

Abstract
Polymer nanogels have gained considerable attention as a potential platform for drug delivery applications. Here we describe the design and synthesis of novel polypeptide-based nanogels with hydrophobic moieties in the cross-linked ionic cores. Diblock copolymer, poly(ethylene glycol)-b-poly(L-glutamic acid), hydrophobically modified with L-phenylalanine methyl ester moieties was used for controlled template synthesis of nanogels with small size (ca. 70 nm in diameter) and narrow particle size distribution. Steady-state and time-resolved fluorescence studies using coumarin C153 indicated the existence of hydrophobic domains in the ionic cores of the nanogels. Stable doxorubicin-loaded nanogels were prepared at high drug capacity (30 w/w%). We show that nanogels are enzymatically-degradable leading to accelerated drug release under simulated lysosomal acidic pH. Furthermore, we demonstrate that the nanogel-based formulation of doxorubicin is well tolerated and exhibit an improved antitumor activity compared to a free doxorubicin in an ovarian tumor xenograft mouse model. Our results signify the point to a potential of these biodegradable nanogels as attractive carriers for delivery of chemotherapeutics.
Keywords: nanogel, PEG-b-Poly(L-glutamic acid), phenylalanine, core-shell morphology, doxorubicin
Introduction
A special type of polymeric “soft” materials, nanosized hydrogels (nanogels), has attracted significant attention as promising pharmaceutical carriers for delivery of therapeutic and diagnostic agents. These materials have several important advantages over other particulate delivery systems such as stability, versatility, flexibility, high loading capacity and biocompatibility (Chacko et al., 2012, Kabanov and Vinogradov, 2009, Vinogradov et al., 2002). They can be designed to facilitate the incorporation of a variety of compounds or even particles through a combination of electrostatic, hydrophobic, and hydrogen bonding interactions. The nanogel composition, size and swelling behavior can be tuned to control the drug-release characteristics of the nanogel carriers. Furthermore, attachment of specific ligands to nanogel surface enables targeted drug delivery (Murphy et al., 2011, Nukolova et al., 2011)
We have previously developed novel type of ionic nanogels with core-shell spatial distribution of polymer chains using controlled template synthesis. The fabrication procedure involved a preparation of micellar templates by the self-assembly of double hydrophilic block copolymers (poly(ethylene glycol)-b-poly(methacrylic acid), PEG-b-PMA) with oppositely charged condensing agent (e.g., Ca2+ or Ba2+). This was followed by chemical cross-linking of ionic blocks in the core and removal of condensing agent (Bronich et al., 2005). The resulting nanogels contained hydrophilic cross-linked PMA ionic cores surrounded by a flexible hydrophilic PEG. Control over the size and pH-dependent swelling behavior was systemically achieved by varying the degree of cross-linking and the chemical structure of cross-linkers (Kim et al., 2009, Oberoi et al., 2011). Such nanogels can entrap diverse chemical and biological agents for cancer therapy with very high loading capacities. Incorporation of cisplatin into the nanogels by polymer-metal complex formation improved drug pharmacokinetics, enhanced its antitumor efficacy, and eliminated cisplatin-mediated nephrotoxicity in a mouse model of ovarian cancer (Oberoi et al., 2012). We demonstrated that the integration of targeting folate moieties onto the surface of nanogels could further facilitate their selective accumulation in tumor tissue and potentiate the anti-cancer efficacy of the drug (Nukolova, et al., 2011). Thus, our findings indicated that nanogel-based anti-cancer therapeutics hold great potential as an effective treatment modality in cancer. However, because these nanogels are not degradable, there is a concern for their long-term accumulation in the body that will impede the translation of such nanomedicines to practice.
Among the recently developed nanomedicine platforms poly(amino acids)-based polymers are particularly interesting because of their biocompatibility, biodegradability and lack of toxicity (Carlsen and Lecommandoux, 2009, Lavasanifar et al., 2002, Li, 2002). OPAXIO™, a poly-L-glutamate-paclitaxel conjugate, showed clinical benefits in women patients with non-small-cell lung cancer (Langer et al., 2008) and is currently under evaluation for esophageal cancer (Ng et al., 2010). Kataoka’s group has developed several micellar formulations of anticancer drugs based on PEG-polyaspartate or PEG-polyglutamate block copolymers that are undergoing phase I/II clinical trials and showing improved antitumor efficacy and reduced systemic toxicity (Bae and Kataoka, 2009, Matsumura, 2008, Matsumura and Kataoka, 2009). In present work, we explored PEG-b-poly(L-glutamic acid) block copolymers for development of biodegradable nanogels. Toward this goal, micellar templates were prepared by using self-assembled aggregates of phenylalanine-modified PEG-b-poly(L-glutamic acid) (PEO-b-PPGA), which were further condensed by addition of Ca2+ ions. Cystamine, a biodegradable cross-linker, was utilized for the cross-linking of nanogels. Our results demonstrate that the presence of hydrophobic moieties in the ionic cross-linked cores of nanogels greatly determine their swelling behavior, doxorubicin loading capacity and release characteristics. In addition, we evaluated an anti-tumor effect of drug-loaded nanogels on cancer cell lines in vitro and in vivo in tumor-bearing mice.
Experimental Section
Materials
Poly(ethylene glycol)-b-poly(L-glutamic acid) (PEG-b-PGA) diblock copolymer (Mw/Mn = 1.38, MW 27,500) was purchased from Alamanda Polymers, Inc (Madison, AL, USA). The block lengths were 114 and 150 repeating units for PEG and PGA, respectively. Doxorubicin hydrochloride was a kind gift from Dong-A Pharmaceutical Company, South Korea. Poly(L-glutamic acid) sodium salt (MW 3,000 – 15,000), L-phenylalanine methyl ester hydrochloride, calcium chloride, cystamine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and coumarin 153 (C153) were obtained from Sigma-Aldrich (St Louis, MO). Lysotracker™ (green), fetal bovine serum (FBS: both dialyzed and heat inactivated) and Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Invitrogen Inc (Carlsbad, CA). Bovine serum albumin (BSA) and NUNC™ chambered glass coverslips for live cell imaging was purchased from Fisher Scientific (Waltham, MA). MTT reagent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, was purchased from Research Products International (Prospect, IL). All other chemicals were of reagent grade and used without further purification.
Synthesis of hydrophobically modified PEG-b-PGA
PEG-b-PGA was hydrophobically modified by the conjugation of L-phenylalanine methyl ester hydrochloride (PME) in the presence of EDC. Copolymers (further denoted as PEG-b-PPGA) with targeted degrees of PME grafting of 25% and 50% were prepared by varying the molar ratio of the glutamic acid residues of PEG-b-PGA to PME. Equimolar amounts of EDC and PME (0.137 mmol or 0.275 mmol) were added to aqueous solution PEG-b-PGA (2 mL, 100mg, 0.545 mmol carboxylate groups) and stirred for 24 h at r.t.. The pH of the reacting solution was ~ 6.0. The resulting copolymers were purified by dialysis against distilled water, freeze-dried and characterized by 1H-NMR spectroscopy (Varian 500 MHz spectrometer, D2O 25°C). The degree of grafting of PME was determined by comparing relative signal intensities of oxymethylene protons of PEG (3.7 ppm) and phenyl group protons of PME (7.1–7.4 ppm). The concentration of carboxylate groups in the copolymer samples was estimated by potentiometric titration.
Synthesis of nanogels with cross-linked ionic cores
Nanogels with cross-linked ionic cores were prepared by using block ionomer complexes of the PEG-b-PPGA and divalent metal cations (Ca2+) as templates by the previously described method with a slight modification. In brief, PEG-b-PPGA/Ca2+ complexes were prepared by mixing an aqueous solution of PEG-b-PPGA with a solution of CaCl2 at a molar ratio of [Ca2+]/[COO−] = 1.5. The EDC (0.2 eq) and cystamine (0.1 eq) were then added to the dispersion of PEG-b-PPGA/Ca2+ complexes (eq are with respect to the amount of carboxylate groups) to achieve 20% of cross-linking degree. This degree represents the maximum theoretical amount of cross-linking that could take place, rather than the precise extent of amidation. The reaction mixture was allowed to stir overnight at r.t. Metal ions and byproducts of the cross-linking reaction were removed by exhaustive dialysis of the reaction mixtures first against 0.5% aqueous ammonia in the presence of EDTA, and then against distilled water. Nanogels composed of double hydrophilic PEG-b-PGA were synthesized using PEG-b-PGA/Al3+ complexes prepared at a molar ratio [Al3+]/[COO−] = 1.35. The chains were cross-linked using EDC and cystamine at 70% targeted degree of cross-linking ([EDC]/[ED] = 2; [COOH]/[EDC] = 1.4).
