

Abstract
Since the discovery of cis-platinum, many transition metal complexes have been synthesized and assayed for antineoplastic activity. In recent years, ruthenium-based molecules have emerged as promising antitumor and anti-metastatic agents with potential uses in platinum-resistant tumors or as alternatives to platinum. Ruthenium compounds theoretically possess unique biochemical features allowing them to accumulate preferentially in neoplastic tissues and to convert to their active state only after entering tumor cells. Intriguingly, some ruthenium agents show significant activity against cancer metastases but have minimal effects on primary tumors. Two ruthenium-based drugs, NAMI-A and KP1019, have reached human clinical testing. This review will highlight the chemical properties, mechanism of action, preclinical data, and early phase clinical results of these two lead ruthenium compounds. Other promising ruthenium agents will also be reviewed with emphasis on the novel ruthenium compound ONCO4417, and DW1/2 that has demonstrated Pim-1 kinase inhibition in preclinical systems. Further development of these and other ruthenium agents may rely on novel approaches including rational combination strategies as well as identification of potential pharmacodynamic biomarkers of drug activity aiding early phase clinical studies.
Keywords: Ruthenium, Chemotherapy, NAMI-A, KP1019, ONCO4417
Introduction
The success of platinum as anticancer agent [1] has stimulated a search for other metallic cytotoxic compounds with equal or greater antitumor activity and lower toxicity. Three platinum-based antineoplastic agents are now in routine clinical practice: cisplatin, carboplatin, and oxaliplatin [2]. Although these heavy metal agents are active against a variety of cancers, their use is associated with severe side effects including gastrointestinal symptoms (nausea, vomiting, diarrhea, abdominal pain), renal tubular injury, neuromuscular complications, and ototoxicity. In addition, the use of platinum is limited in many tumor types by primary and acquired resistance to this agent [3]. This has led to an ongoing quest for the discovery of non-platinum metals that may extend the spectrum of activity of metal-based drugs [4]. Among these, ruthenium (Ru) appears to be the most promising [5].
In the last 30 years, a large number of ruthenium-containing agents have been synthesized and tested for potential antitumor activity [6]. Ruthenium, a transition metal of the platinum group (group 8B of the periodic table), was first hypothesized to exert its anticancer effects by direct interaction with DNA as observed with platinum. However, it is now known that ruthenium shows a number of differences with platinum [7]. First, ruthenium appears to accumulate preferentially in neoplastic masses rather than normal tissues possibly by using transferrin to enter into tumors [8]. It has been proposed that transferrin–ruthenium complexes are actively transported into neoplastic tissues containing high transferrin receptor densities. Once bound by the transferrin receptor, it is assumed that the complex liberates ruthenium that is then internalized by the tumor [9]. Second, ruthenium remains in its relatively inactive Ru(III) oxidation state until it reaches the tumor site. In this environment, where there is a lower oxygen content and higher acidity compared to normal tissues, reduction to the more reactive Ru(II) occurs [10]. This reaction, termed activation-by-reduction, not only results in selective tumor targeting but may also direct cytotoxic activity toward hypoxic tumors that are more likely to be resistant to chemotherapy and radiation [11]. Finally, some ruthenium agents demonstrate greater efficacy against cancer metastases than against primary tumors [12]. This antimetastatic effect is likely mediated by inhibition of tumor cell detachment, invasion/migration, and re-adhesion to a new growth substrate [13]. In view of these properties, ruthenium is predicted to show patterns of antitumor activity and clinical toxicity that are distinct from those of platinum.
To date, two ruthenium agents, NAMI-A and KP1019, have entered human clinical trials (Figs. 1, 2) [14, 15]. Despite their structural and chemical similarities, these two Ru(III) complexes show distinct antitumor behaviors. In preclinical studies, NAMI-A has demonstrated inhibitory effects against the formation of cancer metastases in a variety of tumor animal models but appears to lack direct cytotoxic effects [16, 17], while KP1019 has shown direct antitumor activity against a wide range of primary explants of human tumors by inducing apoptosis [18, 19]. Nevertheless, many aspects of the tumor-inhibitory effects of these ruthenium drugs are not still completely understood. This review will focus on the novelty of chemical and biochemical aspects of ruthenium compounds as well as clinical data available for each of the two lead ruthenium-based agents. We will also touch upon other promising ruthenium agents still in early phases of drug development.
Fig. 1.
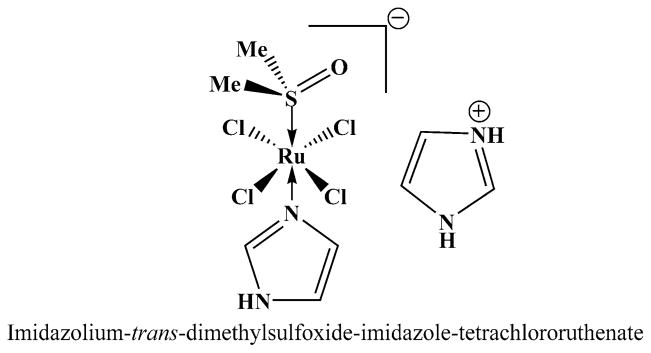
Chemical structure of NAMI-A (Me = methyl group)
Fig. 2.

Chemical structure of KP1019
Chemical and biochemical properties of ruthenium complexes
Ruthenium possesses three properties that make it theoretically suitable for medicinal use. These are (1) slow ligand exchange kinetics, (2) multiple accessible oxidation states, and (3) the ability to mimic iron in binding to certain biologic molecules [20]. Each of these properties is considered in turn.
