

Abstract
Primary sensory afferents and their neighboring host-defense cells are a rich source of lipid-derived mediators that contribute to the sensation of pain caused by tissue damage and inflammation. But an increasing number of lipid molecules have been shown to act in an opposite way, to suppress the inflammatory process, restore homeostasis in damaged tissues and attenuate pain sensitivity by regulating neural pathways that transmit nociceptive signals from the periphery of the body to the central nervous system.
The ‘gate control theory’ of pain postulates that neural circuits in the central nervous system (CNS) dynamically regulate nociceptive signals arising in the periphery of the body. Since its formulation in 19651, this theory has provided a framework in which to interpret the actions of central analgesic circuits, such as those recruited during acute stress or placebo responses. Emerging evidence indicates, however, that nociceptive signals may be subject to a dynamic filtering process even before they reach the spinal cord. Primary sensory neurons and the host-defense cells surrounding them release a variety of analgesic factors that control the traffic of nociceptive information to the CNS (Figure 1). Lipid-derived mediators are crucial players, albeit not the only ones2, in this peripheral gating mechanism. Here, we review the properties of endogenous bioactive lipids that modulate pain initiation by interacting with receptor systems on primary sensory afferents and neighboring cells that are not neurons – including macrophages, mast cells and keratinocytes. We also outline stages of tissue injury and inflammation at which these analgesic lipids may intervene, and speculate on how their actions might be exploited to discover better medicines for pain.
Figure 1. Peripheral gating of nociceptive signals by endogenous lipid mediators.

From the dorsal root ganglia (DRG), where the cell bodies of C and Aδ sensory neurons are located, nociceptive fibers travel both peripherally (to innervate skin, bone, viscera and other internal organs) and centrally (to form synapses with neurons in the dorsal horn of the spinal cord). Peripheral nociceptor terminals respond to acute injury directly, through cell-surface receptors specialized in detecting harmful physical and chemical stimuli, as well as indirectly, through receptors that sense proalgesic (pain-inducing) factors released by cell damage, inflammation, neuropathy or tumor growth (e.g., nucleotides, peptides and various lipid mediators). These sensitizing effects may be modulated by analgesic lipid-derived mediators generated at different stages of the tissue response to damage and under the control of neural and hormonal signals (e.g., autonomic nervous system, circulating corticosteroids). Some of these compounds may be released from the sensory nerve endings themselves, while others may derive from local host-defense cells, such as resident mast cells and macrophages, epidermal keratinocytes and blood-borne leukocytes recruited to the injury site. Nociceptive nerve endings integrate the inputs provided by proalgesic (excitatory) and analgesic (inhibitory) signals, converting them into graded receptor potentials and, eventually, all-or-none action potentials. Alterations in the production of analgesic lipid mediators may occur not only at the nerve endings of primary afferents, but also within the DRG and dorsal horn of the spinal cord, with neurons and other cells (e.g., satellite cells, endoneurial macrophages, microglia) likely contributing to this response.
Proalgesic lipids
The cell bodies of nociceptors – the primary sensory neurons specialized in detecting harmful stimuli – are housed in anatomical structures located outside the CNS, the trigeminal and dorsal root ganglia (DRG), and generate axonal stalks that split into two sets of fibers running in opposite directions. One reaches the skin and most internal organs, with the exception of the brain, where it senses noxious stimuli and converts them into electrical signals. The other set of fibers extends toward the dorsal horn of the spinal cord to form synapses with local neurons, which process the information arriving from the periphery and transmit it to supraspinal sites. Nociceptors are divided into two broad classes with distinct structure and function. ‘Aδ’ nociceptors, which have medium-sized cell bodies and fast-conducting axons, mediate the localized sharp pain sensation that serves as a warning signal of injury. ‘C’ nociceptors, on the other hand, have small-diameter cell bodies and slow-conducting axons, and convey the poorly localized delayed pain that supports tissue repair by inducing defensive behaviors. Many (but not all) nociceptors are considered to be polymodal because they share the ability to recognize as harmful external inputs of widely different modalities – thermal, chemical and mechanical. At the root of this perceptual flexibility is the existence of multiple nociceptor subclasses, each expressing a distinctive repertoire of membrane ion channels, receptors and intracellular signaling proteins. For example, the presence of cation channels of the transient receptor potential (TRP) family renders nociceptors differentially sensitive to heat (TRPV1), cold (TRPM8) and chemical irritants (TRPA1). After tissue damage, this heterogeneous palette of transduction molecules undergoes profound changes, giving rise to a state of hyper-excitability, called peripheral sensitization, in which non-noxious events are perceived as noxious (allodynia) and mildly noxious events are perceived as highly noxious (hyperalgesia). Sensitization is often accompanied by neurogenic inflammation – a local vasodilatory response caused by release of substance P and calcitonin gene-related peptide (CGRP) from primary afferent terminals – and by activation of a group of primary sensory neurons, called silent nociceptors, which becomes responsive to harmful stimuli only after tissue damage has occurred3.
In addition to external stimuli, nociceptors are exquisitely sensitive to a number of endogenous proalgesic (pain-inducing or pain-enhancing) factors that either are quickly released after an injury or produced more slowly during inflammation, peripheral neuropathy and tumor growth (Figure 1). Intracellular nucleotides such as ATP and ADP, which spill out of damaged cells, and peptides such as bradykinin, which is liberated from a plasma globulin during blood clotting, may supply a first wave of proalgesic chemicals by virtue of their ability to activate excitatory receptors on primary afferents. A second, slower wave of sensitizing and proinflammatory agents includes substance P and CGRP as well as a diverse array of bioactive lipid-derived mediators generated by primary afferents and close-by host-defense cells. The key roles that two such mediators, prostaglandin E2 (PGE2) and prostacyclin (PGI2), play in nociceptor sensitization were first identified in the 1970s4 and are now firmly established5. PGE2 and PGI2 are produced at sites of inflammation and activate specific G protein-coupled receptors on sensory neurons to increase membrane excitability and enhance secretion of substance P and CGRP6. The rate-limiting step in this signaling cascade is provided by two functionally similar, but molecularly distinct enzymes – cyclooxygenase (Cox)-1 and Cox-2 – which convert the membrane-derived polyunsaturated fatty acid (PUFA), arachidonic acid, into PGH2, a common precursor for all prostanoids7. By interrupting Cox-1 and Cox-2 activities, non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen suppress primary afferent sensitization and strongly reduce inflammatory pain4. In addition to Cox-1 and Cox-2 products, many other lipid molecules formed through enzymatic or non-enzymatic PUFA oxidation are able to excite nociceptors and cause hyperalgesic states. Recently described examples are the hydroxylated derivatives of linoleic acid8, hepoxilin A39, PGE2-glycerol ester10, and prostamide F2α11. Other lipid-derived proalgesic agents include lysophosphatidic acid6 and lysophosphatidyl inositol12. While the specific functions served by each of these substances are in most cases unclear, the overall significance of lipid-mediated signaling in the induction and maintenance of persistent pain is widely recognized3. Research in the last ten years has unexpectedly revealed, however, the existence of bioactive lipids that contrast the effects of proalgesic agents and modulate pain initiation at sites of injury and inflammation. These analgesic lipid mediators – which include endogenous cannabinoids, lipid-amide agonists of peroxisome proliferator-activated receptor-α (PPAR-α), and various products of oxidative PUFA metabolism (Table 1) – play non-redundant roles in the dynamic modulation of peripheral responses to noxious stimuli.
