yuasa J, Fukuzumi S. Chem Commun. 2006:561–563. doi: 10.1039/b513110c. [DOI] [PubMed] [Google Scholar]
- 28.Sauer J, B3uerlein P, Ebenbeck W, Gousetis C, Sichert H, Troll T, Utz F, Wallfahrer U. Eur J Org Chem. 2001:2629–2638. [Google Scholar]
- 29.Steigel A, Sauer J, Binsch G, Kleier DA. J Am Chem Soc. 1972;94:2770–2779. [Google Scholar]
- 30.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian Inc; Wallingford, CT: 2010. [Google Scholar]
- 31.a) Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. [Google Scholar]; b) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 32.a) Paton RS, Mackey JL, Kim WH, Lee JH, Danishefsky SJ, Houk KN. J Am Chem Soc. 2010;132:9335–9340. doi: 10.1021/ja1009162. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lan Y, Zou LF, Cao Y, Houk KN. J Phys Chem A. 2011;115:13906–13920. doi: 10.1021/jp207563h. [DOI] [PubMed] [Google Scholar]
- 33.a) Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; b) Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199–9208. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J Am Chem Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d. [DOI] [PubMed] [Google Scholar]
- 34.a) Liu F, Paton RS, Kim S, Liang Y, Houk KN. J Am Chem Soc. 2013;135:15642–15649. doi: 10.1021/ja408437u. [DOI] [PubMed] [Google Scholar]; b) Paton RS, Kim S, Ross AG, Danishefsky SJ, Houk KN. Angew Chem. 2011;123:10550–10552. doi: 10.1002/anie.201103998. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:10366–10368. doi: 10.1002/anie.201103998. [DOI] [PubMed] [Google Scholar]
- 35.Houk KN. Acc Chem Res. 1975;8:361–369. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.