Ligand exchange
The key factor explaining why platinum can function as an antineoplastic drug likely relates to its ligand-exchange kinetics, which are in the order of minutes to days rather than microseconds to seconds (as for many other coordination compounds), thereby giving platinum high kinetic stability and preventing rapid equilibration reactions [21]. Ru(II) and Ru(III) have similar ligand exchange kinetics to those of Pt(II) [22]. It has been shown that ligand-exchange processes for the same ligands, but with different metallic ions, can vary by several orders of magnitude. For most small ligands (e.g., water), ligand exchange for metallic ions (e.g., Pt(II), Ru(II), Ru(III), Co(III), and a few others) takes several hours, mimicking the time scale for many cell division processes [23]. In addition, Pt(II) and to a lesser extent Ru(II) have a strong thermodynamic preference for binding to S-donor ligands such as glutathione and methionine, as opposed to O- or N-donor ligands [22].
Oxidation state
Ruthenium is unique in that the oxidation states of II, III, and IV are all accessible under physiologic conditions. In these states, the ruthenium center is predominantly hexacoordinate with octahedral geometry. Although this makes cis-chelation possible, the space required by axial ligands often prevents it from forming structures with DNA as seen with Pt(II). The coordination environment around ruthenium plays an important role in stabilizing its different oxidation states and hence dictates the redox properties of the central metal atoms [24, 25]. Altered metabolism associated with cancer results in a lower oxygen concentration in tumor tissues, promoting a reductive environment. In addition, cancer cells have higher levels of glutathione and a lower pH than normal tissues, further creating a strongly reducing environment [10]. To increase the tropicity of ruthenium toward cancer cells and to minimize harm to normal cells, the redox potential of ruthenium can be modified to improve drug selectivity. For example, these agents can be administered as relatively inert Ru(III) complexes (e.g., pro-drugs) that are then activated by reduction within malignant tissues. In theory if the active Ru(II) complexes leave the low oxygen environment, they will again be converted back to inert Ru(III) compounds by a variety of biologic oxidants.
Iron mimicking
Ruthenium can mimic iron in binding to serum transferrin and albumin [26]. These proteins solubilize and transport iron in plasma. Since rapidly dividing cells (including cancer cells) have a greater requirement for iron, this results in up-regulation of the number of transferrin receptors on the cell surface, resulting in sequestration of more circulating iron-loaded transferrin. To this end, in vivo studies have shown that there is a 2- to 12-fold increase in ruthenium concentration in cancer cells compared to healthy cells, depending on the cell type [9]. Since ruthenium preferentially targets cancer cells, its systemic toxicity, at least in theory, is expected to be reduced. Moreover, it has been shown that ruthenium is transported into cells by both transferrin-dependent and transferrin-independent mechanisms [27]. Transferrin-mediated ruthenium uptake is more efficient when transferrin is saturated with iron to a physiologic degree.
NAMI-A
Clinical chemistry and preclinical studies
NAMI-A [28], [ImH][trans-RuCl4(DMSO)(Im)] (Im = imidazole, DMSO = dimethylsulfoxide), is a Ru(III) complex molecule with DMSO and imidazole coordinated to the ruthenium (Fig. 1). NAMI-A has been shown to possess antitumor and more importantly antimetastatic activities in preclinical studies.
The proposed mechanisms of action of NAMI-A include: (1) interaction with the cell cycle regulation culminating in transient accumulation of cells in the G2/M phase [29, 30], (2) inhibition of matrix metalloproteinases [31], (3) increasing extracellular matrix around tumor vasculature, thereby preventing neoplastic cells from invading nearby tissues and blood vessels [30], and (4) binding through coordination to nucleic acids, therefore, having a direct effect on tumor cell DNA [32]. In preclinical studies, administration of NAMI-A in more frequent smaller dosages showed more prominent antimetastatic effects [12]. Notably, the action of NAMI-A seems to be independent of the type of primary tumor or the stage of growth of metastases [33].
NAMI-A is capable not only of preventing the formation of metastases but also of inhibiting their growth once established [34]. Preclinical animal studies using NAMI-A have shown selective activity against lung metastases from a variety of primary tumors in murine models [33, 35]. NAMI-A reduced the weight of lung metastases more than their number. Since larger concentrations of NAMI-A in the lungs than in other tissues was ruled out, this finding was assumed to be related to the selective interference of NAMI-A with the growth of metastases already established in the lungs [35]. Toxicologic studies in dogs and mice have revealed an acceptable toxicity profile. The calculated half-life was approximately 18 h. Toxicity was observed at concentrations greater than 50 mg/kg/day and in mice that survived treatment was reversed within 3 weeks of the end of the treatment [36].
Clinical trials
NAMI-A was the first ruthenium drug to reach human clinical development. A phase I and pharmacokinetic study using this agent began in 1999 and was reported in 2004 [37]. In that study, 24 patients with a wide variety of metastatic solid tumors (including colorectal, lung, melanoma, ovarian, and pancreatic cancers) refractory to conventional therapies were treated according to a dose escalation protocol. Patients received NAMI-A as a 3-h intravenous infusion daily for 5 days every 3 weeks. Twelve dose levels were administered in two-patient cohorts, ranging from 2.4 mg/m2/day (12 mg/m2/cycle) to 500 mg/m2/day (2500 mg/m2/cycle). At a dose of 400 mg/m2/day, patients developed transient painful blisters on the hands and feet. At 500 mg/m2/day, blisters again developed and persisted for weeks to months. Formation of blisters was considered a dose-limiting toxicity. Because blisters did not appear at a dose of 300 mg/m2/day on this schedule, this was defined as the maximum tolerated dose recommended for phase II studies [37]. Other side effects of NAMI-A included anemia, lymphopenia, fatigue, anorexia, stomatitis, peripheral edema, alopecia, nausea (controlled with granisetron), diarrhea, tinnitus, and infusion-site phlebitis. Biochemical abnormalities included mild creatinine elevations (all reversible in less than 3 weeks) and hyperbilirubinemia. Two patients experienced hypersensitivity reactions that resolved with dexamethasone.