Turbidity measurements
The turbidity measurements were carried out at 420 nm using a Perkin-Elmer Lambda 25 UV/VIS spectrophotometer after equilibration of the system for 3 min, which was proven to be sufficient for equilibration. The data are reported as (100 – T)/100, where T is transmittance (%).
Particle characterization
The effective hydrodynamic diameter (Deff) and ζ-potential of nanogels were determined using a Malvern Zetasizer (Malvern Instruments Ltd., Malvern, UK). All measurements were performed in automatic mode, at 25°C. Software provided by the manufacturer was used to calculate size, polydispersity indices (PDI) and ζ-potential of nanogels. The values were calculated from the measurements performed at least in triplicate.
Atomic Force Microscopy (AFM)
Samples for AFM imaging were prepared by depositing 5 μL of an aqueous dispersion of nanogels (ca. 1.0 mg/mL) onto positively charged 1-(3-aminopropyl)silatrane mica surface (APS-mica) for 2 min, followed by surface drying under argon atmosphere. The AFM imaging in air was performed with regular etched silicon probes (TESP) with a spring constant of 42 N/m using a Multimode NanoScope IV system (Veeco, Santa Barbara, CA) operated in a tapping mode. The images were processed and the widths and heights of the particles were determined by using Femtoscan software (Advanced Technologies Center, Moscow, Russia).
Circular dichroism (CD) spectroscopy
The CD spectra were recorded using Aviv circular dichroism spectrometer (model 202SF, Aviv Associates, Inc., Lakewood, NJ) equipped with a Peltier temperature controller. The scans were taken from 260 to 200 nm at 1 nm intervals with a scan rate of 15 nm/min using a 1 cm pathlength cell at 25, 37 and 50°C. Samples were prepared in 10mM phosphate buffer at pH 7.0. The pH of the solution was adjusted using either a 0.1 M HCl or NaOH solution until the desired pH was obtained. The samples were allowed to equilibrate for 20 min at each temperature. All the spectra were acquired in triplicate and averaged. Mean residual ellipticity ([θMRE], deg cm2/dmol) was calculated as [θMRE] = (θ)/10lcn, where (θ) is the measured ellipticity (mdeg), l is the path length (Zhou et al.), c is the polymer molar concentration and n is the number of residues in the peptide. The α-helix contents were estimated using DICHROWEB software.
Fluorescence measurements
Steady-state fluorescence spectra of pyrene as the fluorescent probe were recorded with a Flourlog3 spectrofluorometer (HORIBA Jobin Yvon Inc., NJ, USA) at λex = 336 nm, λem = 350 – 460 nm with the slit width of 1 nm for excitation and emission. For sample preparation known amounts of stock solution of pyrene in acetone were added to empty vials, followed by acetone evaporation. Aqueous solutions of polymer samples were added to the vials and kept overnight under constant stirring at r.t. The pyrene concentration in the final solution was 6 × 10−7 M, a concentration slightly below the solubility of pyrene in water at 22°C. All measurements were performed at r. t. using air-equilibrated solutions in a quartz cell with 1 cm optical path length. In separate experiments, 25 μl of coumarin 153 (C153) stock solution (1mg/mL in acetone) was added to the vials and solvent was evaporated. Polymer samples (1 mg/mL in 10mM phosphate buffer at pH 7) were added to these vials and incubated overnight at r.t. Final concentration of C153 in solutions was 10 μg/mL. Fluorescence emission spectra of C153 in each solution were recorded at λex = 425 nm and λem = 460 – 600 nm (slit width (ex) = slitwidth (em) = 1 nm). The same samples were further used to determine fluorescence lifetimes of C153 by time-correlated single-photon counting spectroscopy (TCSPC) using NanoLED (Ex = 460 nm) as the excitation source. TCSPC instrumental response profiles were obtained by scattering excitation light from an aqueous suspension of nondairy creamer. The C153 fluorescenece decays were measured at different emission (522 –552 nm) wavelengths depending on copolymer sample. The TCSPC transients were acquired over 4096 channels with up to 10,000 counts at the peak maximum. Data were collected at less than 2% of the source repetition rate to avoid photon pile up effects. Decay curves were analyzed by nonlinear least-squares fitting algorithm using DAS6 decay analysis software (Ng, Fontaine).
Drug loading and release
Nanogel dispersions were mixed with DOX (2 mg/mL) at a feeding ratio of R = 0.5 (R is a molar ratio of DOX to carboxylate groups of the nanogels) at pH 7.0, followed by incubation for 24 h at r.t. The unbound DOX was removed by ultrafiltration using Amicon YM-30 centrifugal filter devices (MWCO 30,000 Da, Millipore) pretreated with DOX. DOX was assayed by measuring the absorbance at 485 nm using Lambda 25 UV/VIS spectrophotometer. Drug loading capacity was calculated as percent ratio of mass of incorporated drug to total mass of drug-loaded nanogels without water. Drug release (minimum of 200 μg DOX) was examined in phosphate buffered saline (PBS, pH 7.4, 0.14 M NaCl), acetate buffered saline (ABS, pH 5.5, 0.14 M NaCl), and ABS in the presence of cathepsin B (10 units/mL) at 37°C by equilibrium dialysis method using a membrane 3,500 Da cutoff and expressed as a percentage of the total DOX and plotted as a function of time.
Confocal microscopy on live cells
MCF-7 human breast cancer cells (1×106/chamber) were grown in live cell chambers (Fischer Scientific, Waltham, MA) in DMEM media for 2 days (37°C, 5% CO2) and exposed to DOX-loaded PEG-b-PPGA nanogels for 45 min followed by incubation with Lysotracker Green® for 5 min. After exposure cells were washed with PBS and kept in DMEM media for live cell confocal imaging (Carl Zeiss LSM 510 Meta, Carl Zeiss Inc., Thornwood, NY, USA).
In vitro cytotoxicity studies
Cells seeded in 96-well plates (5,000 cells/well) 24 h before the experiments were exposed to various doses of DOX alone (0–50 μg/ml), nanogels alone and DOX-loaded nanogels for 24 h and then cultured for additional 72 h in drug-free media 37 °C. Cytotoxicity was determined by a standard MTT assay (Ferrari et al., 1990) and the IC50 values (dose which kill 50% of cells) were calculated by using GraphPad Prism Software (GraphPad Software, San Diego California, USA).
Anti-tumor efficacy
Anti-tumor activity was evaluated in four-week-old female athymic (Ncr-nu/nu) mice bearing subcutaneous A2780 cell ovarian xenografts. Mice with 100–200 mm3 tumors (4–7 mm in each dimension, approximately 2 weeks after inoculation) were randomized to four treatment groups with similar mean tumor volumes of each group (n = 6). Treatments (5% dextrose, empty nanogel, DOX alone, DOX-loaded nanogel) were administered via tail vein injections at 4-day intervals at an equivalent dose of 4 mg-DOX/kg. Animal body weight and tumor volume were monitored every second day. Tumor volume (V = 0.5 x L x W2) was estimated by measuring two orthogonal diameters (longer dimension: L, and smaller dimension: W) of the tumor using electronic calipers. Animals were sacrificed when greatest tumor dimension exceeded 20 mm, tumor became necrotic, or animal exhibited a body weight loss of more than 20%. All other animals were sacrificed by day 20. Protocols were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Statistical differences were determined using a one-way ANOVA followed by Tukey’s test for comparison of treatment. All statistical analyses were carried out using GraphPad Prism Software (Version 5.0, GraphPad Software, CA, USA). The p-values less than 0.05 were considered statistically significant.