Table 1.
Analgesic lipid-derived mediators produced at sites of acute tissue injury, inflammation and neuropathy.
| Lipid Mediator | Metabolic Precursor | Biosynthetic Enzyme | Molecular Target | Select Ref. |
|---|---|---|---|---|
| Anandamide | N-arachidonoyl-PE | NAPE-PLD | CB1 | 30 |
| 2-Arachidonoyl-sn-glycerol | PIP2 | PLC-β and DGL-α | CB1 and CB2 | 5 |
| Palmitoyl ethanolamide | N-palmitoyl-PE | NAPE-PLD | PPAR-α | 30, 72 |
| Oleoyl ethanolamide | N-oleoyl-PE | NAPE-PLD | PPAR-α | 69, 72 |
| Resolvin D1 | DHA | 15-lipoxygenase and 5-lipoxygenase | GPR32 | 93 |
| Resolvin E1 | EPA | Cytochrome P450 and 5-lipoxygenase | ChemR23 | 93 |
| Lipoxin A4 | Arachidonic acid | 15-lipoxygenase and 5-lipoxygenase | ALX, CB1 | 88, 92 |
| Epoxyeicosa-trienoic acids | Arachidonic acid | Cytochrome P450 | Unknown | 89 |
Abbreviations: ALX, lipoxin A4 receptor; CB1 and CB2, type-1 and -2 cannabinoid receptors; ChemR23, Chemerin receptor-23; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; GPR32, G protein-coupled receptor-32; NAPE-PLD, N-acyl-phosphatidylethanolamine-selective phospholipase D; PE, phosphatidylethanolamine; PIP2, phosphatidylinositol-4,5-bisphosphate; PPAR-α, peroxisome proliferator-activated receptor type-α; TRPV1, transient receptor potential vanilloid-1 channel.
Endogenous cannabinoids: antinociceptive response to injury
Neural and non-neural cells in damaged and inflamed tissues produce a family of arachidonic acid derivatives, called endocannabinoids, which suppress sensitization and neurogenic inflammation by activating CB1 and CB2 cannabinoid receptors – the same Gi/o protein-coupled receptors that are targeted by Δ9-tetrahydrocannabinol in marijuana13.
Cannabinoid receptors
CB1 receptors are highly expressed in the CNS, where they are concentrated in axon terminals of inhibitory interneurons and excitatory projection neurons14. Thanks to this presynaptic localization and to their ability to modulate calcium and potassium channel activities, CB1 receptors have important functions in the regulation of neurotransmitter release in the brain and spinal cord14. Substantial levels of CB1 expression are also found in nociceptive and non-nociceptive sensory neurons of the DRG15,16,17,18 and trigeminal ganglion19, as well as in host-defense cells that make contact with those neurons (e.g., macrophages, mast cells and epidermal keratinocytes)20,21 (Figure 2). Experimental interventions that evoke persistent hyperalgesia in animals are associated with profound changes in CB1 expression in spinal and supraspinal structures of the CNS22. Similar alterations have been shown to occur in the DRG. For example, prolonged peripheral inflammation elicited by injection of complete Freund’s adjuvant (CFA) in the rat hind paw, is accompanied by elevation of CB1 receptor density in dermal nerve endings and cell bodies of nociceptive neurons that project to that paw23. Similarly, growth of painful histolytic tumors in the mouse heel bone is associated with increased CB1 expression in nociceptors that innervate the bone and its surrounding tissues24.
Figure 2. Endocannabinoid control of peripheral nociception.

Acute tissue injury and inflammation stimulate the localized release of anandamide (AEA) and 2-arachidonoyl-sn-glycerol (2-AG) from primary sensory afferents and neighboring non-neural cells – including macrophages, mast cells and blood-derived platelets. AEA release from its membrane precursor, N-arachidonoyl-phosphatidylethanolamine (NArPE), may be initiated by calcium entry in nociceptors and/or stimulation of Toll-like receptor-4 (TLR4) in macrophages. 2-AG mobilization from phosphatidylinositol-4,5-bisphosphate (PIP2) may be stimulated by activation of B2 bradykinin receptors in nociceptors, platelet activating factor (PAF) receptors in macrophages, and/or TLR4 in platelets. Newly formed AEA and 2-AG activate CB1 and/or CB2 cannabinoid receptors modulating membrane excitability and calcium signals in primary afferent terminals, stimulating the secretion of β-endorphin (β-endo) from keratinocytes (which binds to μ-opioid receptors [μOR] on nociceptors), and inhibiting macrophage activation and mast-cell degranulation. FcεRI is the high affinity immunoglobulin E receptor.
Studies in primary cultures of DRG neurons have shown that cannabinoid receptor agonists – dual CB1/CB2 ligands such as CP-55940 and Win-55212-2 or CB1-selective ligands such as arachidonyl-2-chloroethylamide (ACEA) – inhibit voltage-gated calcium channels25 and acid-sensing ion channels (ASICs)26, reduce calcium transients evoked by capsaicin activation of TRPV124,27, and block nerve growth factor-induced TRPV1 sensitization28. Selective CB1 receptor antagonists (e.g., rimonabant and AM251) prevent these effects27,28,29. Consistent with those results, in vivo pharmacological studies have demonstrated that cannabinoid agonists suppress nociceptive behaviors and CGRP release in rodents through a peripheral CB1-mediated mechanism30,31,32,33. The peripheral nature of these actions is strikingly illustrated by results obtained in mutant mice that specifically lack CB1 in nociceptive neurons17,18. Compared to their wild-type littermates, these mice show decreased sensitivity to the analgesic effects exerted by local or systemic (but not intrathecal) administration of cannabinoid agents. Importantly, these mice also display an exaggerated hyperalgesic response to CFA injection, suggesting that CB1 receptors on primary afferents modulate the initiation of inflammatory pain.
The second cannabinoid receptor subtype, CB2, has limited structural similarity with CB1 (the two proteins are only 40% identical) and a distinctive distribution in mammalian tissues13. Substantial CB2 receptor levels are found in cells of hematopoietic origin, including those interacting with primary afferents during tissue injury (e.g., macrophages and mast cells)20,34 (Figure 2). CB2 receptors are sparsely expressed in the brain, spinal cord and DRG of healthy rats and mice, but their numbers increase markedly after peripheral nerve damage35,36. For example, ligation of the L5–L6 spinal nerve and transection or compression of the sciatic nerve all heighten CB2 expression in sensory neurons of rodent DRG37,38. Moreover, CB2 receptors have been identified in TRPV1-positive nociceptive neurons of human DRG, where their expression is also stimulated by nerve damage39. In line with these morphological findings, CB2 activation by selective agonists (e.g. AM1241 and JWH-133) exerts profound anti-nociceptive effects in animal models of acute, inflammatory and neuropathic pain36,40,41. That such effects may be primarily due to a peripheral mechanism is not only suggested by the localization of CB2 receptors, outlined above, but also by experiments showing that administration of CB2 agonists into the rat paw suppresses hyperalgesia evoked by injection of capsaicin and reduces the response of spinal neurons to mechanical stimulation42,43. Both neural and non-neural cell populations are likely to contribute to these effects, as indicated by the ability of CB2 agonists to attenuate calcium transients elicited by capsaicin in rat and human DRG neurons27,39, enhance release of β-endorphin from keratinocytes44, and reduce the secretion of immune mediators from macrophages and mast cells45 (Figure 2).