Pharmacokinetic analysis from the same study revealed a linear relationship between NAMI-A dose and area under the concentration–time curve for total plasma ruthenium (R2 = 0.75) and unbound plasma ruthenium (R2 = 0.96) over the entire dose range [38]. NAMI-A also demonstrated linear elimination kinetics. Mean plasma clearance of total ruthenium was 0.17 l/h, and the mean terminal half-life was 50 h. The mean volume of distribution at steady-state of total ruthenium was 10.1 l. The majority of the drug was protein-bound in plasma. Ruthenium was found to accumulate in white blood cells, but its accumulation was not directly proportional to the daily dose or total exposure of NAMI-A [37].
Twenty of 24 patients in this study (83%) were evaluable for tumor responses. One heavily pretreated patient with progressive metastatic non-small cell lung cancer achieved stable disease for 21 weeks. The remainder of the patients showed disease progression. A phase II study evaluating NAMI-A has not yet been conducted.
KP1019
Clinical chemistry and preclinical studies
KP1019, [InH][trans-RuCl4(In)2] (In = indazole), is a stable Ru(III) complex containing two indazole heterocycles coordinated to the metal center via nitrogen atoms (Fig. 2). Unlike NAMI-A, KP1019 is thought to possess direct cytotoxic activity by promoting apoptosis in a number of cancer cell lines as well as in a range of tumor models (especially colorectal cancers) including primary explanted human tumors [39, 40].
KP1019 induced apoptosis in colorectal cancer cell lines through interference with the electron transport chain, and depolarization of mitochondrial membranes and activation of caspase-3 as well as down-modulation of the antiapoptotic factor bcl-2 [41]. Notably, KP1019-induced cell death appears independent of the p53 status of tumor cells, suggesting that DNA strand breaks are not a dominant mechanism for this agent [42]. However, the formation of reactive oxygen species in tumor cell lines has been reported and this may possibly contribute to DNA damage, albeit to a small extent [41]. KP1019 has also shown activity in primary explants of human tumors that were resistant to a variety of standard chemotherapeutic agents [43].
In vivo, KP1019 revealed strong cytotoxic activity in transplantable murine tumors of the chemoresistant colon carcinoma model MAC15A [39]. Autochthonous colorectal tumors of the rat closely resemble human colon cancer in histological appearance, metastatic progression, tumor-host interactions, and chemosensitivity profile. In this model, KP1019 induced complete or near-complete tumor responses in the majority of lesions while 5-fluorouracil only produced partial responses in about half the tumors and cisplatin had no activity [43]. These effects of KP1019 were achieved with mild adverse events, including suppression of erythropoiesis.
Clinical trials
KP1019 was the second ruthenium-based agent that was tested in humans. To this end, a pharmacokinetic and phase I dose-escalation study was recently reported [19, 44, 45]. In this study, 8 patients with advanced and refractory solid tumors (including colorectal, endometrial, melanoma, and bladder carcinomas) received KP1019 intravenously (10 ml/min) at doses ranging from 25 to 600 mg twice weekly over 3 weeks (days 1, 4, 8, 11, 15, and 18). Infusion duration was 0.1–4.25 h, depending on the dose of the drug administered (higher doses required larger volumes for drug solubilization). Only mild (grade 1) treatment-related toxicities were observed, although two patients dropped out of the study after only one treatment cycle due to adverse events unrelated to the study drug. The maximum tolerated dose could not be reached even when KP1019 was given at 600 mg twice weekly over 3 weeks. The dose could not be escalated further due to problems with drug solubility requiring increasingly large infusion volumes. No dose-limiting toxicities were observed [19]. The optimal dose for phase II studies was set at 400 mg (flat dose) on days 1, 4, 8, 11, 15, and 18 of a 3-week cycle. However, given the results of the phase I study, a reliable dose recommendation for this agent is probably not possible.
After intravenous administration, KP1019 was highly bound to plasma proteins (mainly albumin and to a lesser extent transferrin). The unbound fraction was less than 1% [45]. The area under the concentration–time curve increased proportionally with KP1019 dose, indicating linear pharmacokinetics. The terminal half-life of KP1019 was quite long with a mean of about 100 h (range, 51–290 h). The total clearance was low (range, 0.024–0.036 l/h), and the volume of distribution at steady-state was small (range, 4.0–10.5 l). Renal excretion of ruthenium was 1–5% of the administered dose [44]. Ruthenium–DNA adducts were detectable in peripheral leucocytes from all patients irrespective of dose. Thus, the determination of Ru–DNA adduct levels may be a feasible biomarker of efficacy and/or toxicity in phase II studies.
Out of six evaluable patients all of which had progressive disease at trial entry, five had disease stabilization that lasted for 8–10 weeks. Stable disease was achieved even at the lowest dose level [19]. A phase II study evaluating KP1019 in patients with advanced colorectal cancer is now being planned.
Other ruthenium compounds
Besides NAMI-A and KP1019, a large number of other ruthenium complexes have been prepared and tested for antitumor activity in cultured tumor cells and animal models. Some notable examples are discussed here.
In 2001, a novel group of arene ruthenium(II) diamines was developed [46]. These agents demonstrated strong cytotoxicity in cancer cells in vitro associated with a parallel DNA interaction [47, 48]. RM175 (Fig. 3), one of the lead compounds in this class, demonstrated a p53 and p21/WAF1-dependent early growth arrest in HCT116 colorectal cancer cells. In these cells, RM175 also resulted in an apoptotic response through p53 and Bax as well as a long-term loss of clonogenicity [49]. Other investigators using different ruthenium(II) complexes have shown both p53-dependent and p53-independent mechanisms of cytotoxicity in glioblastoma and neuroblastoma cell lines with G1 arrest and apoptosis induction [50]. In these cell lines, accumulation of p53 and p73 correlated with an increase in p21/WAF1 and Bax expression.
Fig. 3.