Results and Discussion
Design and Synthesis of Cross-linked Nanogels
We extended our synthetic approach using a template-assisted procedure in order to develop biodegradable cross-linked nanogels (Figure 1). The proposed design implicates a replacement of the PMA core segment of the previously reported nondegradable PEG-b-PMA nanogels with enzymatically degradable poly(L-glutamic acid). However, the condensation of block copolymer precursor, PEG-b-PGA, with Ca2+ ions did not result in the formation of micellar templates. To address this issue, hydrophobically modified PEG-b-PGA derivatives (PEG-b-PPGA) were synthesized by carbodiimide mediated grafting of PGA segments with L-phenylalanine methyl ester (PME) moieties. Two PEG-b-PPGA copolymers with different degrees of PME grafting were prepared by varying the molar ratio of the glutamic acid residues of PEG-b-PGA to PME. The degrees of PME grafting were 17% and 30% as was determined by 1H-NMR analysis. These copolymers are further denoted as PEG-b-PPGA17 and PEG-b-PPGA30, respectively.
Figure 1.

Scheme for the synthesis of cross-linked PEG-b-PPGA nanogels.
Hydrophobically modified water-soluble polymers and polyelectrolytes exhibit unusual aqueous solution behavior due to hydrophobic associations that occur in order to minimize water-hydrophobe contacts (McCormick CL, 1989). The tendency of intra- or intermolecular association strongly depends on macromolecular architecture, in particular, on the number and distribution of hydrophobic groups along the polymer backbone. Fluorescent technique using pyrene as a probe is widely used for characterization of the self-organization of hydrophobically modified polymers and the nature of thus formed hydrophobic domains. This method is based on the sensitivity of the spectroscopic properties of pyrene to the polarity of its microenvironment. The partitioning of the pyrene probe into the less polar environment results in a characteristic decrease of the intensity ratio of the third and first vibrational peaks (I1/I3) along with increasing fluorescence intensity. Steady-state fluorescence spectra of pyrene in the presence of PEG-b-PPGA copolymers were utilized to qualitatively characterize the association of phenylalanine groups or lack thereof. Figure 2A depicts the dependence of I1/I3 values of pyrene as a function of PEG-b-PPGA concentration in aqueous solutions (10 mM phosphate buffer, pH 7.0). In aqueous or similarly polar environment I1/I3 ratio is found between 1.6 and 1.9 (Dong and Winnik, 1982, Kalyanasundaram and Thomas, 1977). As expected, I1/I3 value measured for pyrene in solutions of double hydrophilic PEG-b-PGA copolymer of various concentrations was approximately 1.8 reflecting a polarity of bulk water (Figure 2A). Remarkably, no changes in spectroscopic characteristics of pyrene probe were detected in the solutions of PEG-b-PPGA17. I1/I3 remained approximately equal, within experimental error, to its value in water in the entire range of concentrations studied (up to 3 mg/mL). These data can indicate an absence of hydrophobic associations in the PEG-b-PPGA17 solutions. In contrast, for PEG-b-PPGA30 as the copolymer concentration increased, the I1/I3 decreased and leveled off at a value of 1.45–1.49 at concentrations above 0.2 mg/mL. The polarity of the local microenvironment of pyrene resembled that in the cores of block copolymer micelles formed by hydrophobic blocks of moderate polarity such as poly(ε-caprolactone) (Wang et al., 2005), poly(n-butyl acrylate) (Colombani et al., 2007). These observations suggest that pyrene molecules reside inside the hydrophobic domains formed through association of pendant phenylalanine groups in solutions of PEG-b-PPGA30 copolymer. No macroscopic aggregation was detected by dynamic light scattering (DLS) in PEG-b-PPGA30 solutions in this range of concentrations (up to 0.2 mg/mL). It appears that at higher degree of grafting the random modification of the carboxylic groups of PGA segment leads to the formation of PME-rich regions that may serve as domains for pyrene solubilization. However, we do not exclude the possibility that some loose pre-aggregates of copolymer chains stabilized by intermolecular hydrophobic associations may exist in diluted PEG-b-PPGA30 solutions. Indeed, a slight change in the slope of concentration dependence of fluorescence intensity I1 was observed at PEG-b-PPGA30 concentration of 0.3 mg/mL (Figure 2B) and may be attributed to onset of intermolecular self-assembly. Notably, the formation of small (intensity-average diameter of approximately 71 nm) particles with relatively narrow particle size distribution (PDI = 0.13) was detected in PEG-b-PPGA30 solutions at higher concentration (1 mg/mL). This observation also implies that hydrophobic interactions at the microscopic level may take place at much lower concentration than reflected by macroscopic properties.
Figure 2.
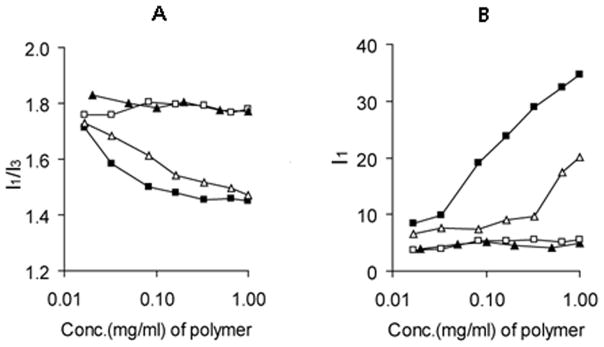
(A) Variation of the I1/I3 ratio (I373nm/I383nm). (▲) PEG-b-PGA, (□) PEG-b- PPGA17, (△) PEG-b-PPGA30 and (■) cl-PEG-b-PPGA30 nanogels. (B) The normalized emission fluorescence intensity at 373 nm (I1) for various PGA-based polypeptides and cross-linked nanogels as a function of polymer concentration at 25°C and pH 7.0. Concentration of pyrene is 6 × 10−7 M.
Complexes of PEG-b-PPGA with Ca2+ were prepared by simple mixing of an aqueous solution of the corresponding copolymer with a solution of CaCl2 (Bellomo et al., 2004). The BIC formation was monitored by turbidimetic titration. Figure 3 presents the data on turbidity of PEG-b-PPGA/Ca2+ mixtures as a function of the charge ratio in the mixture, Z. The latter was calculated as Z = Cmn/Ci, where Cm is Ca2+ molar concentration, n is the valence of the metal ion (= 2), and Ci is the molar concentration of the carboxylate groups of PPGA chains at a given pH. The experiments were carried out at pH 8.0, when the most of the carboxyl groups of the PPGA are ionized (pKa of PGA is 4.4 (Li, 2002). A turbidimetric titration curve for PEG-b-PGA/Ca2+ mixture is also presented in Figure 3. Contrary to PEG-b-PGA/Ca2+ mixtures that were transparent in the entire range of the charge ratios studied, the formation of slightly opalescent dispersion was observed in PEG-b-PPGA30/Ca2+ mixtures in the vicinity of Z = 1.7. At this critical ratio and above the nanosized particles (30–40 nm in diameter) were detected in the dispersions by DLS. It appears that hydrophobic and π-π stacking interactions of the multiple phenylalanine moieties played a major role in driving self-assembly in these systems. Notably, formation of aggregates was not observed for PEG-b-PPGA17 copolymer with lower degree of PME grafting even at significant excess of Ca2+ ions. This indicates that specific self-assembly behavior of PEG-b-PPGA/Ca2+ complexes is determined by a fine interplay between screened electrostatic and hydrophobic interactions. A certain critical content of relatively hydrophobic PME groups needs to be grafted to polar and highly hydrated PGA segment to trigger the formation of BIC nanoaggregates.
Figure 3.

(A) Turbidity in the PEG-b-PGA/Ca2+ mixtures as a function of the charge ratio in the mixture, Z: (▲) PEG-b-PGA/Ca2+, (□) PEG-b-PPGA17/Ca2+ and (■) PEG-b-PPGA30/Ca2+.