In addition to CB1 and CB2, endocannabinoid lipids are also known to interact with non-cannabinoid sites, such as TRPV1 channels and G protein-coupled GPR55 receptors. These interactions have attracted a great deal of attention and are discussed in several recent reviews46,47, but their relevance to the peripheral control of nociception is still unclear.
Endogenous cannabinoids
The two best-known members of the endocannabinoid family of lipid mediators, anandamide and 2-arachidonoyl-sn-glycerol (2-AG), are produced through distinct biochemical pathways, each starting with the enzyme-mediated hydrolysis of a specific phospholipid precursor in cell membranes48. Anandamide is formed by cleavage of a relatively uncommon and still poorly understood lipid molecule, in which phosphatidylethanolamine (PE) is covalently linked to arachidonic acid via an amide bond (Figure 3). In neurons, this reaction is stimulated by calcium ions and is catalyzed by a structurally unique phospholipase D (PLD) that exclusively recognizes N-acyl-substituted PE species49. In macrophages, on the other hand, the same reaction may occur in two separate steps: a molecularly uncharacterized phospholipase C (PLC) converts N-arachidonoyl-PE into phospho-anandamide, which then loses its phosphate group by the action of a phosphatase29. Newly formed anandamide acts near its sites of production, as an autocrine or paracrine messenger (Figure 2), and is rapidly eliminated through a process consisting of carrier-mediated transport into cells (the molecular mechanism of which has so far resisted elucidation)50 followed by intracellular breakdown to arachidonic acid and ethanolamine, catalyzed by the serine hydrolase fatty acid amide hydrolase (FAAH)48,49. Alternatively, anandamide may be directly transformed by Cox-2 (but not Cox-1) into proalgesic prostamides11 (Figure 3). A novel class of Cox-2 inhibitors that specifically stop this reaction, without affecting the oxygenation of arachidonic acid to prostaglandins, has been recently disclosed51.
Figure 3. Formation and deactivation of endocannabinoid lipids.

(A) Anandamide is generated by hydrolysis of the membrane lipid, N-arachidonoyl-phosphatidylethanolamine (NArPE), which is catalyzed in neurons by a phospholipase D that selectively recognizes N-acylated species of PE (NAPE-PLD). The concentrations of NArPE and anandamide are very low in resting neurons, but quickly increase in response to neural activity and/or neurotransmitter receptor occupation. In macrophages, activation of Toll-like receptor 4 (TLR4) by the bacterial endotoxin lipopolysaccharide stimulates an as-yet-uncharacterized phospholipase C (PLC), which converts NArPE into phospho-anandamide. This intermediate is dephosphorylated by a phosphatase (P-ase) to form anandamide. Endogenously produced anandamide modulates nociception by binding to CB1 cannabinoid receptors. In both neurons and macrophages, anandamide is converted into arachidonic acid and ethanolamine by fatty acid amide hydrolase (FAAH), which is selectively blocked by inhibitors such as the O-arylcarbamate derivative, URB597. Anandamide can also be transformed by cyclooxygenase-2 (Cox-2) into proalgesic prostamides, a reaction that is prevented by substrate-selective inhibitors such as LM-4131. Non-steroidal anti-inflammatory drugs (NSAIDs) stop the conversion of arachidonic acid into prostaglandins by inhibiting both Cox-1 and Cox-2 activities.
(B) 2-arachidonyl-sn-glycerol (2-AG). Gq-coupled receptors and Toll-like receptor-4 (TLR4) stimulate phospholipase C-β (PLC-β), which converts phosphatidylinositol-4,5-bisphosphate (PIP2) into 1,2-diacylglycerol (DAG). DAG is hydrolyzed by diacylglycerol lipase-α (DGL-α) to yield 2-AG, which inhibits nociceptive responses by activating CB1 and CB2 cannabinoid receptors. Monoacylglycerol lipase (MGL) terminates 2-AG signaling by hydrolyzing this glycerol ester into arachidonic acid and glycerol. 2-AG and arachidonic acid may be transformed by cyclooxygenase (Cox-1) and/or Cox-2 into various families of proalgesic lipid mediators, which include PGE2 glycerol ester (derived from 2-AG) and PGE2 (derived from arachidonic acid).
Inflammation and nerve injury cause marked changes in anandamide mobilization. For example, in mice and rats, intraplantar injection of CFA17 or ligation of the sciatic nerve increases anandamide levels in the affected paw40, whereas an opposite effect is produced by tumor implantation in the heel bone52 or topical skin application of a pro-inflammatory phorbol ester53. Importantly, marked changes in anandamide levels have also been documented in biopsies of human subjects with acute pancreatitis54. The molecular mechanisms underlying such alterations are still incompletely understood. Heightened anandamide mobilization may be due to enhanced calcium entry in nociceptors and/or to stimulation of Toll-like receptor-4 (TLR4) in macrophages29 (Figure 2) [TLR4 is a member of the pattern recognition family of receptors, which recognize exogenous pathogen-associated molecules, such as lipopolysaccharide (LPS), as well as endogenous damage-associated molecules, such as uric acid]. Conversely, lowered anandamide availability may result from injury-induced changes in FAAH expression. In the DRG of healthy rats, FAAH is localized to small-diameter C nociceptors, but peripheral nerve damage (sciatic nerve axotomy or lumbar spinal nerve transection) induces its ectopic expression in large-diameter sensory neurons55. Moreover, growth of painful bone tumors is associated with enhanced FAAH expression in the paw bearing the tumor and the DRG innervating it24. This effect – along with a parallel, but independent increase in carrier-mediated anandamide uptake by neurons and/or other cells of the DRG56 – may be responsible for the observed deficit in anandamide mobilization52.
Irrespective of the regulatory mechanisms involved, anandamide released at sites of injury may modulate nociceptive signals by activating local CB1 receptors. Experiments with pharmacological agents that block intracellular FAAH activity support this idea. For example, intraplantar delivery of the brain-penetrant FAAH inhibitor URB597, at doses that presumably do not interfere with FAAH activity in the CNS, reduces mechanically evoked responses of rat spinal neurons through a mechanism that requires CB1 activation35. Furthermore, systemic administration of the peripherally restricted FAAH inhibitor, URB937, which selectively interrupts anandamide degradation outside the brain and spinal cord57, causes striking anti-hyperalgesic effects in rodent models of visceral and inflammatory pain57,58. Such effects are exclusively dependent on peripheral FAAH blockade and are prevented by the CB1 antagonists, rimonabant and AM251, but not by the CB2 antagonist AM63057,58. Interestingly, FAAH inhibitors act in a synergistic manner with NSAIDs to attenuate pain-related behaviors elicited, in mice, by carrageenan or sciatic nerve constriction58,59. This super-additive interaction underscores the deep functional links existing between endocannabinoids and prostanoids, as well as the interest in developing dual inhibitors that target both FAAH and Cox activities.