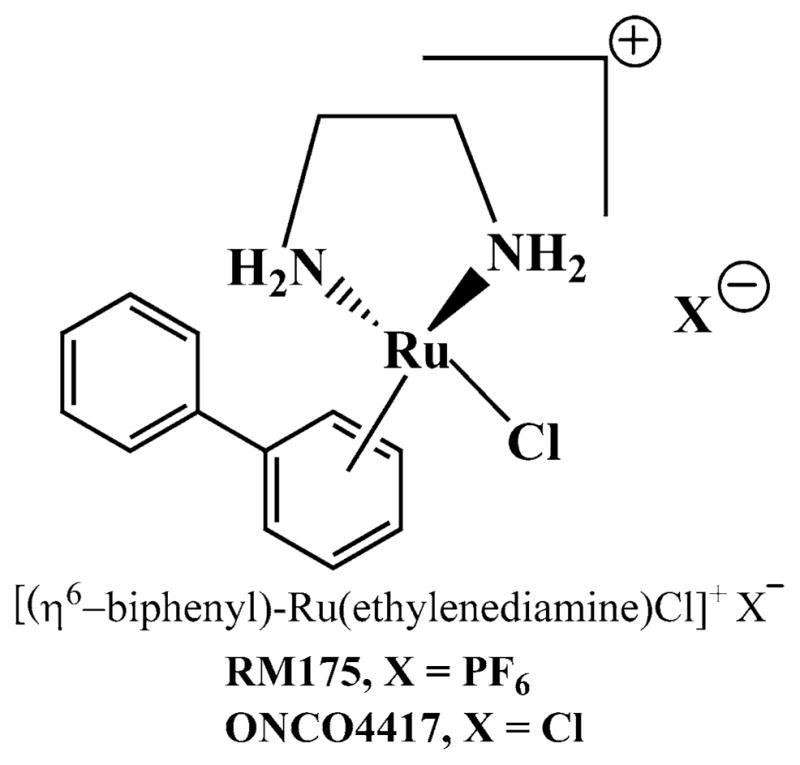
Chemical structures of two arene ruthenium(II) diamines
Substituting the hexafluorophosphate (PF6) anion in RM175 for chloride results in another ruthenium salt, ONCO4417 (Fig. 3), possessing antineoplastic activities [51]. This agent was shown to induce apoptosis in cancer cells and to arrest cell cycle at the G2/M phase. In addition, ONCO4417 caused DNA damage at levels similar to cisplatin [51]. In vitro studies have demonstrated that ONCO4417 has comparable efficacy to platinum in a number of tumor cell lines (including ovarian, lung, esophageal, pancreatic, melanoma, and colorectal). Moreover, ONCO4417 retained its antitumor efficacy in a cisplatin-resistant ovarian cancer cell line (A2780cis), suggesting that this drug does not share cross-resistance mechanisms with cisplatin [52].
A distinct series of ruthenium(II) arenes characterized by the presence of phosphoadamantane coordinates, called RAPTAs, were developed in 2004 (Fig. 4) [53, 54]. These compounds are weakly cytotoxic against tumor cells in vitro and usually free of toxicity to healthy cells even with prolonged exposure at millimolar concentrations [55, 56]. In vitro, RAPTA-T inhibited some steps of the metastatic process such as detachment from the primary tumor cell mass, migration/invasion, and re-adhesion to a new growth substrate in breast cancer cell lines. These antimetastatic effects appear to be mediated through interactions with extracellular matrix components (collagen IV, fibronectin). In vivo, RAPTA-T selectively reduced the growth of lung metastases [13].
Fig. 4.
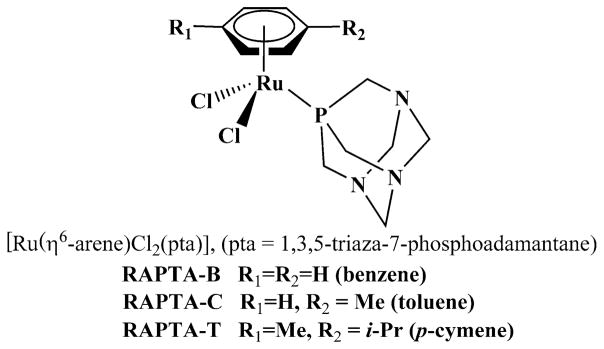
Chemical structures of three ruthenium(II) arenes (RAPTA)
In an effort to investigate the molecular mechanism of RAPTA-C, the molecule inhibited cell growth effectively by triggering G2/M phase arrest and apoptosis in Ehrlich ascites carcinoma cells lines. Cell cycle arrest was associated with increased levels of p21 and reduced amounts of cyclin E. RAPTA-C treatment also enhanced the levels of p53 and triggered the mitochondrial apoptotic pathway, as shown by the change in Bax to bcl-2 ratios, resulting in cytochrome c release and caspase-9 activation. c-Jun NH2-terminal kinase (JNK) was proven to be a critical mediator in RAPTA-C-induced cell growth inhibition. Activation of JNK by RAPTA-C increased significantly during apoptosis [57].
A series of ruthenium(II) polypyridyl complexes exhibited in vitro cytotoxic activity against two cancer cell lines, MCF-7 (breast cancer) and HT-29 (colon cancer), at micro-molar IC50 values. Their mechanism of action includes a formation of a stable intercalative binding into DNA and a modification of cell membrane function and cell adhesion properties. The cytotoxicity of these ruthenium agents relies on the size of the polypyridyl ligands [58, 59].
Ruthenium complexes that mimic organic enzyme inhibitors also have been synthesized. The natural product staurosporine is a highly potent inhibitor for various kinases, and it was shown that its effect can be mimicked by an octahedral ruthenium complex (Fig. 5) [60]. The relevance of these organometallic inhibitors as anticancer agents has been demonstrated recently. The organometallic glycogen synthase kinase (GSK)-3β inhibitor DW1/2 is a potent activator of p53 and thus induces p53-activated apoptosis via the mitochondrial pathway through down-regulation of Mdm2 and Mdm4 in highly chemoresistant melanoma cells [61]. The anticancer agent DW1/2 works by specifically targeting a protein, rather than DNA. This is one of the first demonstrations of a ruthenium drug targeting a signal transduction pathway. In vivo studies evaluating DW1/2 or other compounds in this group have not yet been conducted. The development of novel drugs with non-classical protein targets is becoming a major new theme in metal-based drug development. More complete chemical work may be needed to give these compounds a pharmacokinetic profile suitable for drug administration.
