

Abstract
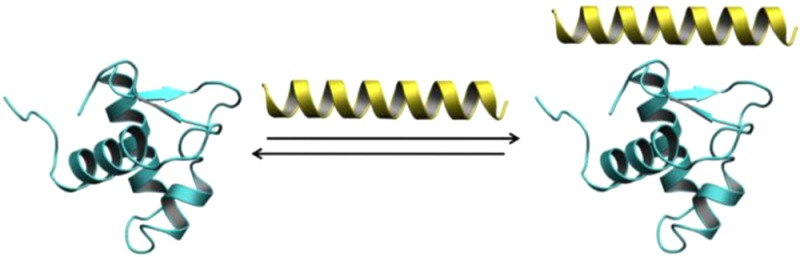
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like1 (SMARCAL1) is a recently identified DNA damage response protein involved in remodeling stalled replication forks. The eukaryotic single-strand DNA binding protein replication protein A (RPA) recruits SMARCAL1 to stalled forks in vivo and facilitates regression of forks containing leading strand gaps. Both activities are mediated by a direct interaction between an RPA binding motif (RBM) at the N-terminus of SMARCAL1 and the C-terminal winged-helix domain of the RPA 32 kDa subunit (RPA32C). Here we report a biophysical and structural characterization of the SMARCAL1–RPA interaction. Isothermal titration calorimetry and circular dichroism spectroscopy revealed that RPA32C binds SMARCAL1-RBM with a Kd of 2.5 μM and induces a disorder-to-helix transition. The crystal structure of RPA32C was refined to 1.4 Å resolution, and the SMARCAL1-RBM binding site was mapped on the structure on the basis of nuclear magnetic resonance chemical shift perturbations. Conservation of the interaction surface to other RBM-containing proteins allowed construction of a model for the RPA32C/SMARCAL1-RBM complex. The implications of our results are discussed with respect to the recruitment of SMARCAL1 and other DNA damage response and repair proteins to stalled replication forks.
Stalling of DNA replication forks results from insufficient nucleotide precursors, damaged template DNA, collisions between replisome and transcriptional complexes, or difficult to replicate genomic regions.1,2 In some cases, fork stalling leads to uncoupling of helicase and polymerase activities, generating an excess of RPA-coated ssDNA and activating the DNA damage response (DDR). The DDR is mediated by ATM, ATR, and DNA-dependent protein kinase activity and recruitment of a number of fork remodeling and DNA repair proteins.3
One such protein is SMARCAL1, a SNF2 ATP-dependent fork remodeler that facilitates rescue of stalled DNA replication forks.2,4−7 SMARCAL1 is activated by phosphorylation by ATR, and its deficiency increases cellular sensitivity to replication stress agents and leads to accumulation of ssDNA and double-strand breaks.2,8,9 Mutations in SMARCAL1 lead to the autosomal-recessive pleiotropic disorder Schimke Immunoosseous Dysplasia (SIOD).10 SMARCAL1 is recruited to stalled forks through a direct interaction with RPA.6 Binding is mediated by the 32 N-terminal residues of SMARCAL1,2,6,11 which is homologous to previously characterized RPA interaction motifs of the human uracil-DNA glycosylase (UNG2), RAD52, XPA, and Timeless-interacting protein (TIPIN).2,11,12 In addition to localizing SMARCAL1 to damaged forks, RPA enforces a preference of SMARCAL1 for ssDNA regions on the leading strand template.13
RPA is the predominant eukaryotic single-stranded DNA binding protein and essential for virtually all DNA transactions, including the DDR.14,15 RPA protects ssDNA from nucleases and prevents the formation of aberrant structures and reannealing,16−19 while also serving as a scaffold that links the substrate to the succession of enzymes that process the DNA. RPA is a modular heterotrimer composed of 70, 32, and 14 kDa subunits that together contain seven globular domains and one disordered domain (Figure 1). Binding of ssDNA is mediated by four OB-fold domains: the RPA70A, -70B, and -70C domains and the RPA32D domain. In addition to binding ssDNA, RPA interacts with numerous DNA-processing proteins.11,12,20−22 These interactions have been mapped by our laboratory and others to the RPA70N, -70A, -70B, and -32C domains.11,15,23−25 The RPA70N and -32C domains are the primary mediators of protein interactions.12,20 RPA32C serves as the primary contact point for multiple DDR and repair proteins, including XPA, UNG2, RAD52, TIPIN, and SMARCAL1.2,6,11,26−29 Although RPA32C is known to recruit SMARCAL1 through an N-terminal RBM, the details and strength of this RPA32C–SMARCAL1 interaction are not known.
Figure 1.