Unlike anandamide, 2-AG derives from the hydrolysis of a well-known inositol-containing phospholipid, phosphatidylinositol-4,5-bisphosphate (PIP2), the centerpiece of a lipid pathway that generates multiple intracellular and transcellular messengers48 (Figure 3). The first reaction in this pathway is catalyzed by the β-isoform of PLC (PLC-β), which is activated by Gq-coupled receptors and converts PIP2 into the second messenger 1,2-diacylglycerol (DAG). DAG regulates protein kinase C and other cellular effectors, but also serves as substrate for the hydrolytic activity of diacylglycerol lipase-α (DGL-α), which cleaves DAG into 2-AG48. The serine hydrolase, monoacylglycerol lipase (MGL), is the main enzyme involved in terminating 2-AG-mediated endocannabinoid signaling, which it accomplishes by hydrolyzing this glycerol ester and releasing free arachidonic acid. Like anandamide, 2-AG may be metabolized by Cox-2 to produce proalgesic derivatives such as PGE2 glycerol ester10.
In the brain and spinal cord, 2-AG mobilization is controlled by receptors linked to transducing Gq proteins46,60 and is crucially involved in the descending modulation of pain during acute stress61. Similarly, in cultures of rat DRG neurons, the proalgesic peptide bradykinin causes a rapid release of 2-AG, presumably through activation of B2 bradykinin receptors62, while platelet-activating factor (PAF) produces a similar effect in Raw264.7 macrophages by engaging PAF receptors63. In addition to these Gq-mediated responses, generation of 2-AG may be initiated, in rat platelets, by TLR4 activation64. Mechanisms such as those described above probably underlie the localized increase in 2-AG levels observed in CFA-inflamed mouse paws17. The relevance of localized 2-AG release to nociceptive signaling is confirmed by the finding that intraplantar injections of MGL inhibitors (URB602 or JZL-184) counteract the pain-related behaviors evoked by formalin administration in rats65 or tumor growth in mice52. The dependence of these effects on CB2 receptor activity52,66 distinguishes MGL inhibitors from FAAH inhibitors, and indicates that 2-AG and anandamide may influence pain initiation through complementary, rather than overlapping mechanisms (Figure 3).
In summary, many lines of evidence suggest that endocannabinoid lipids are rapidly recruited during tissue injury to provide a first line of control over arising nociceptive signals. The therapeutic opportunity offered by this peripheral gating mechanism – the possibility of achieving effective analgesia without causing untoward central effects – has fueled intense efforts to discover new agents that influence endocannabinoid signaling outside the CNS (Table 2). Such efforts are not without challenges, however. Among the problems to be faced are the selection of specific endocannabinoid mechanisms that must be targeted to achieve adequate analgesic efficacy (e.g., peripheral CB1 vs CB2 agonism, global vs peripheral FAAH inhibition), and the identification of clinical pain conditions that would most likely respond to pharmacological interference with those mechanisms.
Table 2. Peripheral lipid-mediated signaling as a source of new analgesic medicines.
The current therapy of chronic pain relies heavily on opiates (e.g., morphine and oxycodone) and anticonvulsants (e.g., gabapentin and pregabalin), two classes of drugs that primarily act on neural circuits of the brain and spinal cord. These agents help many, but their use is associated with central side effects and abuse potential. The existence of peripheral lipid-mediated mechanisms that regulate the flow of nociceptive signals to the central nervous system (CNS) offers the opportunity to develop medications that control pain without producing undesired central effects. Activation of CB1 cannabinoid receptors in the brain is psychotropic, but peripherally restricted CB1 agonists or globally active CB2 agonists (e.g., AM1241) reduce nociceptive responses in rodent models without eliciting overt signs of cannabinoid intoxication. Marked anti-nociceptive actions, mediated by CB1 and/or CB2 receptors, are also observed with inhibitors of the endocannabinoid-hydrolyzing enzymes, fatty acid amide hydrolase (FAAH, e.g. URB597 or URB937) and monoacylglycerol lipase (MGL, e.g. JZL184). PPAR-α-mediated antinociception can be achieved either using direct PPAR-α agonists, such as the endogenous ligand palmitoylethanolamide (PEA), or inhibitors of the PEA-hydrolyzing enzyme N-acylethanolamine acid amidase (NAAA, e.g. ARN077). Similar approaches may be used to exploit the analgesic properties of endogenous bioactive lipids generated by oxygenation of polyunsaturated fatty acids (PUFAs): inhibition of soluble epoxide hydrolase (sEH), which converts epoxygenated PUFAs into inactive dihydroxy-acids, and administration of resolvins both produce marked antinociception in rodent models.
| Molecular Target | Representative Probe | Chemical Structure | Select Ref. |
|---|---|---|---|
| Peripheral CB1 receptor | Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone |
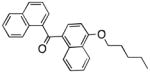
|
32 |
| CB2 receptor | AM1241 |
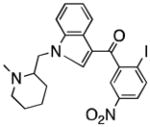
|
41 |
| FAAH | URB597 |
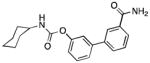
|
98 |
| Peripheral FAAH | URB937 |
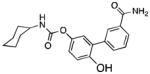
|
57 |
| MGL | JZL184 |
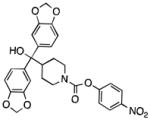
|
99 |
| PPAR-α | Palmitoylethanolamide |
|
76 |
| NAAA | ARN077 |
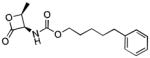
|
100 |
| Soluble EH | t-AUCB |
|
89 |
| CB1 receptor (Allosteric Modulation) | Lipoxin A4 |
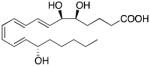
|
92 |
| ChemR32 | Resolvin E1 |
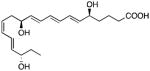
|
88 |
Endogenous PPAR-α agonists: homeostatic control of nociception
PPAR-α is a ligand-operated transcription factor that plays essential roles in energy metabolism and inflammation67. An unusually spacious ligand-binding pocket allows this receptor to recognize, albeit with different affinities, a variety of endogenous fatty-acid derivatives. These include low-potency agonists (e.g., free fatty acids), which act at mid-micromolar to high-micromolar concentrations68, and high-potency agonists [e.g., 8(S)-hydroxyeicosatetraenoic acid and amides of ethanolamine with saturated and monounsaturated fatty acids (FAE)], which act at submicromolar to single-digit micromolar concentrations69,70,71. The FAEs, oleoylethanolamide (OEA) and palmitoylethanolamide (PEA) (Figure 4), engage PPAR-α with relatively high potency (median effective concentration, EC50: 0.12 μM for OEA and 3 μM for PEA)69,70 and have emerged as potential homeostatic regulators of nociception.
Figure 4. Formation and deactivation of OEA and PEA in macrophages.

The fatty acid ethanolamides (FAEs), palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), are generated by cleavage of two distinct membrane phospholipids, N-palmitoyl-phosphatidylethanolamine (PE) and N-oleoyl-PE. This reaction is catalyzed by the same N-acyl PE-specific phospholipase D (NAPE-PLD) that mediates anandamide release in neurons. Under basal conditions, macrophages produce substantial amounts of N-palmitoyl-PE, N-oleoyl-PE and the corresponding FAEs, which inhibit inflammatory responses and increases nociceptive thresholds by activating PPAR-α. In macrophages and, possibly, primary sensory neurons of the DRG, FAEs are converted into fatty acid and ethanolamine by the cysteine hydrolase, N-acylethanolamine acid amidase (NAAA), which is selectively inhibited by the threonine β-lactone ARN077. In neurons and other cells, FAE hydrolysis is primarily mediated by fatty acid amide hydrolase (FAAH), which is blocked by various classes of inhibitors, including the O-aryl carbamate URB597.