Fig. 5.

Chemical structure of DW1, mimicking the protein kinase inhibitor staurosporine
Another novel concept in the design of bioactive molecules is taking advantage of transition metals in coordination complexes as a platform for organizing the organic ligands in the 3-D space, thus providing access to areas of the chemical space that may not be easily reachable with purely organic scaffolds. The co-crystal structures of both the R and the S enantiomers of the ruthenium complex DW1/2 (Fig. 5) with the human protein kinase Pim-1 clearly show that these ruthenium complexes bind to their target in a fashion typical of organic enzyme inhibitors. In this example, the metal center is not involved in any direct interactions with the active site of Pim-1. Instead, the metal controls the orientation of the organic ligands in the receptor space, yielding 3-D structures that are complementary in shape and functional group presentation to the active site of Pim-1 [62]. Pim-1 is a proto-oncogene of the serine–threonine kinase family with increased expression in a variety of murine and human acute leukemias. Expression of Pim-1 increases cell mitogenesis and survival independently of growth factor stimulation. Pim-1 also synergizes with c-Myc in leukemogenesis and enhances transcriptional activity of the cellular proliferation factor c-Myc. Pim-1 may also protect hematopoietic cells from apoptosis induced by genotoxic stress or growth factor withdrawal, perhaps by directly targeting nuclear effectors [63]. Targeting the Pim kinase family could provide a new avenue in cancer chemotherapy specifically intended to overcome resistance against small-molecule tyrosine kinase inhibitors [64].
Discussion
Ruthenium has been studied deeply and broadly as a potential anticancer chemotherapeutic in the last two decades from both a chemical and a biologic perspective. The unique characteristics of ruthenium that (at least in theory) make it an attractive anticancer agent are: (1) the selective accumulation in tumor cells via interaction with transferrin receptors, (2) activation-by-reduction from inactive Ru(III) to active Ru(II) in the more hypoxic and acidic tumor microenvironment, (3) favorable ligand-exchange kinetics, (4) antimetastatic effects (of some ruthenium agents) by inhibition of tumor cell detachment, invasion/migration, and re-adhesion, and (5) unique DNA binding patterns different from those of platinum owing to special ligand geometries.
These distinctive behaviors offer ruthenium the theoretical potential for non-cross-resistance with platinum as well as a different spectrum of activity with lower toxicity. However, in spite of large amounts of preclinical data, none of the ruthenium compounds that have reached clinical testing to date have been shown to mediate their effects through these putative mechanisms. For example, the main reported dose-limiting toxicity for NAMI-A in the phase I clinical trial was blister formation on the hands and feet. These blisters were poorly reversible and often lasted for several weeks. Intriguingly, ruthenium concentrations in these blisters were below the limit of detection. It is therefore not clear whether ruthenium itself or its ligands have the tendency to accumulate in the epidermis or other skin layers, causing temporary or permanent toxicity. This finding seems to argue against the selective tropicity of NAMI-A toward tumor cells. By contrast, in the phase I trial of KP1019, an agent that does not contain a sulfur-bearing ligand (i.e., DMSO), no dose-limiting toxicities were observed although that study only included eight patients and there were problems with drug solubility.
Despite a wealth of preclinical data on a variety of ruthenium agents, it is still not clear whether and when this new class of compounds will enter later-phase clinical testing. Significant improvements have been achieved in preclinical drug discovery tools in the last two decades. However, a novel agent entering pharmaceutical development had only an 8% chance of making it to the market in 2000, a decrease from the historical success rate of 14% in 1985 [65]. To address this problem, in 2004, the US Food and Drug Administration (FDA) published an Executive Summary Statement outlining that expenditure for the development of novel therapies had disproportionately increased relative to major drug and biologic product submissions [65]. To overcome stagnation in the critical pathway from phase I to phase III clinical trials, the FDA introduced the concept of exploratory investigational new drug (IND) studies. These are clinical trials conducted early in phase I that involve limited human exposure (ten or fewer patients receiving very small doses of an experimental drug for 7 days or less) and have no therapeutic intent. The purpose of such “phase 0” studies is to assist in the go/no-go decision of a drug’s fate early in its development, using relevant human models instead of relying on sometimes inconsistent or artificial cell-based and animal data, thus helping to confirm end points such as mechanism of action, pharmacology, bioavailability, pharmacodynamics, and metabolic microdose assessments [66]. However, in order to evaluate target drug effects, a biomarker specific to an investigational agent must be known and an assay to measure it must be developed and validated before study initiation. This type of strategy may not be ideal for the development of ruthenium compounds, which are generally not considered agents that target tumor-specific cellular pathways and for which biologic markers of activity do not currently exist. Indeed, since the vast majority of ruthenium drugs (with the possible exception of DW1/2) are DNA-targeting agents, it is difficult to imagine that relevant cellular pharmacodynamic biomarkers of drug effect will be discovered soon.
The future clinical development of ruthenium drugs may therefore rely on the performance of combination trials, where a particular ruthenium agent is given in combination with a taxane, an antimetabolite, or a topoisomerase II inhibitor (akin to the combination strategies of platinum drugs). It will first be necessary, however, to assay for synergy between ruthenium compounds and conventional chemotherapy drugs in cancer cell lines and preclinical animal models. Once preclinical rationale exists, the path from bench to bedside will further be expedited through support from government agencies and trial orchestration through cooperative groups, as well as by the development of a biomarker research facility focusing on the discovery of ruthenium-related tissue or plasma biomarkers.