The PEG-b-PPGA30/Ca2+ BIC (Z = 3) were further utilized as templates for synthesis of the nanogels as outlined in Figure 1. The cross-linking of the PPGA30/Ca2+ cores was achieved via condensation reactions between the carboxylic groups of PPGA segments and the amine groups of cystamine in the presence of a water-soluble carbodiimide, EDC. The targeted extent of cross-linking (20%) was controlled by the molar ratio of cross-linker to carboxylic acid groups of the glutamic acid residues. After completion of the cross-linking reaction the size of the PEG-b-PPGA30/Ca2+ micelles in the dispersion was similar to that of the precursor complexes (37 nm vs. 34 nm), confirming that the micelles retained their integrity and that no observable intermicellar fusion can be detected. After exhaustive dialysis against water cross-linked nanogels (cl-PEG-b-PPGA) were isolated and characterized. The resulting nanogels were uniform (PDI = 0.11), had net negative charge and displayed an effective diameter of about 72 nm (pH 7). Noteworthy, the size of formed nanogel was significantly larger than the size of the original PEG-b-PPGA30/Ca2+ template (ca. 34 nm). This corresponded to the 2.1-fold increase in the diameter and 9.3-fold increase in the volume of the particles. Such an expansion was consistent with the removal of the metal ions and swelling of the nanogels.
The success of cross-linking reactions was further confirmed by testing the stability of the nanogels in the presence of urea. The ability of aqueous urea to act as a solvent for both nonpolar and polar groups of proteins plays a vital role in protein unfolding and stabilization of the denatured forms (Rossky, 2008). Therefore, it was expected that urea is able to destabilize PEG-b-PPGA30 micellar aggregates by weakening the hydrophobic interactions among phenylalanine pendant groups in the core region as well as by disrupting hydrogen-bonding interactions between polypeptide chains. Indeed, significant increase in the size along with the drastic increase of polydispersity index (PDI = 0.88) was detected by DLS in the dispersion of non-cross-linked micelles after addition of 8 M urea suggesting their structural disintegration. In the meantime, cl-PEG-b-PPGA nanogels remained stable and exhibited only little changes in average size in the presence of urea (Figure S1).
The dimensions and morphology of cl-PEG-b-PPGA nanogels were further characterized by tapping-mode AFM in air. The typical topographic image of the nanogels showed round nanoparticles with a narrow distribution in size (Figure 4). As expected the number-average particle height (10.3 ± 0.2 nm) and diameter (27.7 ± 0.2 nm) values of dehydrated nanogels were reduced compared to the hydrodynamic diameters determined by DLS. The low height values relative to the diameters indicate that significant deformation occurred upon absorption onto the mica surface, the substrate for AFM characterization, which is commonly observed for soft objects measured with tapping-mode AFM in air due to the loading force of the tip and dehydration. Furthermore, the electrostatic interactions of the negatively charged nanogels with the positively charged amino-modified mica surface might force additional flattering of the nanogel particles. Interestingly, cl-PEG-b-PPGA nanogels containing hydrophobic moieties in the cross-linked cores were characterized by a significantly lower aspect ratio (ca. 2.7) compared to PEG-poly(methacrylic acid) (PEG-PMA) nanogels containing entirely hydrophilic PMA cores stabilized by cystamine cross-links (aspect ratio of 12.2) (Kim et al., 2010). These data suggest that the presence of hydrophobic domains formed by phenylalanine moieties within the cores of the cl-PEG-b-PPGA nanogels provided substantial reinforcement of the core and then limit the deformation of the nanogels at the surface. It is also possible that “macromolecular crowding” effect within the volume of the cross-linked PPGA cores of the nanogels might further provoke intermolecular association of the hydrophobic groups. To test this hypothesis we examined nanogels using the hydrophobic and solvatochromic fluorescent probe coumarin 153 (C153). Photophysical properties of C153 are extremely sensitive to the environment of the dye molecule, especially the local polarity and microviscosity, and, therefore, have been used for dynamic probing of different complex environments (Jin et al., 2007, Kumbhakar, 2007, Steege et al., 2007). Specifically, the blue shift of the C153 emission maximum is occurred upon transfer of the probe from polar to the nonpolar environment. Steady-state fluorescence emission spectra of C153 in the solutions of various PGA-based copolymers and cl-PEG-b-PPGA nanogels are presented in Figure 5A and the wavelengths of the maximum emission are listed in Table 1. It is evident that the emission maximum of C153 in a solution of PEG-b-PPGA17 (λmax = 551.5 nm) is essentially identical to those in buffer and unmodified PEG-b-PGA aqueous solution (λmax = 552 nm). This indicates that C153 experiences a highly polar environment in PEG-b-PPGA17 solution, which is consistent with an absence of the hydrophobic association of grafted phenylalanine groups of PEG-b-PPGA17. An observed shift to shorter wavelength in the emission spectrum of C153 in the dispersion of PEG-b-PPGA30 aggregates (λmax = 541.5 nm) is a manifestation of the decrease in the local solvent polarity of the environment suggesting incorporation of the probe into the phenylalanine hydrophobic domains. These results are thus in accordance with the pyrene solubilization studies discussed above. Interestingly, when C153 was solubilized within the cl-PEG-b-PPGA nanogels, it exhibited a drastic blue shift of the emission maximum to 522.5 nm. This result implies that the local environment of the probe in the phenylalanine domains in the cores of the nanogels is more hydrophobic in comparison to those in non-crosslinked PEG-b-PPGA30 aggregates. It should be noted that a similar spectral blue shift was observed for C153 during aggregation of Pluronic block copolymers undergoing the unimer-to-micelle phase transition (Kumbhakar et al., 2006). It has been shown that exclusion of the water molecules and burying of poly(propylene oxide) blocks in the micelle cores led to a significant reduction in local solvent polarity of the probe. Therefore, we can infer that the local environment of C153 in PEG-b-PPGA30 nanogels corresponds to presumably “dry” surroundings much like the cores of Pluronic micelles. We can further compare the polarity of local environment in nanogels with that of common organic solvents using empirical π* solvatochromic polarity scale (Horng et al., 1995). It has been demonstrated that there is a very good correlation between the π* values of the solvent and the frequency of C153 emission maximum given as νem [10−3 cm−1] = 21.217–3.505π* (Horng, et al., 1995). According to this relationship, the π* value for C153 incorporated into PEG-b-PPGA30 aggregates is about 0.78, close to the polarity of dichloromethane (π* = 0.73) and nitromethane (π* = 0.75) (Horng, Gardecki, 1995). In nanogels, the local environment of C153 has π* value of 0.58 that corresponds to the polarity similar to benzene or tetrahydrofuran (π* = 0.55). This drop in the effective polarity may reflect the rearrangements of phenylalanine domains and thus water molecules associated with nanogel cores. The phenylalanine domains in the cross-linked cores of nanogels are likely to become more hydrophobic and do not contain polar water molecules to the extent that the PEG-b-PPGA30 aggregates.
Figure 4.
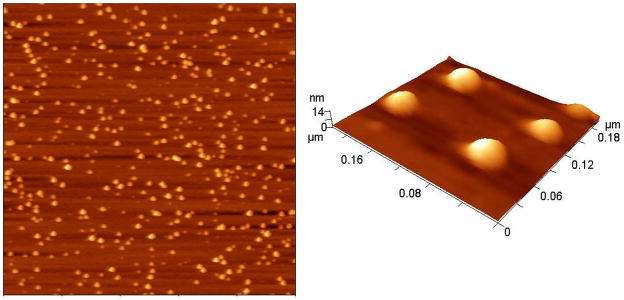
Tapping mode AFM images of cl-PEG-b-PPGA nanogels in air. Targeted degree of cross-linking was 20%. Scan size was 2 μm.
Figure 5.

(A) Normalized steady-state emission spectra and (B) lifetime measurement of C153 in aqueous solutions of various PGA and nanogels. (1) Phosphate buffer (10mM, pH 7.0) (2) PEG-b-PGA, (3) PEG-b-PPGA17, (4) PEG-b-PPGA30, (5) cl-PEG-b-PPGA nanogels and (6) instrumental response. Concentration of polymers is 1 mg/ml at pH 7.0.