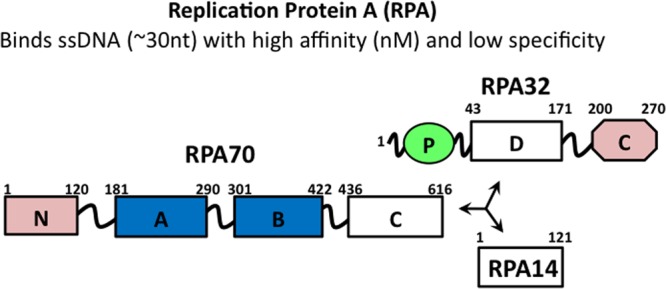
Subunit and domain structure of RPA. OB-fold domains are depicted as rectangles, and the winged helix–turn–helix domain is depicted as an octagon and the disordered phosphorylation domain as a green oval. The high-affinity ssDNA binding domains are colored blue and the primary protein recruitment domains pink. Trimerization is mediated by RPA70C, -32D, and -14.
We report here the characterization of the RPA32C–SMARCAL1 interface at the molecular level. The X-ray crystal structure of RPA32C was determined, and the interaction with SMARCAL1-RBM peptides was characterized by isothermal titration calorimetry, circular dichroism (CD), nuclear magnetic resonance (NMR) spectroscopy, and computational modeling. These results provide insight into the molecular basis for the recruitment of SMARCAL1 to stalled replication forks and how the interaction of DDR and repair proteins with RPA32C is fine-tuned by the selection of residues at the binding interface.
Methods
RPA32C Expression and Purification
The previously reported RPA32C construct, RPA32172–270, was expressed in Escherichia coli BL21-DE3 and purified as described previously.11 The optimized RPA32C construct, RPA32202–270, was cloned into pBG100 (Vanderbilt Center for Structural Biology) and transformed into BL21-DE3 cells (New England Biolabs) for expression. Overnight cultures were prepared from single colonies and used to inoculate 1 L of Luria-Bertani (LB) broth. The cultures were grown at 37 °C to an OD600 of 0.5–0.6, at which time isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM and cultures were grown for an additional 5 h. Cells were harvested by centrifugation and sonicated in buffer A [50 mM Hepes, 500 mM NaCl, 2 mM β-mercaptoethanol (BME), and 10 mM imidazole (pH 7.5)]. The supernatant was applied to a Ni2+-NTA Sepharose column (GE Healthcare) equilibrated with buffer A and washed with buffer A. Following elution with buffer B [50 mM Hepes, 500 mM NaCl, 2 mM BME, and 250 mM imidazole (pH 7.5)], (His)6-tagged H3C protease was added to the eluate and the mixture dialyzed overnight at 4 °C against buffer A and subjected to Ni2+-NTA chromatography to remove the cleaved (His)6 tag. The RPA32202–270 protein obtained was >95% pure as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis. The production of 15N-labeled RPA32172–270 and RPA32202–270 for NMR studies was performed using the same protocols except that cells were grown in M9 medium containing 0.5 g of 15NH4Cl per liter as the sole nitrogen source.
SMARCAL1-RBM Expression and Purification
The cDNA encoding SMARCAL1 residues 1–32 containing the RBM was cloned into pBG101 (Vanderbilt Center for Structural Biology) and the protein expressed in E. coli BL21-DE3 cells as an N-terminally tagged (His)6-GST fusion containing an H3C protease recognition sequence. SMARCAL11–32 was expressed and purified as described above for RPA32202–270, with an additional S75 gel filtration (Amersham) step in 25 mM Tris (pH 7.0), 75 mM NaCl, 5 mM DTT buffer. The SMARCAL17–32 peptide was purchased from Genescript at >95% purity as determined by high-performance liquid chromatography and used without further purification.
Isothermal Titration Calorimetry
RPA32172–270 and SMARCAL11–32 were exchanged into a buffer containing 20 mM sodium phosphate (pH 7.0), 50 mM NaCl, and 5 mM DTT, and ITC data were acquired using a MicroCal VP isothermal titration calorimeter. An initial injection of 2 μL of 800 μM SMARCAL11–32 into 60 μM RPA32172–270 in the sample cell was followed by additional 10 μL injections. The data were analyzed using the Origin software provided by MicroCal. The binding constant (Kd) and thermodynamic parameters were calculated from the average of three separate titrations by fitting the data to a single-site binding model using nonlinear least-squares fitting.