Peroxisome proliferator-activated receptor-α
Like other members of the nuclear receptor superfamily, PPAR-α is activated through ligand binding, which causes the formation of a multi-protein complex that comprises an obligatory partner, the retinoic acid receptor (RXR), along with a variable set of protein co-activators67. In its active form, PPAR-α binds to responsive elements on DNA, enhancing the transcription of various anti-inflammatory proteins, such as inhibitor of κB-α (IκB-α), while concomitantly interrupting the activity of pro-inflammatory transcription factors through direct protein-protein interactions (a process called ‘transrepression’)67. Transcription regulators modulated by PPAR-α include nuclear factor κB (NF-κB), signal transducers and activators of transcription (STATs), activator protein 1 (AP1), and nuclear factor of activated T cells (NFAT)67. In addition to inducing changes in gene expression, which unfold over a period of hours or even days, PPAR-α activation may also produce rapid non-genomic effects, as briefly discussed below.
PPAR-α is expressed in many organs and tissues67 – including DRG neurons72, macrophages67 and other host-defense cells67,73. Consistent with this localization, local or systemic administration of synthetic PPAR-α agonists (e.g., GW7647 and Wy-14643) exerts profound anti-inflammatory effects in mice70,74,75, suppresses pain-related behaviors elicited in rats and mice by injection of carrageenan, formalin or magnesium sulfate72,74, and prevents formalin-induced firing of rat spinal cord neurons72. The endogenous PPAR-α agonist, PEA, has similar anti-hyperalgesic properties, which are synergistic with those of CB1 receptor agonists30,72,76. Additionally, PPAR-α activation strongly reduces thermal and mechanical hyperalgesia evoked in mice by nerve injury (constriction of the sciatic nerve) or inflammation (CFA injection)72. These anti-hyperalgesic effects are absent in PPAR-α–deficient mice72, which also show enhanced responses to several pro-inflammatory and proalgesic stimuli77,78, an important indication that PPAR-α participates in the tonic control of inflammation and nociception.
The analgesic actions of PPAR-α ligands appear within minutes of drug administration, suggesting that they may be mediated, at least initially, by a transcription-independent mechanism. In support of this idea, studies with selective inhibitors have identified two subtypes of calcium-activated potassium channels that may be rapidly regulated by PPAR-α: large-conductance BKca (Kca1.1) and intermediate-conductance IKca channels (Kca3.1)72 (Figure 5). BKca and IKca are known to modulate the excitability of primary sensory neurons79, but the molecular steps connecting PPAR-α activation to the gating of these channels remain to be defined. Anti-hyperalgesic activity may also result from the interaction of PPAR-α with transcription factors of the NFκB complex, which suppress the expression of genes targeted by immune-derived proalgesic mediators, including tumor necrosis factor-α (TNF-α) and interleukin 1β (IL1β) (Figure 5). Lastly, it is worth noting that PPAR-α agonists can also attenuate hyperalgesia after infusion into the cerebral ventricles80. This finding underscores the growing awareness about the role of the PPAR family of receptors (including PPAR-γ and PPAR-α) in the central modulation of pain signals81.
Figure 5. Control of peripheral nociception by endogenous PPAR-α agonists.

In the absence of stimulation, macrophages and dorsal root ganglia (DRG) neurons produce substantial amounts of oleoylethanolamide and palmitoylethanolamide. These fatty acid ethanolamides (FAEs) exert a tonic inhibitory control over macrophage activity and nociceptor excitability by recruiting PPAR-α-dependent mechanisms, which may include opening of BKca and IKca potassium channels and regulation of the nuclear factor-κB (NFκB) transcription complex (composed of p50, p65 and inhibitor of κB-α [IκB-α]). After tissue damage or during inflammation, microbial toxins, such as lipopolysaccharide (LPS), and/or immune-derived cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin 1β (IL1β), suppress FAE biosynthesis and stop FAE signaling at PPAR-α, temporarily disabling its inhibitory function. These molecular events may take place both in (A) neurons and other cells of the DRG, and (B) peripheral nerve endings of primary sensory neurons. In the periphery, FAEs may also influence tissue repair, by engaging PPAR-α in keratinocytes, and mast cell degranulation, possibly by interacting with G protein-coupled GPR55 receptors. Abbreviation: NFκBRE: NFκB responsive elements.
Endogenous agonists of PPAR-α
OEA and PEA are chemically related to anandamide and, in neurons, they are produced by the activity of the same PLD that catalyzes anandamide release82 (Figure 4). Nevertheless, the mechanisms governing the formation of these FAEs differ from those involved in the production of anandamide in two important aspects. First, many cell types in the body – including DRG neurons and skin cells – generate substantial amounts of OEA and PEA even in the absence of external stimuli, which are required instead to trigger on-demand anandamide release. Because of this tonic production, the levels of OEA and PEA in healthy tissues are much higher than those of anandamide. For example, the baseline concentration of PEA in mouse ear skin is approximately 3.5 μM, more than 175 times greater than that of anandamide53. Thus, based on the known affinities of PPAR-α for OEA and PEA (EC50: 0.12–3 μM), it is reasonable to assume that a substantial fraction of PPAR-α is constitutively bound to FAEs in non-stimulated cells. Second, many pro-inflammatory stimuli dampen the production of OEA and PEA even as they trigger anandamide formation in the same cell. For example, macrophages exposed to the bacterial toxin LPS respond with a rapid burst of anandamide release29 followed by a slower persistent decrease in FAE content83,84. Experiments with Raw264.7 macrophages suggest that the PLC/phosphatase pathway mediates the initial spike in anandamide production29 (Figure 3), whereas the delayed decline in OEA and PEA results from a down-regulation in NAPE-PLD transcription due to reduced acetylation of histone proteins bound to the NAPE-PLD promoter84. The ability of tissue damage to suppress FAE mobilization has been documented using multiple animal models of inflammation and peripheral neuropathy53,56,70,83. Adding clinical relevance to these observations, one study found that the concentrations of OEA and PEA in synovial fluid are lower in subjects with rheumatoid arthritis and osteoarthritis than in healthy controls85.
The results outlined above suggest that endogenous FAE signaling at PPAR-α exerts a tonic inhibitory control over the induction of inflammatory and nociceptive responses. This idea was tested using pharmacological agents that selectively block the cysteine hydrolase, N-acylethanolamine acid amidase (NAAA)53,56,83, which catalyzes the deactivating hydrolysis of OEA and PEA in macrophages49 (Figure 4). Topical administration of the NAAA inhibitor, N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide, in mice reinstates normal PEA levels in activated leukocytes and blunts inflammatory responses induced by LPS or carrageenan83. Exogenous PEA mimics these effects while PPAR-α deletion abolishes them. Importantly, under the same conditions, the FAAH inhibitor URB597 fails to normalize PEA levels, highlighting the critical role of NAAA in PEA and OEA degradation by innate-immune cells. A second NAAA inhibitor, the compound ARN077, attenuates heat hyperalgesia and mechanical allodynia elicited in mice and rats by carrageenan injection, sciatic nerve ligation or ultraviolet B-radiation53. These anti-nociceptive effects are absent in PPAR-α-deficient mice and are prevented, in rats, by the PPAR-α antagonist GW6471. Similarly, ARN077 reduces mechanical hyperalgesia in fibrosarcoma-bearing mice and corrects, through a mechanism mediated by PPAR-α abnormal calcium signaling in DRG neurons co-cultured with fibrosarcoma cells86. Confirming its proposed mechanism of action, ARN077 restores baseline FAE levels in diseased tissues of neuropathic and tumor-bearing mice53,86.