Table 1.
Steady state maximum fluorescence wavelength and fluorescence lifetime of C153 in various PGA-based polymer and nanogels.
| Samples | buffer | PEG-b-PGA | PEG-b-PPGA25 | PEG-b-PPGA50 | cl-PEG-b-PPGA nanogels |
|---|---|---|---|---|---|
| Max. Emission Wavelength (nm) | 552.0 | 552.0 | 551.5 | 541.5 | 522.5 |
| T1 (ns) | 1.57 | 1.59 | 1.54 | 1.66 | 3.15 |
| T2 (ns) | 2.20 | 2.27 | 2.29 | 4.56 | 6.70 |
| T1 amplitude (%) | 85.2 | 79.1 | 80.2 | 57.4 | 19.8 |
| T2 amplitude (%) | 14.8 | 20.9 | 19.8 | 42.6 | 80.2 |
Time-resolved fluorescence measurements were carried out to further substantiate the observed changes in the steady-state fluorescence of C153 incorporated into nanogels. The fluorescence decays of C153 as measured at its respective emission maxima peak in various PGA-based copolymers and cl-PEG-b-PPGA nanogels are shown in Figure 5B. All emission decays were best fitted into a bi-exponential function and the fluorescence lifetime parameters summarized in Table 1. It was observed that the probe lifetimes do not show significant changes in the cases of unmodified PEG-b-PGA and PEG-b-PPGA17 copolymers, giving the values comparable to those in phosphate buffer. On the contrary, the long component of C153 decay was shifted from 2.3 ns to 4.6 ns in the dispersion of PEG-b-PPGA30 aggregates indicating the association of the probes with the hydrophobic domains of PEG-b-PPGA30 aggregates. The increase in lifetime of the longer component of C153 emission decay (~6.7 ns) as well as in its fractional contribution was even more pronounced in cl-PEG-b-PPGA nanogels. Thus, C153 probe reported a substantial decrease in the polarity of the interior of the nanogels, which in turn can reflect the changes of the nanogel internal structure. Perhaps, the formation of denser polymer network in the cores of the nanogels results in the rearrangements of the hydrophobic domains and causes a less hydrated microenvironment around the probe. It is likely that the more hydrophobic, rigid core of cl-PEG-b-PPGA nanogels can have implications for the loading and retention of the encapsulated guest molecules.
It is important to note, that the cross-linking and restricted penetration of water molecules toward the cores of nanogels did not prevent their degradation by proteolytic enzymes. The enzymatic biodegradability of PGA-based nanogels was determined by incubating the nanogels with cathepsin B at pH 5.5, followed by analysis of the reaction mixture using size exclusion chromatography (SEC) and DLS (Figure S2). Nanogels were hydrolyzed relatively slowly: a noticeable decrease in the UV absorption of the nanogel peak and simultaneous appearance of secondary peak at increased elution times corresponding to products of lower molecular masses were observed after 48 h of incubation. Furthermore, a drastic increase in size and polydispersity index was detected by DLS in nanogel dispersions under these conditions suggesting enzymatically-driven nanogel destabilization. It is likely that the observed slow degradation of nanogels is due to the steric hindrances imposed by the compact structure of hydrophobically modified PPGA core, which prevented easy enzyme access to polymer substrate. Likewise, PME modification of γ-carboxylic group in the side chains of PGA may render the formation of enzyme-substrate complex more difficult, decreasing the probability of backbone cleavage. One can also speculate that initial hydrolysis of amide bonds of nanogels may mainly occur at the interface region between the core and the shell, resulting in partial detachment of PEG chains and potentially increased accessibility of enzymes to susceptible bonds in the polymer. On the other hand, hydrophobic interactions between the exposed PPGA core and products of their degradation will in turn lead to the formation of large aggregates over time. However, further studies will be necessary to characterize the degradation products and determine whether drug incorporation can alter the degradation pattern of the nanogels. Overall, it is believed that enzymatic degradability of cl-PEG-b-PPGA nanogels would be advantageous due to specific intracellular drug release triggered by disassembly of the delivery carrier and reduced risk of polymer accumulation inside the cells.
Swelling behavior of cl-PEG-b-PPGA nanogels
The nanogels studied in this work are composed of PGA, a weak polyelectrolyte (pKa 4.4). Since ionization degree of PGA increased at higher pH, dissociation of the glutamic acid carboxylic groups within the core induced intramolecular electrostatic repulsions and, thus, caused the overall swelling of the nanogel particles. Additionally, it is well-known that PGA chains can undergo a pH-dependent random-coil-to-helix transitions with apparent pKa of 5.4 (Abbruzzetti et al., 2000) and these conformational changes can also influence the swelling behavior of cl-PEG-b-PPGA nanogels. The pH-induced dimensional changes of nanogels were studied by DLS and electrophoretic mobility measurements, and the results are presented in Figure 6. No significant changes in size and ζ-potential of the nanogels were observed above pH 7 where the ionization of the PGA chains was essentially complete. A sharp decrease of hydrodynamic diameter with a concomitant increase in ζ-potential was determined below pH 7. The loss of the polyelectrolyte behavior, reduced osmotic pressure and transition to an ordered conformation upon protonation of acid residues of the cross-linked PPGA chains led to the collapse of the network that comprise the cores of the nanogels. It should be pointed out that the observed changes were completely reversible and the size distribution of nanogels remained relatively narrow (PDI < 0.14). In order to shed additional insight into self-organization properties of the peptide segments in the cores of nanogels, the effect of pH on the conformational behavior of PGA-based copolymers and cl-PEG-b-PPGA nanogels was studied using CD spectroscopy (Figure S3). Figure 7 depicts typical CD spectra for the prepared block copolymers and nanogels at pH 5 and pH 7. The CD spectra of the unmodified PEG-b-PGA copolymer showed the typical pattern of a random coil conformation at pH 7 and that of an α-helix with characteristic two negative minima at 208 and 222 nm at pH 5 (Figure 7A, B). The helicity value estimated using mean residue ellipticity at 222 nm was approximately 59% at pH 5 and was decreasing with increasing pH. These results are consistent with the pH-dependent coil-to-helix transition reported for PGA homopolymer and other PGA-based copolymers (Kukula et al., 2002). To highlight the effect of cross-linking on the ability of PEG-b-PGA to form ordered secondary structures, we also synthesized unmodified PEG-b-PGA nanogels (cl-PEG-b-PGA). Since no condensation of double hydrophilic PEG-b-PGA can be achieved using Ca2+ ions, PEG-b-PGA/Al3+ complexes were utilized as the templates for the synthesis of nanogels (70% targeted degree of cross-linking). The resulting cl-PEG-b-PGA nanogels had hydrodynamic diameter ca. 175 nm and broad size distribution (PDI = 0.29) at pH 7 as determined by DLS. The CD spectra of the cl-PEG-b-PGA were essentially identical to that of the parent PEG-b-PGA copolymer (Figure 7C). Interestingly, however, the coil-to-helix transition of the cross-linked nanogels was shifted to a higher pH value (~5.6) compared to that of linear copolymer (pH ~5.2) (Figure S3). This shift of the transition point can be attributed to the modulation of the apparent dissociation constant of the carboxylic acid groups in more compact internal structure of the PGA core of the nanogel: a higher density of the dissociable groups can cause a shift of their apparent pKa to higher values and therefore can stabilize α-helix conformation. Despite of the observed shift in transition the estimated helix content for cl-PEG-b-PGA at pH 5 was lower (~ 42%) than for PEG-b-PGA, which might be explained by the decreased conformational freedom of PGA segments due to high number of cross-links in the core. Another feature of CD spectra for both PEG-b-PGA and cl-PEG-b-PGA samples was the greater ellipticity values at 222 nm than at 208 nm. The mean residue ellipticity ratio, [θ]222nm/([θ]208nm, is usually used to distinguish whether the helices are monomeric ([θ]222nm/([θ]208nm < 0.9) or are adopting coiled coil conformation ([θ]222nm/([θ]208nm ≥ 1) (Zhou, Kay, 1992). The ellipticity ratio in the range of 1.06 – 1.1 for PEG-b-PGA and cl-PEG-b-PGA suggests that the formed helices can be further associated as in coiled coil systems presumably due to intermolecular hydrogen-bonding and hydrophobic interactions. However the exact structural changes resulting in the increase of ellipticity ratios is not fully understood at present. As is seen in Figure 7A the hydrophobic modification of PGA blocks caused a significant decrease of relative helical content in PEG-b-PPGA copolymers at pH 5, which can be judged from attenuation of the ellipticity at 222 nm. An increased proportion of unordered conformations in the PGA blocks of PEG-b-PPGA copolymers can be explained by the fact that bulky phenylalanine groups in the side chains of the PGA backbone might restrict the compact packing needed for the formation of α-helix that is densely coiled structure held by intramolecular hydrogen bonding (Adams et al., 2008). Although the polypeptide backbone dominates the far-UV CD spectra, the contribution of the aromatic residues can become significant when the content of these residues is high and the estimation of secondary structure can be complicated. Moreover, the CD spectra of hydrophobically modified copolymers showed features which is not observed in PEG-b-PGA. In particular, the increase of the degree of modification minima at 208 nm gradually disappeared while the band corresponding to n - π* transition is shifted from 222 nm to 225 nm. It is likely that the processes of aggregation of the helical PGA segments are more pronounced in the case of PEG-b-PPGA copolymers due to an increase in hydrophobic interactions with phenylalanine residues or domains.