In conclusion, ruthenium compounds are second generation (post-platinum) transition metal chemotherapeutics that possess unique properties granting them, at least in pre-clinical studies, more selective entry into tumor cells with fewer toxic effects to normal cells. A wealth of laboratory data now demonstrates that numerous ruthenium(II) and (III) complexes show antitumor activity in a variety of cancer cell lines and animal models. While some of these agents appear to have direct cytotoxic effects on primary tumors, others exhibit their activity primarily through inhibition of metastatic progression. To date, two ruthenium(III) compounds (KP1019 and NAMI-A) have reached phase I clinical testing, and progression to phase II clinical trials is under consideration. Finally, the agent DW1/2 has shown promising in vitro activity as a signal transduction inhibitor by interference with various protein kinases. Further disease-specific phase II studies will be needed to elucidate the real-world therapeutic potential of ruthenium complexes in hematologic and solid tumor oncology.
Footnotes
Conflict of interest statement The authors indicate no financial or other conflicts of interest.
References
- 1.Galanski M, Jakupec MA, Keppler BK. Update of the pre-clinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 2.Galanski M. Recent developments in the field of anticancer platinum complexes. Recent Pat Anticancer Drug Discov. 2006;1:285–295. doi: 10.2174/157489206777442287. [DOI] [PubMed] [Google Scholar]
- 3.Brabec V, Kasparkova J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum anti-tumor drugs. Drug Resist Updat. 2005;8:131–146. doi: 10.1016/j.drup.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Ott I, Gust R. Non platinum metal complexes as anti-cancer drugs. Arch Pharm (Weinheim) 2007;340:117–126. doi: 10.1002/ardp.200600151. [DOI] [PubMed] [Google Scholar]
- 5.Sava G, Bergamo A. Ruthenium drugs for cancer chemotherapy: an ongoing challenge to treat solid tumours. In: Bonetti A, et al., editors. Platinum and other heavy metal compounds in cancer chemotherapy. Humana Press; New York: 2009. pp. 57–66. [DOI] [Google Scholar]
- 6.Kostova I. Ruthenium complexes as anticancer agents. Curr Med Chem. 2006;13:1085–1107. doi: 10.2174/092986706776360941. [DOI] [PubMed] [Google Scholar]
- 7.Heffeter P, Jungwirth U, Jakupec M, Hartinger C, Galanski M, et al. Resistance against novel anticancer metal compounds: differences and similarities. Drug Resist Updat. 2008;11:1–16. doi: 10.1016/j.drup.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Sava G, Zorzet S, Giraldi T, Mestroni G, Zassinovich G. Antineoplastic activity and toxicity of an organometallic complex of ruthenium(II) in comparison with cis-PDD in mice bearing solid malignant neoplasms. Eur J Cancer Clin Oncol. 1984;20:841–847. doi: 10.1016/0277-5379(84)90223-2. [DOI] [PubMed] [Google Scholar]
- 9.Sava G, Bergamo A. Ruthenium-based compounds and tumour growth control (review) Int J Oncol. 2000;17:353–365. doi: 10.3892/ijo.17.2.353. [DOI] [PubMed] [Google Scholar]
- 10.Schluga P, Hartinger CG, Egger A, Reisner E, Galanski M, et al. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006;14:1796–1802. doi: 10.1039/b511792e. [DOI] [PubMed] [Google Scholar]
- 11.Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009;9:442–458. doi: 10.2174/156652409788167087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagliardi R, Sava G, Pacor S, Mestroni G, Alessio E. Antimetastatic action and toxicity on healthy tissues of Na[trans-RuCl4(DMSO)Im] in the mouse. Clin Exp Metastasis. 1994;12:93–100. doi: 10.1007/BF01753975. [DOI] [PubMed] [Google Scholar]
- 13.Bergamo A, Masi A, Dyson PJ, Sava G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int J Oncol. 2008;33:1281–1289. [PubMed] [Google Scholar]
- 14.Sava G, Alessio E, Bergamo A, Mestroni G. Sulfoxide ruthenium complexes. Top Biol Inorg Chem. 1999;1:143–169. [Google Scholar]
- 15.Pieper T, Borsky K, Keppler BK. Non-platinum antitumor compounds. Top Biol Inorg Chem. 1999;1:171–199. [Google Scholar]
- 16.Sava G, Pacor S, Mestroni G, Alessio E. Na[trans-RuCl4(DMSO)Im], a metal complex of ruthenium with antimetastatic properties. Clin Exp Metastasis. 1992;10:273–280. doi: 10.1007/BF00133563. [DOI] [PubMed] [Google Scholar]
- 17.Sava G, Pacor S, Bergamo A, Cocchietto M, Mestroni G, et al. Effects of ruthenium complexes on experimental tumors: irrelevance of cytotoxicity for metastasis inhibition. Chem Biol Interact. 1995;95:109–126. doi: 10.1016/0009-2797(94)03350-1. [DOI] [PubMed] [Google Scholar]
- 18.Jakupec MA, Arion VB, Kapitza S, Reisner E, Eichinger A, et al. KP1019 (FFC14A) from bench to bedside: preclinical and early clinical development—an overview. Int J Clin Pharmacol Ther. 2005;43:595–596. doi: 10.5414/cpp43595. [DOI] [PubMed] [Google Scholar]
- 19.Hartinger CG, Jakupec MA, Zorbas-Seifried S, Groessl M, Egger A, et al. KP1019, a new redox-active anticancer agent–preclinical development and results of a clinical phase I study in tumor patients. Chem Biodivers. 2008;5:2140–2155. doi: 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- 20.Clarke MJ, Zhu F, Frasca DR. Non-platinum chemotherapeutic metallopharmaceuticals. Chem Rev. 1999;99:2511–2534. doi: 10.1021/cr9804238. [DOI] [PubMed] [Google Scholar]