To sum up, a plausible interpretation of the available data is that endogenous FAEs acting at PPAR-α help maintain host-defense homeostasis by preventing the launch of inappropriate inflammatory and nociceptive reactions (Figure 5). Thus, the function of these lipid mediators may be compared to that of an electronic high-pass filter, which attenuates signals that fail to reach a set threshold. Two strategies may be used to exploit this mechanism for therapeutic purposes (Table 2). The first is to directly activate PPAR-α using synthetic or endogenous ligands67,76. Though theoretically possible, this approach is riddled with practical problems due to the safety risks posed by excessive direct-agonist activation of PPAR-α87, and the inherent complexity of adapting natural lipid molecules to therapeutic usage (e.g., quick metabolism, high plasma protein binding, low solubility). An alternative strategy is to create small drug-like molecules that enhance intrinsic PPAR-α signaling by interrupting the NAAA-mediated degradation of OEA and PEA 53,83,86. Since the main effect of NAAA inhibitors is to normalize FAE signaling in injured or inflamed tissues, rather than indiscriminately activating PPAR-α throughout the body, this approach may offer a degree of cellular selectivity that might translate into greater efficacy and safety.
Bioactive products of PUFA oxygenation: return to nociceptive homeostasis
The lipid profile of an exudate – the fluid that filters from the blood into a site of inflammation – undergoes dramatic changes in the course of a self-limited inflammatory reaction. As this reaction phases out into resolution, the initial complement of potent proalgesic and proinflammatory lipid mediators – prostanoids such as PGE2 and PGI2, and leukotrienes such as LTB4 – is replaced by a different set of lipid molecules that promote the return of the inflamed tissue to homeostasis by stopping neutrophil infiltration, accelerating the clearance of cellular debris, enhancing the antimicrobial activity of epithelial cells, and stimulating wound healing88. As part of their effort to restore homeostasis, these bioactive lipids may also normalize nociceptive signaling by binding to selective receptors present on the surface of neurons and immune cells.
Epoxygenated fatty acids
The cytochrome P450 family of enzymes catalyzes the oxygenation of various long-chain PUFAs – including arachidonic, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) – each giving rise to a distinct group of bioactive epoxide-containing fatty acids (EpFAs), which act near their site of synthesis to regulate vascular tone, kidney function and inflammation89 (Figure 6). The EpFAs are quickly converted by the enzyme soluble epoxide hydrolase (sEH) into 1,2-dihydroxy-fatty acids, which are either less active or have different activities than the parent molecules. The discovery of potent and selective inhibitors of intracellular sEH activity has allowed researchers to explore the functions of endogenously produced EpFAs in animal models of inflammatory and neuropathic pain. These investigations have shown that systemic administration of sEH inhibitors or local injection of synthetically prepared EpFAs attenuate pain-related responses evoked by LPS or carrageenan in rats89. A more recent study has examined the impact of three structurally different sEH inhibitors in rats rendered hypersensitive to pain stimuli by treatment with streptozotocin (STZ), a toxin that causes type-1 diabetes by selectively killing insulin-secreting β-cells in the pancreas90. The inhibitors reduce mechanical allodynia in STZ-treated rats, without influencing normal nociceptive responding in healthy animals, and concomitantly increase the levels of non-metabolized EpFAs in plasma and spinal cord. Even though the receptor(s) responsible for these effects are still unknown, the marked anti-hyperalgesic properties of sEH inhibitors support a role for endogenous EpFAs in the modulation of nociceptive signaling, and identify sEH as a potential target for analgesic drugs89 (Table 2).
Figure 6. Biosynthetic pathways for analgesic PUFA-derived mediators.

This simplified scheme illustrates the biosynthetic pathways for representative polyunsaturated fatty acid (PUFA)-derived mediators involved in the peripheral control of nociception. (A) Biosynthesis and degradation of 5,6-epoxyeicosatrienoic acid (5,6-EET), a member of the epoxygenated fatty acid (EpFA) family of lipids. Cytochrome P450 converts arachidonic acid into 5,6-EET, which is subsequently hydrolyzed by soluble epoxide hydrolase (sEH) into 5,6-dihydroxyeicosatrienoic acid. (B–D) Biosynthetic pathways for (B) lipoxin A4, (C) resolvin E1, and (D) maresin 1. Abbreviations: LOX, lipoxygenase, Cox-2, cyclooxygenase-2.
Lipoxins, resolvins and allied compounds
The lipoxins are generated through the sequential enzyme-mediated oxygenation of membrane-derived arachidonic acid88 (Figure 6). Their biosynthetic routes are multiple and complex, and have been reviewed in detail elsewhere91. One important point to mention here is that several key pathways of lipoxin biosynthesis involve the cooperation among different cell types that physically contact one another during an inflammatory response. For example, neutrophils produce lipoxin A4 (LXA4), one of the best-known members of this class, utilizing as substrate a short-lived metabolite of arachidonic acid released from nearby macrophages88. This unusual mode of production, called transcellular biosynthesis, reflects the physiological need to match the complement of bioactive lipid mediators present at an inflammatory site with the varying cellular profiles progressively recruited to that site. The anti-inflammatory and pro-resolution effects of LXA4 are mediated by G protein-coupled ALX/FPR2 receptors (also called formyl peptide receptor-like 1, FPRL-1)88, but the attenuated nociception that accompanies such effects might also involve CB1 cannabinoid receptors. Pharmacological studies suggest that LXA4 acts as a positive allosteric CB1 modulator, potentiating CB1-mediated responses evoked by anandamide in vitro and in vivo and increasing the ability of this compound to displace the binding of a radiolabeled CB1 ligand to mouse brain membranes92.
Resolvins and protectins are produced, like the lipoxins, through the multi-step oxygenation of membrane-derived PUFAs; however, they do not utilize as precursor arachidonic acid, an omega-6 PUFA, but rather the omega-3 PUFAs, EPA and DHA (Figure 6). EPA gives rise to E-series resolvins such as resolvin E1 (RvE1), while DHA is converted into D-series resolvins, such as resolvin D1 (RvD1), as well as protectin D191. Additionally, in human macrophages and platelets, DHA may be transformed by 12-lipoxygenase to form maresin 1 (Figure 6)91. The pro-resolving and immune-modulating properties displayed by these bioactive lipids are impressive88, but are not sufficient to explain the array of anti-nociceptive effects exerted by some of them. For example, intraplantar administration of RvE1 or RvD1 in mice suppresses pain-related responses evoked by a broad variety of proalgesic stimuli – including formalin, capsaicin, CFA, TNF-α and PGE293,94. RvE1 and RvD1 are also highly effective at reducing nociceptive behaviors caused by skin incision or muscle retraction in mice and rats, two models of human post-operative pain94. The receptor mechanisms that mediate these analgesic actions are only partially understood. There is evidence that the Gi-coupled receptor ChemR23, which also binds the adipokine chemerin, is involved in the effects of RvE1. This idea is consistent with the presence of ChemR23 in DRG neurons and the ability of chemerin to mimic the anti-nociceptive actions of RvE193. RvD1 activates the LXA4 receptor, ALX/FPR2, but the functional significance of this interaction is still unclear95.