Figure 6.

Physicochemical characterization of cl-PEG-b-PPGA nanogels. (A) Effective diameter (Deff) and (B) ζ-potential as a function of pH. Targeted degree of cross-linking was 20%.
Figure 7.

Mean residual ellipticity of PGA-based polymers (A, B) and nanogels (C, D) at pH 5 (A, C) and at pH 7.0 (B, D) at 25°C. In A and B, (■) PEG-b-PGA, (▲) PEG-b-PPGA17 and (△) PEG-b-PPGA30. In C and D, (□) PEG-b-PGA nanogels and (■) cl-PEG-b-PPGA nanogels.
The aforementioned changes in CD spectra were even more distinct for cl-PEG-b-PPGA nanogels (Figure 7C). It is likely that both the decreased conformational freedom of PGA segments and presence of hydrophobic domains in the cross-linked core of the nanogels promote the segregation of the ordered structures that may further contribute to the collapse of the nanogels. To assess the relative stability of these self-organized ordered superstructures we carried out thermal denaturation experiments at pH 5. As shown in the temperature-dependent CD spectra in Figure S4, the helix content in nonmodified PEG-b-PGA decreased with increasing temperature from 25°C to 50°C, which suggests a gradual denaturation/unfolding of the helical aggregates into partially ordered unimers. In contrast, practically no changes were observed in the CD spectra of either PEG-b-PPGA30 copolymer or cl-PEG-b-PPGA nanogels in response to temperature increase. These observations may be explained by the stabilizing influence of hydrophobic phenylalanine domains, presumably by increasing the likelihood of both intra- and interchain hydrophobic interactions within the helical aggregate structures to resist unfolding.
DOX loading and release from cl-PEG-b-PPGA nanogels
We previously demonstrated that DOX can be efficiently encapsulated into the cores of anionic nanogels at pH 7 when both the DOX molecule and the carboxylic groups of the nanogels are completely ionized and oppositely charged (Kim, et al., 2010). In the present study DOX was incorporated into cl-PEG-b-PPGA nanogels using a similar procedure. As expected, drug loading was accompanied by a decrease in both the size (from ca. 72 nm to ca. 60 nm) and net negative charge (−50.7 mV to −22.7 mV) of the nanogels, which was consistent with the neutralization of the PPGA segments upon DOX binding to carboxylate groups. Considering the amphiphilic nature of DOX, the interactions between anthraquinone moiety of DOX and phenylalanine hydrophobic domains of nanogels are also contributed to the formation of drug-polymer complexes. Under these conditions DOX loading capacity of cl-PEG-b-PPGA nanogels (the net amount of drug loaded into a carrier) was about 30.4 w/w% as measured by UV-vis spectroscopy. Nonmodified hydrophilic cl-PEG-b-PGA nanogels exhibited lower drug loading capacity of ca. 27 w/w% despite the higher total content of carboxylic groups and large volume of the PGA core assessable to the drug molecules. Interestingly, the loading capacity of non-crosslinked PEG-b-PPGA30 micellar aggregates was much lower (18.3 w/w%) compared to nanogels. In addition, drug loading led to a substantial increase of the particle size (137 nm vs. 71 nm) and broader particle size distribution of DOX-loaded micelles, suggesting that the drug binding to PEG-b-PPGA30 copolymer induced structural rearrangement of micellar aggregates. It is well known for regular polymeric amphiphiles forming core-shell aggregates that a decrease in the weight fraction of shell-forming block shifts aggregate structures toward small mean curvature and larger size (Jain and Bates, 2003). Similarly, the reduced electrostatic repulsion and increased hydrophobicity of PPGA segments being neutralized by drug molecules led to formation of larger PEG-b-PPGA30/DOX aggregates. Thus, while the encapsulation of DOX into studied PGA-based nanostructures is mainly governed by electrostatic interactions, it seems that the cross-linked core of cl-PEG-b-PPGA nanogels provides a more favorable environment for the entrapment of DOX molecules. Notably, DOX-loaded cl-PEG-b-PPGA nanogels were stable in aqueous dispersions, exhibiting no aggregation or precipitation for a prolonged period of time (weeks).
Of particular interest was the finding that the structure of nanogel cross-linked core affected the DOX release profiles. The release of the entrapped DOX from nanogels was studied by equilibrium dialysis at 37 °C at either pH 7.4 (PBS) or pH 5.5 (ABS), which reflect conditions encountered in plasma and in intracellular compartments (lysosomes), respectively. DOX release profiles are presented in Figure 8. As evident from Figure 8A, at pH 7.4 nonmodified cl-PEG-b-PGA nanogels exhibited a burst release of over 85% of the incorporated drug within 8 h. In contrast, the release rates of DOX from hydrophobically modified carriers were substantially less. For instance, at 8 h, non-crosslinked PEG-b-PPGA30 micellar analogues released about 45% of the drug whereas only ~ 20% of the incorporated DOX was released from cl-PEG-b-PPGA nanogels. Intermolecular interactions in combination with more compact cross-linked core could account for the delayed and controlled release of DOX from of cl-PEG-b-PPGA nanogels.
Figure 8.

(A) Release profiles of DOX in PBS buffer (pH 7.4, 0.14M NaCl) at 37 °C. (■) cl-PEG-b-PPGA nanogels, (□) cl-PEG-b-PGA nanogels, (●) non-cross-linked PEG-b-PPGA30 micelles, and (○) free DOX.
(B) Release profiles of DOX from cl-PEG-b-PPGA nanogels and non-cl-PEG-b-PPGA micelles. in PBS buffer (pH 7.4, 0.14M NaCl) and ABS buffer (pH 5.5, 0.14M NaCl) at 37°C. (■) cl-PEG-b-PPGA nanogels, pH 7.4, (□) cl-PEG-b-PPGA nanogels, pH 5.5, (◆) cl-PEG-b-PPGA nanogels in the presence of cathepsin, pH 5.5, The loading amount of DOX for each sample is 200 μg. Data are expressed as the mean ± S.D. of three independent measurements.
DOX release from cl-PEG-b-PPGA nanogels was also a pH-dependent process. Indeed, drug molecules were liberated from the nanogels faster at pH 5.5 than at pH 7.4 (Figure 8B). This was presumably due to protonation of carboxylic groups of PGA, which weakens the DOX and nanogel electrostatic coupling as was discussed previously (Nukolova, et al., 2011). Importantly, significant acceleration of DOX release from cl-PEG-b-PPGA was observed at the acidic pH in presence of cathepsin B in release media due to degradation of the polypeptide backbone. Cathepsin B is a lysosomal thiol-dependent protease (Otto and Schirmeister, 1997) and is also extracellularly present in pathological tissues such as tumors and sites of inflammation (Hashimoto et al., 2001, Koblinski et al., 2000). It should be noted that cystamine, which is used as a cross-liker for synthesis of the nanogels, contains a reductively labile disulfide bonds prone to cleavage by the lysosomal cysteine proteases. We recently demonstrated that nanogels with disulfide bonds in the ionic cores were rapidly degraded in the presence of the reducing agent, which in turn accelerated the release of the incorporated drug (Kim, et al., 2010). Therefore, these results suggest that enzymatic degradation of cl-PEG-b-PPGA nanogels can further facilitate the drug release once located within targeted tumor tissue and tumor cells.