- 21.Bloemink MJ, Reedijk J. Cisplatin and derived anticancer drugs: mechanism and current status of DNA binding. Met Ions Biol Syst. 1996;32:641–685. [PubMed] [Google Scholar]
- 22.Reedijk J. New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc Natl Acad Sci USA. 2003;100:3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada H, Koike T, Hurst JK. Water exchange rates in the diruthenium μ-oxo ion cis, cis-[(bpy)2Ru(OH)2]2O4+ J Am Chem Soc. 2001;123:12775–12780. doi: 10.1021/ja010594l. [DOI] [PubMed] [Google Scholar]
- 24.Chakravarty J, Bhattacharya S. Ruthenium phenolates. Synthesis, characterization and electron-transfer properties of some salicylaldiminato and 2-(arylazo)phenolato complexes of ruthenium. Polyhedron. 1996;15:1047–1055. [Google Scholar]
- 25.Baitalik S, Adhikary B. Heterochelates of ruthenium(II): electrochemistry, absorption spectra, and luminescence properties. Polyhedron. 1997;16:4073–4080. [Google Scholar]
- 26.Kratz F, Messori L. Spectral characterization of ruthenium(III) transferrin. J Inorg Biochem. 1993;49:79–82. doi: 10.1016/0162-0134(93)85017-3. [DOI] [PubMed] [Google Scholar]
- 27.Pongratz M, Schluga P, Jakupec MA, Arion VB, Hartinger CG, et al. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J Anal At Spectrom. 2004;19:46–51. [Google Scholar]
- 28.Mestroni G, Alessio E, Sava G. International Patent PCT C 07F 15/00. 98/0043 A61 K 31/28, WO. 1998
- 29.Bergamo A, Gagliardi R, Scarcia V, Furlani A, Alessio E, et al. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J Pharmacol Exp Ther. 1999;289:559–564. [PubMed] [Google Scholar]
- 30.Zorzet S, Bergamo A, Cocchietto M, Sorc A, Gava B, et al. Lack of in vitro cytotoxicity, associated to increased G2-M cell fraction and inhibition of matrigel invasion, may predict in vivo-selective antimetastasis activity of ruthenium complexes. J Pharmacol Exp Ther. 2000;295:927–933. [PubMed] [Google Scholar]
- 31.Vacca A, Bruno M, Boccarelli A, Coluccia M, Ribatti D, et al. Inhibition of endothelial cell functions and of angiogenesis by the metastasis inhibitor NAMI-A. Br J Cancer. 2002;86:993–998. doi: 10.1038/sj.bjc.6600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pluim D, van Waardenburg RC, Beijnen JH, Schellens JH. Cytotoxicity of the organic ruthenium anticancer drug NAMI-A is correlated with DNA binding in four different human tumor cell lines. Cancer Chemother Pharmacol. 2004;54:71–78. doi: 10.1007/s00280-004-0773-6. [DOI] [PubMed] [Google Scholar]
- 33.Sava G, Gagliardi R, Bergamo A, Alessio E, Mestroni G. Treatment of metastases of solid mouse tumours by NAMI-A: comparison with cisplatin, cyclophosphamide and dacarbazine. Anticancer Res. 1999;19:969–972. [PubMed] [Google Scholar]
- 34.Sava G, Capozzi I, Clerici K, Gagliardi G, Alessio E, et al. Pharmacological control of lung metastases of solid tumours by a novel ruthenium complex. Clin Exp Metastasis. 1998;16:371–379. doi: 10.1023/a:1006521715400. [DOI] [PubMed] [Google Scholar]
- 35.Sava G, Clerici K, Capozzi I, Cocchietto M, Gagliardi R, et al. Reduction of lung metastasis by ImH[trans-RuCl4(DM-SO)Im]: mechanism of the selective action investigated on mouse tumors. Anticancer Drugs. 1999;10:129–138. doi: 10.1097/00001813-199901000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Cocchietto M, Sava G. Blood concentration and toxicity of the antimetastasis agent NAMI-A following repeated intravenous treatment in mice. Pharmacol Toxicol. 2000;87:193–197. doi: 10.1034/j.1600-0773.2000.d01-73.x. [DOI] [PubMed] [Google Scholar]
- 37.Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JH. A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin Cancer Res. 2004;10:3717–3727. doi: 10.1158/1078-0432.CCR-03-0746. [DOI] [PubMed] [Google Scholar]
- 38.Brouwers EE, Tibben MM, Rosing H, Schellens JH, Beijnen JH. Determination of ruthenium originating from the investigational anti-cancer drug NAMI-A in human plasma ultrafiltrate, plasma, and urine by inductively coupled plasma mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:1521–1530. doi: 10.1002/rcm.2985. [DOI] [PubMed] [Google Scholar]
- 39.Galanski M, Arion VB, Jakupec MA, Keppler BK. Recent developments in the field of tumor-inhibiting metal complexes. Curr Pharm Des. 2003;9:2078–2089. doi: 10.2174/1381612033454180. [DOI] [PubMed] [Google Scholar]
- 40.Kapitza S, Pongratz M, Jakupec MA, Heffeter P, Berger W, et al. Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells. J Cancer Res Clin Oncol. 2005;131:101–110. doi: 10.1007/s00432-004-0617-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapitza S, Jakupec MA, Uhl M, Keppler BK, Marian B. The heterocyclic ruthenium(III) complex KP1019 (FFC14A) causes DNA damage and oxidative stress in colorectal tumor cells. Cancer Lett. 2005;226:115–121. doi: 10.1016/j.canlet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Heffeter P, Pongratz M, Steiner E, Chiba P, Jakupec MA, et al. Intrinsic and acquired forms of resistance against the anti-cancer ruthenium compound KP1019 [indazolium trans-[tetra-chlorobis(1H-indazole)ruthenate (III)] (FFC14A) J Pharmacol Exp Ther. 2005;312:281–289. doi: 10.1124/jpet.104.073395. [DOI] [PubMed] [Google Scholar]