With their high pharmacological potencies and short life spans, lipoxins, resolvins and allied molecules fit the profile of ‘soft drugs’ – agents that undergo quick and predictable deactivating metabolism after having accomplished their therapeutic goal. Not surprisingly, therefore, pharmaceutical development in this area is focused on the use of resolvin analogs for the topical treatment of localized inflammatory conditions, such as dry eye (http://WWW.RESOLVYX.COM/products/index.asp). Nevertheless, there may be several pain-related applications for resolvin-based soft drugs, including the intra-articular treatment of pain arising from knee surgery or osteoarthritis.
Conclusions
The evidence presented in this article allows us to conjecture that bioactive lipid mediators regulate the access of nociceptive information to the CNS at three distinct stages. In healthy tissues, the tonic release of endogenous PPAR-α agonists, such as PEA and OEA, may help set the threshold for nociception by regulating the baseline transcriptional activity of the NFκB complex and the opening of membrane ion channels in primary sensory afferents and nearby host-defense cells. After an injury has occurred, the temporary interruption of OEA and PEA biosynthesis caused by cell damage and infection may disable the inhibitory influence exerted by these lipid messengers, allowing inflammation to unfold and nociceptive thresholds to decrease. At the same time, the localized on-demand formation of the endocannabinoids, anandamide and 2-AG, may mitigate the effects of exogenous and endogenous proalgesic agents by attenuating nociceptor excitability and contrasting local pro-inflammatory signals. Lastly, as the response to tissue damage moves toward its resolution phase, a wave of analgesic products of oxidative PUFA metabolism, such as lipoxins and resolvins, may help normalize nociceptive responses in the healing tissue. It is likely, but remains to be proven, that the correct deployment of this signaling program is subject to local and systemic regulatory mechanisms. At the local level, lipid mediators released in the early phases of injury may govern the generation of substances acting at later stages: this has been demonstrated for proresolving lipoxins, for example, the production of which is partly controlled by proalgesic Cox-2-derived prostaglandins88. At the system level, neural and hormonal signals may help integrate peripheral and central gating mechanisms during conditions of altered nociception, such as acute stress or body-wide inflammation. The autonomic nervous system, which modulates the biosynthesis of endogenous PPAR-α agonists in some peripheral tissues (e.g., white adipose)96, might play an important role in this context97. In addition to addressing such issues, future studies will also need to determine whether errors in the correct unfolding of the intrinsic analgesic mechanism outlined here might contribute to the development and maintenance of pathological pain states. Aided by continuing advances in mass spectrometry-based lipidomics, such studies may lead to the discovery of novel analgesic lipids and the development of innovative medicines that effectively control pain without interfering with central neurotransmission.
Acknowledgments
The authors thank Drs. Bruce D. Hammock, Andrea Hohmann and Charles N. Serhan for thoughtful discussions. Work in DP’s lab is funded in part by grants from the American Asthma Foundation and the National Institute on Drug Abuse, one of the National Institutes of Health.
References
- 1.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 2.Stein C, Machelska H. Modulation of peripheral sensory neurons by the immune system: implications for pain therapy. Pharmacol Rev. 2011;63:860–881. doi: 10.1124/pr.110.003145. [DOI] [PubMed] [Google Scholar]
- 3.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira SH. Prostaglandins, aspirin-like drugs and analgesia. Nat New Biol. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- 5.Murata T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 6.Park KA, Vasko MR. Lipid mediators of sensitivity in sensory neurons. Trends Pharmacol Sci. 2005;26:571–577. doi: 10.1016/j.tips.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50 (Suppl):S29–34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 2009;106:18820–18824. doi: 10.1073/pnas.0905415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregus AM, et al. Spinal 12-lipoxygenase-derived hepoxilin A3 contributes to inflammatory hyperalgesia via activation of TRPV1 and TRPA1 receptors. Proc Natl Acad Sci U S A. 2012;109:6721–6726. doi: 10.1073/pnas.1110460109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatta L, et al. Discovery of prostamide F2alpha and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS One. 2012;7:e31111. doi: 10.1371/journal.pone.0031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gangadharan V, et al. A novel biological role for the phospholipid lysophosphatidylinositol in nociceptive sensitization via activation of diverse G-protein signalling pathways in sensory nerves in vivo. Pain. 2013 doi: 10.1016/j.pain.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Yao B, Mackie K. Endocannabinoid receptor pharmacology. Curr Top Behav Neurosci. 2009;1:37–63. doi: 10.1007/978-3-540-88955-7_2. [DOI] [PubMed] [Google Scholar]
- 14.Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- 16.Mitrirattanakul S, et al. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114. doi: 10.1016/j.pain.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal N, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veress G, et al. Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct Funct. 2013;218:733–750. doi: 10.1007/s00429-012-0425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stander S, Schmelz M, Metze D, Luger T, Rukwied R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177–188. doi: 10.1016/j.jdermsci.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Sugawara K, et al. Cannabinoid receptor 1 controls human mucosal-type mast cell degranulation and maturation in situ. J Allergy Clin Immunol. 2013;132:182–193. doi: 10.1016/j.jaci.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Palazzo E, et al. Changes in cannabinoid receptor subtype 1 activity and interaction with metabotropic glutamate subtype 5 receptors in the periaqueductal gray-rostral ventromedial medulla pathway in a rodent neuropathic pain model. CNS Neurol Disord Drug Targets. 2012;11:148–161. doi: 10.2174/187152712800269731. [DOI] [PubMed] [Google Scholar]
- 23.Amaya F, et al. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain. 2006;124:175–183. doi: 10.1016/j.pain.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Khasabova IA, et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci. 2008;28:11141–11152. doi: 10.1523/JNEUROSCI.2847-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu YQ, et al. Cannabinoids inhibit acid-sensing ion channel currents in rat dorsal root ganglion neurons. PLoS One. 2012;7:e45531. doi: 10.1371/journal.pone.0045531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sagar DR, et al. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci. 2005;22:371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- 28.McDowell TS, Wang ZY, Singh R, Bjorling D. CB1 cannabinoid receptor agonist prevents NGF-induced sensitization of TRPV1 in sensory neurons. Neurosci Lett. 2013;551:34–38. doi: 10.1016/j.neulet.2013.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 31.Richardson JD, Kilo S, Hargreaves KM. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- 32.Dziadulewicz EK, et al. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone: a potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J Med Chem. 2007;50:3851–3856. doi: 10.1021/jm070317a. [DOI] [PubMed] [Google Scholar]
- 33.Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckley NE. The peripheral cannabinoid receptor knockout mice: an update. Br J Pharmacol. 2008;153:309–318. doi: 10.1038/sj.bjp.0707527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beltramo M, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;23:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- 38.Svizenska IH, Brazda V, Klusakova I, Dubovy P. Bilateral changes of cannabinoid receptor type 2 protein and mRNA in the dorsal root ganglia of a rat neuropathic pain model. J Histochem Cytochem. 2013;61:529–547. doi: 10.1369/0022155413491269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anand U, et al. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain. 2008;138:667–680. doi: 10.1016/j.pain.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Jhaveri MD, Sagar DR, Elmes SJ, Kendall DA, Chapman V. Cannabinoid CB2 receptor-mediated anti-nociception in models of acute and chronic pain. Mol Neurobiol. 2007;36:26–35. doi: 10.1007/s12035-007-8007-7. [DOI] [PubMed] [Google Scholar]