In vitro and in vivo anti-tumor efficacy
Our previous work demonstrated that nanogels based on PEG-poly(methacrylic acid) enter epithelial cancer cells via endocytosis and are translocated into the lysosomes (Sahay et al., 2010). Similarly, DOX-loaded cl-PEG-b-PPGA nanogels were taken up by the MCF-7 breast cancer cells and were co-localized with the lysosomes within 45 min (Figure 9). The lysosomal trapping of DOX-loaded cl-PEG-b-PPGA nanogels is expected to modulate the release of the drug as well as control the degradation of the carrier. The cytotoxicity of DOX-loaded cl-PEG-b-PPGA nanogels was assessed in human MCF-7 breast and A2780 ovarian cancer cells using MTT assay. Calculated IC50 values are summarized in Table 2. Importantly, cl-PEG-b-PPGA nanogels alone were not toxic at concentrations used for the treatment by DOX-loaded nanogels formulations. As expected, DOX-loaded cl-PEG-b-PPGA nanogels displayed lower cytotoxic activities than free DOX. The reduction in cytotoxicity was consistent with the corresponding sustained manner of DOX release from the nanogels.
Figure 9.
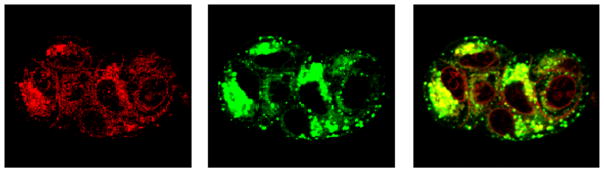
Cellular localization of DOX-loaded cl-PEG-b-PPGA nanogels in MCF-7 cells. MCF-7 cells were exposed for 45 min at 37°C to DOX-loaded cl-PEG-b-PPGA nanogels and Lysotracker® (Green) for 10 min. Images of live MCF-7 cells show significant co-localization of cl-PEG-b-PPGA nanogels within the lysosomes.
Table 2.
In vitro cytotoxicity of DOX-loaded cl-nanogels in cancer cells at 24h
| Cell line | IC50a | |
|---|---|---|
| Free Dox | DOX-loaded nanogelsb | |
| A2780 | 0.024±0.012 | 0.725±0.079 |
| MCF7 | 0.046±0.021 | 0.17±0.058 |
IC50 (μg/mL) represents the concentration of a drug for 50% inhibition in vitro (n=4).
DOX-loaded cl-PEG-b-PPGA nanogels with targeted degree of cross-linking of 20% were prepared at R = 0.5 and pH 7.0.
An in vivo anti-tumor efficacy of DOX-loaded cl-PEG-b-PPGA nanogels was examined in mice bearing subcutaneous ovarian human cancer xenografts. Free DOX, DOX-loaded cl-PEG-b-PPGA nanogels and empty nanogels were injected 4 times at 4-day intervals at an equivalent dose of 4 mg-DOX/kg. Changes in tumor volume and body weight are shown in Figure 10A and B, respectively. Both DOX and DOX/nanogel treatments exhibited moderate antitumor effect within this experimental setting and delayed tumor growth (p<0.05) compared to controls (5% dextrose and empty nanogels). Nevertheless, tumors in the animals treated with DOX-loaded cl-PEG-b-PPGA nanogels remained significantly smaller (p<0.05) than in animals treated with free DOX. We found the tumor inhibition by DOX-loaded cl-PEG-b-PPGA nanogels to be around 65–75% as compared to 40–50% in the DOX group between days 4 and 12 (a control group of animals was euthanized at this time point). Moreover, no significant changes in body weight were observed for control and treatment groups, indicating that all treatments were well tolerated (Figure 10B). These proof-of-concept data demonstrate that biodegradable PEG-polypeptide nanogels delivered sufficient concentration of DOX to inhibit tumor growth. It appears that nanogel particles were capable to accumulate in solid tumors due to enhanced permeability and retention (EPR) effect. The increased circulation time of nanogels (Oberoi, et al., 2012) could also enhance exposure of the tumor to the drug. However, additional studies are required to evaluate pharmacokinetic properties of cl-PEG-b-PPGA nanogel formulations and the drug exposure in tumor and normal tissues. Given the lack of toxicity of cl-PEG-b-PPGA carrier we hypothesize that antitumor efficacy can be further improved by using a higher dose of DOX in nanogel formulation as well as by incorporating tumor-targeting ligands into nanogels.
Figure 10.

Effect of DOX-loaded cl-PEG-b-PPGA nanogels on (A) tumor growth and (B) body weight in A2780 human ovarian cancer xenograft-bearing female nu/nu mice (n=6/group). Administrations of each formulation were given at four times at four-day intervals. The dose of DOX was 4 mg/kg in all preparations. Control (PBS) (▲), empty nanogels (△), free DOX (□) and DOX-loaded cl-PEG-b-PPGA nanogels (■). The data represent mean ± SEM. *p<0.05
Conclusions
In this contribution, we have reported the design, synthesis and characterization of well-defined biodegradable polypeptide-based nanogels. Diblock copolymer poly(ethylene glycol)-b-poly(L-glutamic acid) hydrophobically modified with L-phenylalanine methyl ester moieties (PEG-b-PPGA) was used for controlled template synthesis of nanogels. The resulting nanogels showed the hydrogel-like behavior due to the protonation of carboxylic groups and pH-dependent helix-to-coil transition of PPGA segments. Nanogels maintained their robust structure in strong destabilization conditions (urea), but could be rapidly disrupted by enzymatic biodegradation. These nanogels were able to effectively incorporate DOX up to 30 w/w%. We demonstrated that microenvironment formed by the hydrophobic domains in the nanogel cores influences solubilization capacity and release characteristics of the nanogels. Fluorescent probe studies also suggest that hydrophobic domains inside nanogels can also solubilize hydrophobic drugs and, thus, provide unique opportunities for combinational drug delivery. Our preliminary in vivo studies, treating highly aggressive A2780 tumor, showed improved anti-tumor effect for the DOX-loaded nanogel versus free DOX. Considering the high stability of the materials, simple and mild preparation procedure, high loading capacity, sustained-release property, and biodegradability, hydrophobically modified nanogels should be promising carriers for delivery of chemotherapeutics.
Acknowledgments
This work was supported by National Institutes of Health grants CA116590 (T.K.B.). The authors acknowledge the assistance of the Nanomaterials Core facility (supported by the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103480). We would like to thank the NMR, Confocal Microscopy and Nanoimaging Core Facilities at UNMC for excellent technical assistance, and Dr. Daria Alakhova for the help in the preparation of illustration for this paper.