- 43.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, et al. From bench to bedside–preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlor-obis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A) J Inorg Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 44.Lentz F, Drescher A, Lindauer A, Henke M, Hilger RA, et al. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs. 2009;20:97–103. doi: 10.1097/CAD.0b013e328322fbc5. [DOI] [PubMed] [Google Scholar]
- 45.Henke MM, Richly H, Drescher A, Grubert M, Alex D, et al. Pharmacokinetic study of sodium trans[tetrachlorobis(1H-indazole)-ruthenate (III)]/-indazole hydrochloride (1:1.1) (FFC14A) in patients with solid tumors. Int J Clin Pharmacol Ther. 2009;47:58–60. doi: 10.5414/cpp47058. [DOI] [PubMed] [Google Scholar]
- 46.Morris RE, Aird RE, del Murdoch PS, Chen H, Cummings J, et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J Med Chem. 2001;44:3616–3621. doi: 10.1021/jm010051m. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Parkinson JA, Parsons S, Coxall RA, Gould RO, et al. Organometallic ruthenium(II) diamine anticancer complexes: arene-nucleobase stacking and stereospecific hydrogen-bonding in guanine adducts. J Am Chem Soc. 2002;124:3064–3082. doi: 10.1021/ja017482e. [DOI] [PubMed] [Google Scholar]
- 48.Chen H, Parkinson JA, Morris RE, Sadler PJ. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: novel recognition mechanisms. J Am Chem Soc. 2003;125:173–186. doi: 10.1021/ja027719m. [DOI] [PubMed] [Google Scholar]
- 49.Hayward RL, Schornagel QC, Tente R, Macpherson JS, Aird RE, et al. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother Pharmacol. 2005;55:577–583. doi: 10.1007/s00280-004-0932-9. [DOI] [PubMed] [Google Scholar]
- 50.Gaiddon C, Jeannequin P, Bischoff P, Pfeffer M, Sirlin C, et al. Ruthenium (II)-derived organometallic compounds induce cytostatic and cytotoxic effects on mammalian cancer cell lines through p53-dependent and p53-independent mechanisms. J Pharmacol Exp Ther. 2005;315:1403–1411. doi: 10.1124/jpet.105.089342. [DOI] [PubMed] [Google Scholar]
- 51.Aird RE, Cummings J, Ritchie AA, Muir M, Morris RE, et al. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br J Cancer. 2002;86:1652–1657. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster RE, Cole DA, Mead S, Sadler PJ, Grimshaw KM. Investigation into the mechanism of action of the ruthenium(II) organometallic complex, ONCO 4417. Proceedings of the American association for cancer research; April 18–22; 2009. p. Abstract 889. [Google Scholar]
- 53.Scolaro C, Bergamo A, Brescacin L, Delfino R, Cocchietto M, et al. In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J Med Chem. 2005;48:4161–4171. doi: 10.1021/jm050015d. [DOI] [PubMed] [Google Scholar]
- 54.Vock CA, Scolaro C, Phillips AD, Scopelliti R, Sava G, et al. Synthesis, characterization, and in vitro evaluation of novel ruthenium(II) η6-arene imidazole complexes. J Med Chem. 2006;49:5552–5561. doi: 10.1021/jm060495o. [DOI] [PubMed] [Google Scholar]
- 55.Scolaro C, Geldbach TJ, Rochat S. Influence of hydrogen-bonding substituents on the cytotoxicity of RAPTA compounds. Organometallics. 2006;25:756–765. [Google Scholar]
- 56.Scolaro C, Chaplin AB, Hartinger CG, Bergamo A, Cocchietto M, et al. Tuning the hydrophobicity of ruthenium(II)-arene (RAPTA) drugs to modify uptake, biomolecular interactions and efficacy. Dalton Trans. 2007;43:5065–5072. doi: 10.1039/b705449a. [DOI] [PubMed] [Google Scholar]
- 57.Chatterjee S, Kundu S, Bhattacharyya A, Hartinger CG, Dyson PJ. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53-JNK pathways. J Biol Inorg Chem. 2008;13:1149–1155. doi: 10.1007/s00775-008-0400-9. [DOI] [PubMed] [Google Scholar]
- 58.Schäfer S, Ott I, Gust R, Sheldrick WS. Influence of the polypyridyl (pp) ligand size on the DNA binding properties, cytotoxicity and cellular uptake of organoruthenium(II) complexes of the type [(η6-C6Me6)Ru(L)(pp)]n+ [L = Cl, n = 1; L = (NH2)2CS, n = 2] Eur J Inorg Chem. 2007;19:3034–3046. [Google Scholar]
- 59.Schatzschneider U, Niesel J, Ott I, Gust R, Alborzinia H, et al. Cellular uptake, cytotoxicity, and metabolic profiling of human cancer cells treated with ruthenium(II) polypyridyl complexes [Ru(bpy)2(N–N)]Cl2 with N–N = bpy, phen, dpq, dppz, and dppn. Chem Med Chem. 2008;3:1104–1109. doi: 10.1002/cmdc.200800039. [DOI] [PubMed] [Google Scholar]
- 60.Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, et al. Exploring chemical space with organometallics: ruthenium complexes as protein kinase inhibitors. Synlett. 2007;8:1177–1189. [Google Scholar]
- 61.Smalley KS, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 62.Debreczeni JE, Bullock AN, Atilla GE, Williams DS, Bregman H, et al. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew Chem Int Ed Engl. 2006;45:1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 63.Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105:1759–1767. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- 64.Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006;66:3828–3835. doi: 10.1158/0008-5472.CAN-05-2309. [DOI] [PubMed] [Google Scholar]
- 65.US Department of Health and Human Services Food and Drug Administration. [last accessed 02/05/2010];Innovation or stagnation: challenges and opportunity on the critical path to new medical products. 2004 http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html.
- 66.LoRusso PM. Phase 0 clinical trials: an answer to drug development stagnation? J Clin Oncol. 2009;27:2586–2588. doi: 10.1200/JCO.2008.21.5798. [DOI] [PubMed] [Google Scholar]