- 41.Malan TP, Jr, et al. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 42.Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- 43.Elmes SJ, Jhaveri MD, Smart D, Kendall DA, Chapman V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naive rats and in rat models of inflammatory and neuropathic pain. Eur J Neurosci. 2004;20:2311–2320. doi: 10.1111/j.1460-9568.2004.03690.x. [DOI] [PubMed] [Google Scholar]
- 44.Ibrahim MM, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50:193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akopian AN, Ruparel NB, Jeske NA, Patwardhan A, Hargreaves KM. Role of ionotropic cannabinoid receptors in peripheral antinociception and antihyperalgesia. Trends Pharmacol Sci. 2009;30:79–84. doi: 10.1016/j.tips.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao P, Abood ME. GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci. 2013;92:453–457. doi: 10.1016/j.lfs.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 48.Piomelli D, Astarita G, Rapaka R. A neuroscientist’s guide to lipidomics. Nat Rev Neurosci. 2007;8:743–754. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- 49.Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J. 2013;280:1874–1894. doi: 10.1111/febs.12152. [DOI] [PubMed] [Google Scholar]
- 50.Piomelli D. More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hermanson DJ, et al. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16:1291–1298. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacol Res. 2011;64:60–67. doi: 10.1016/j.phrs.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasso O, et al. Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain. 2013;154:350–360. doi: 10.1016/j.pain.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michalski CW, et al. Cannabinoids ameliorate pain and reduce disease pathology in cerulein-induced acute pancreatitis. Gastroenterology. 2007;132:1968–1978. doi: 10.1053/j.gastro.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lever IJ, et al. Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. J Neurosci. 2009;29:3766–3780. doi: 10.1523/JNEUROSCI.4071-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khasabova IA, et al. Increased anandamide uptake by sensory neurons contributes to hyperalgesia in a model of cancer pain. Neurobiol Dis. 2013;58:19–28. doi: 10.1016/j.nbd.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clapper JR, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sasso O, et al. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gregg LC, et al. Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-alpha initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. J Neurosci. 2012;32:9457–9468. doi: 10.1523/JNEUROSCI.0013-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hohmann AG, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 62.Gammon CM, Allen AC, Morell P. Bradykinin stimulates phosphoinositide hydrolysis and mobilization of arachidonic acid in dorsal root ganglion neurons. J Neurochem. 1989;53:95–101. doi: 10.1111/j.1471-4159.1989.tb07299.x. [DOI] [PubMed] [Google Scholar]
- 63.Liu J, et al. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J Biol Chem. 2003;278:45034–45039. doi: 10.1074/jbc.M306062200. [DOI] [PubMed] [Google Scholar]
- 64.Varga K, Wagner JA, Bridgen DT, Kunos G. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 1998;12:1035–1044. doi: 10.1096/fasebj.12.11.1035. [DOI] [PubMed] [Google Scholar]
- 65.Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Desroches J, Guindon J, Lambert C, Beaulieu P. Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br J Pharmacol. 2008;155:913–924. doi: 10.1038/bjp.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gervois P, Mansouri RM. PPARalpha as a therapeutic target in inflammation-associated diseases. Expert Opin Ther Targets. 2012;16:1113–1125. doi: 10.1517/14728222.2012.715633. [DOI] [PubMed] [Google Scholar]
- 68.Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu J, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 70.LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci. 2005;77:1685–1698. doi: 10.1016/j.lfs.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 71.Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids. 2007;42:491–497. doi: 10.1007/s11745-007-3054-4. [DOI] [PubMed] [Google Scholar]
- 72.LoVerme J, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 73.Dubrac S, Schmuth M. PPAR-alpha in cutaneous inflammation. Dermatoendocrinol. 2011;3:23–26. doi: 10.4161/derm.3.1.14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taylor BK, Dadia N, Yang CB, Krishnan S, Badr M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation. 2002;26:121–127. doi: 10.1023/a:1015500531113. [DOI] [PubMed] [Google Scholar]
- 75.Kostadinova R, Wahli W, Michalik L. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 2005;12:2995–3009. doi: 10.2174/092986705774462905. [DOI] [PubMed] [Google Scholar]
- 76.Petrosino S, Iuvone T, Di Marzo V. N-palmitoyl-ethanolamine: Biochemistry and new therapeutic opportunities. Biochimie. 2010;92:724–727. doi: 10.1016/j.biochi.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 77.Devchand PR, et al. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 78.Ruiz-Medina J, et al. Alteration of neuropathic and visceral pain in female C57BL/6J mice lacking the PPAR-alpha gene. Psychopharmacology (Berl) 2012;222:477–488. doi: 10.1007/s00213-012-2662-8. [DOI] [PubMed] [Google Scholar]
- 79.Sachs D, Cunha FQ, Ferreira SH. Peripheral analgesic blockade of hypernociception: activation of arginine/NO/cGMP/protein kinase G/ATP-sensitive K+ channel pathway. Proc Natl Acad Sci U S A. 2004;101:3680–3685. doi: 10.1073/pnas.0308382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.D’Agostino G, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-kappaB nuclear signalling in dorsal root ganglia. Eur J Pharmacol. 2009;613:54–59. doi: 10.1016/j.ejphar.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 81.Morgenweck J, et al. Activation of peroxisome proliferator-activated receptor gamma in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology. 2010;58:337–345. doi: 10.1016/j.neuropharm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsuboi K, et al. Biosynthetic pathways of bioactive N-acylethanolamines in brain. CNS Neurol Disord Drug Targets. 2013;12:7–16. doi: 10.2174/1871527311312010005. [DOI] [PubMed] [Google Scholar]
- 83.Solorzano C, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu C, et al. Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Mol Pharmacol. 2011;79:786–792. doi: 10.1124/mol.110.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Richardson D, et al. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res Ther. 2008;10:R43. doi: 10.1186/ar2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khasabova IA, Xiong Y, Coicou LG, Piomelli D, Seybold V. Peroxisome proliferator-activated receptor alpha mediates acute effects of palmitoylethanolamide on sensory neurons. J Neurosci. 2012;32:12735–12743. doi: 10.1523/JNEUROSCI.0130-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bortolini M, Wright MB, Bopst M, Balas B. Examining the safety of PPAR agonists - current trends and future prospects. Expert Opin Drug Saf. 2013;12:65–79. doi: 10.1517/14740338.2013.741585. [DOI] [PubMed] [Google Scholar]
- 88.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Inceoglu B, et al. Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type I diabetes. Proc Natl Acad Sci U S A. 2012;109:11390–11395. doi: 10.1073/pnas.1208708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801:1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pamplona FA, et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci U S A. 2012;109:21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011;34:599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu ZZ, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med. 2010;16:592–597. doi: 10.1038/nm.2123. 591p following 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krishnamoorthy S, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A. 2010;107:1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.LoVerme J, Guzman M, Gaetani S, Piomelli D. Cold exposure stimulates synthesis of the bioactive lipid oleoylethanolamide in rat adipose tissue. J Biol Chem. 2006;281:22815–22818. doi: 10.1074/jbc.M604751200. [DOI] [PubMed] [Google Scholar]
- 97.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kathuria S, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 99.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16:744–53. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Armirotti A, et al. β-Lactones inhibit N-acylethanolamine acid amidase by S-Acylation of the catalytic N-terminal cysteine. ACS Med Chem Lett. 2012;3:422–426. doi: 10.1021/ml300056y. [DOI] [PMC free article] [PubMed] [Google Scholar]