References
- Abbruzzetti S, Viappiani C, Small JR, Libertini LJ, Small EW. Kinetics of local helix formation in poly-L-glutamic acid studied by time-resolved photoacoustics: neutralization reactions of carboxylates in aqueous solutions and their relevance to the problem of protein folding. Biophys J. 2000;79:2714–21. doi: 10.1016/S0006-3495(00)76509-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DJ, Atkins D, Cooper AI, Furzeland S, Trewin A, Young I. Vesicles from peptidic side-chain polymers synthesized by atom transfer radical polymerization. Biomacromolecules. 2008;9:2997–3003. doi: 10.1021/bm8006693. [DOI] [PubMed] [Google Scholar]
- Bae Y, Kataoka K. Intelligent polymeric micelles from functional poly (ethylene glycol)-poly (amino acid) block copolymers. Adv Drug Deliv Rev. 2009;61:768–84. doi: 10.1016/j.addr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nat Mater. 2004;3:244–8. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- Bronich TK, Keifer PA, Shlyakhtenko LS, Kabanov AV. Polymer micelle with cross-linked ionic core. J Am Chem Soc. 2005;127:8236–7. doi: 10.1021/ja043042m. [DOI] [PubMed] [Google Scholar]
- Carlsen A, Lecommandoux S. Self-assembly of polypeptide-based block copolymer amphiphiles. Curr Opin Colloid Interface Sci. 2009;14:329–39. [Google Scholar]
- Chacko RT, Ventura J, Zhuang J, Thayumanavan S. Polymer nanogels: A versatile nanoscopic drug delivery platform. Adv Drug Deliv Rev. 2012;64:836–51. doi: 10.1016/j.addr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani O, Ruppel M, Schubert F, Zettl H, Pergushov DV, Müller AH. Synthesis of poly (n-butyl acrylate)-block-poly (acrylic acid) diblock copolymers by ATRP and their micellization in water. Macromolecules. 2007;40:4338–50. [Google Scholar]
- Dong DC, Winnik MA. The Py scale of solvent polarities. Solvent effects on the vibronic fine structure of pyrene fluorescence and empirical correlations with ET and Y values. Photochem Photobiol. 1982;35:17–21. [Google Scholar]
- Ferrari M, Fornasiero MC, Isetta AM. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J Immunol Methods. 1990;131:165–72. doi: 10.1016/0022-1759(90)90187-z. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kakegawa H, Narita Y, Hachiya Y, Hayakawa T, Kos J, et al. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem Biophys Res Commun. 2001;283:334–9. doi: 10.1006/bbrc.2001.4787. [DOI] [PubMed] [Google Scholar]
- Horng M, Gardecki J, Papazyan A, Maroncelli M. Subpicosecond measurements of polar solvation dynamics: coumarin 153 revisited. J Phys Chem. 1995;99:17311–37. [Google Scholar]
- Jain S, Bates FS. On the origins of morphological complexity in block copolymer surfactants. Science. 2003;300:460–4. doi: 10.1126/science.1082193. [DOI] [PubMed] [Google Scholar]
- Jin H, Baker GA, Arzhantsev S, Dong J, Maroncelli M. Solvation and rotational dynamics of coumarin 153 in ionic liquids: comparisons to conventional solvents. J Phys Chem B. 2007;111:7291–302. doi: 10.1021/jp070923h. [DOI] [PubMed] [Google Scholar]
- Kabanov AV, Vinogradov SV. Nanogels as pharmaceutical carriers: finite networks of infinite capabilities. Angew Chem Int Ed Engl. 2009;48:5418–29. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanasundaram K, Thomas J. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J Am Chem Soc. 1977;99:2039–44. [Google Scholar]
- Kim JO, Nukolova NV, Oberoi HS, Kabanov AV, Bronich TK. Block ionomer complex micelles with cross-linked cores for drug delivery. Polym Sci A 2009. 2009;51:708–18. doi: 10.1134/S0965545X09060169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JO, Sahay G, Kabanov AV, Bronich TK. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules. 2010;11:919–26. doi: 10.1021/bm9013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–35. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- Kukula H, Schlaad H, Antonietti M, Förster S. The Formation of Polymer Vesicles or “Peptosomes” by Polybutadiene-b lock-poly (l-glutamate) s in Dilute Aqueous Solution. J Am Chem Soc. 2002;124:1658–63. doi: 10.1021/ja012091l. [DOI] [PubMed] [Google Scholar]
- Kumbhakar M. Effect of ionic surfactants on the hydration behavior of triblock copolymer micelles: A solvation dynamics study of coumarin 153. J Phys Chem B. 2007;111:12154–61. doi: 10.1021/jp0746188. [DOI] [PubMed] [Google Scholar]
- Kumbhakar M, Goel T, Nath S, Mukherjee T, Pal H. Microenvironment in the corona region of triblock copolymer micelles: Temperature dependent solvation and rotational relaxation dynamics of coumarin dyes. J Phys Chem B. 2006;110:25646–55. doi: 10.1021/jp063839v. [DOI] [PubMed] [Google Scholar]
- Langer CJ, O’Byrne KJ, Socinski MA, Mikhailov SM, Lesniewski-Kmak K, Smakal M, et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:623–30. doi: 10.1097/JTO.0b013e3181753b4b. [DOI] [PubMed] [Google Scholar]
- Lavasanifar A, Samuel J, Kwon GS. Poly (ethylene oxide)-block-poly (l-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54:169–90. doi: 10.1016/s0169-409x(02)00015-7. [DOI] [PubMed] [Google Scholar]
- Li C. Poly (L-glutamic acid)–anticancer drug conjugates. Adv Drug Deliv Rev. 2002;54:695–713. doi: 10.1016/s0169-409x(02)00045-5. [DOI] [PubMed] [Google Scholar]
- Matsumura Y. Polymeric micellar delivery systems in oncology. Jpn J Clin Oncol. 2008;38:793–802. doi: 10.1093/jjco/hyn116. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–9. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick CLBJ, Schulz DN. Encyclopedia of Polymer Science and Engineering. 2. New York: John Wiley; 1989. [Google Scholar]
- Murphy EA, Majeti BK, Mukthavaram R, Acevedo LM, Barnes LA, Cheresh DA. Targeted nanogels: a versatile platform for drug delivery to tumors. Mol Cancer Ther. 2011;10:972–82. doi: 10.1158/1535-7163.MCT-10-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Fontaine J, Suntharalingam M, Dipetrillo T, Horiba M, Oldenburg N, et al. Neoadjuvant paclitaxel poliglumex (PPX), cisplatin, and radiation (RT) for esophageal cancer. J Clin Oncol (Meeting Abstracts) 2010:4085. doi: 10.1097/COC.0b013e318201a126. [DOI] [PubMed] [Google Scholar]
- Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials. 2011;32:5417–26. doi: 10.1016/j.biomaterials.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi HS, Laquer FC, Marky LA, Kabanov AV, Bronich TK. Core cross-linked block ionomer micelles as pH-responsive carriers for cis-diamminedichloroplatinum (II) J Control Release. 2011;153:64–72. doi: 10.1016/j.jconrel.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi HS, Nukolova NV, Laquer FC, Poluektova LY, Huang J, Alnouti Y, et al. Cisplatin-loaded core cross-linked micelles: comparative pharmacokinetics, antitumor activity, and toxicity in mice. Int J Nanomedicine. 2012;7:2557. doi: 10.2147/IJN.S29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto H-H, Schirmeister T. Cysteine proteases and their inhibitors. Chem Rev. 1997;97:133. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Rossky PJ. Protein denaturation by urea: Slash and bond. Proc Natl Acad Sci. 2008;105:16825–6. doi: 10.1073/pnas.0809224105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay G, Kim JO, Kabanov AV, Bronich TK. The exploitation of differential endocytic pathways in normal and tumor cells in the selective targeting of nanoparticulate chemotherapeutic agents. Biomaterials. 2010;31:923–33. doi: 10.1016/j.biomaterials.2009.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steege KE, Wang J, Uhrich KE, Castner EW. Local polarity and microviscosity in the hydrophobic cores of amphiphilic star-like and scorpion-like macromolecules. Macromolecules. 2007;40:3739–48. [Google Scholar]
- Vinogradov SV, Bronich TK, Kabanov AV. Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. Adv Drug Deliv Rev. 2002;54:135–47. doi: 10.1016/s0169-409x(01)00245-9. [DOI] [PubMed] [Google Scholar]
- Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J. Synthesis and evaluation of a star amphiphilic block copolymer from poly (ε-caprolactone) and poly (ethylene glycol) as a potential drug delivery carrier. Bioconjug Chem. 2005;16:397–405. doi: 10.1021/bc049784m. [DOI] [PubMed] [Google Scholar]
- Zhou N, Kay C, Hodges R. Synthetic model proteins. Positional effects of interchain hydrophobic interactions on stability of two-stranded alpha-helical coiled-coils. J Biol Chem. 1992;267:2664–70. [PubMed] [Google Scholar]