X-ray Crystallography
RPA32C202–270 (RPA32C) was dialyzed into a buffer containing 20 mM Tris (pH 7.5) and concentrated to 15 mg/mL. Crystals were observed by hanging drop vapor diffusion at 21 °C from drops containing a 1:1 ratio of RPA32C and 50 mM sodium acetate (pH 4.5) and 20% PEG 3350. Initial attempts to crystallize the protein were not successful, and crystals were obtained only after the protein was dialyzed against the low-ionic strength buffer. Following optimization of pH and PEG concentration, hexagonal crystals were obtained and grew to full size in ∼3 days. Prior to data collection, MPD was added to a final concentration of 20% and crystals were flash-frozen in liquid nitrogen. X-ray diffraction data were collected at sector 21 (Life Sciences Collaborative Access Team, LS-CAT) of the Advanced Photon Source (Argonne, IL). All data were processed with HKL-2000.30 RPA32C crystallized in space group P65 and contained one molecule in the asymmetric unit. Initial phases were obtained by molecular replacement with PHASER31 using the NMR structure of RPA32C [Protein Data Bank (PDB) entry 1DPU] as a search model. Iterative cycles of model building and refinement were performed using COOT and PHENIX.32,33 The structure was deposited in the PDB as entry 4OU0. PyMOL (Schrödinger) was used for structure visualization and analysis. Data collection and structure refinement statistics are listed in Table S1 of the Supporting Information.
NMR Spectroscopy
All NMR studies were performed on Bruker Advance III 500 or 600 MHz NMR spectrometers equipped with 5 mm single-axis z gradient inverse cryogenic probes. Spectra were recorded using band-selective, optimized flip angle short transient, 15N–1H heteronuclear multiple-quantum coherence (SOFAST-HMQC) spectra.34 Spectra were recorded with 1024 data points in the direct proton, 96 points in the indirect nitrogen dimension, and a recycle delay of 200 ms. All data were processed and analyzed with NMRpipe35 and Sparky (University of San Francisco, San Francisco, CA). The previously assigned backbone 1H and 15N NMR chemical shifts for RPA32172–270 were transferred to RPA32C.11
NMR spectra were acquired at 25 °C in a buffer containing 25 mM Tris buffer (pH 7.0) with 75 mM NaCl and 5 mM DTT. The concentration of [15N]RPA32C was adjusted to 250 μM for titrations with the SMARCAL1 peptides. Data points were collected at SMARCAL1 concentrations of 0, 20, 60, 120, 200, 360, and 600 μM. Titrations of 250 μM [15N]SMARCAL1 were performed with RPA32C at concentrations of 0, 25, 50, 100, 250, and 500 μM. The chemical shift perturbations in a titration of labeled RPA32C with SMARCAL17–32 were analyzed using a weighted average of the change in chemical shift (Δδ) upon binding based on the net perturbations in both the 1H and 15N dimensions calculated using the standard equation (eq 1):36
| 1 |
For in situ proteolysis experiments, 1 μL of Proteinase K (Clontech) dissolved at a ratio of 1/100 (w/v) was added to NMR samples and SOFAST-HMQC spectra were collected at 10 min intervals. The starting concentration in these experiments was 200 μM [15N]RPA32172–270 or 250 μM [15N]SMARCAL11–32.
CD Spectroscopy
CD spectra of RPA32C, SMARCAL17–32, and SMARCAL17–32-bound RPA32C were collected using a Jasco J-810 spectrometer outfitted with a Peltier temperature control module. All spectra were collected at 25 °C in 5 mM Hepes (pH 7.0) buffer containing 75 mM NaCl and 5 mM DTT. Spectra were collected using a 2 nm bandwidth at 1 nm intervals with each data point averaged for 5 s. The RPA32C and SMARCAL17–32 concentrations were 15 and 20 μM, respectively.
Homology Modeling
Homology models were generated using Modeler version 9.13,37−40 the HEX (http://hexserver.loria.fr/)41 server, and the RosettaDock server (http://rosie.rosettacommons.org/docking)42−44 using default settings. The structure of RPA32C in complex with the UNG2 peptide (PDB entry 1DPU)11 was used as a template for Modeler and RosettaDock. Our structure of RPA32C and a Pymol-generated SMARCAL17–32 α-helix were used for docking with HEX.
Results
Interaction of the SMARCAL1 RPA Binding Motif with RPA32C
The primary interaction of SMARCAL1 with RPA has been previously mapped to a motif in the N-terminal region (SMARCAL1-RBM) and the C-terminal globular domain of the RPA32 subunit (RPA32C).2,6,11 The interaction was identified by pull-down assays, but the affinity of the interaction was not quantified. Here, we employed isothermal titration calorimetery (ITC) to measure the affinity of RPA32172–270 for SMARCAL1-RBM (Figure 2). A dissociation constant (Kd) of 2.5 ± 0.1 μM was measured, which was stronger than all previously characterized interactions of RPA32C with target protein fragments except for that with TIPIN, which was measured to be 0.5 μM.12 Binding of SMARCAL1-RBM to RPA32C was exothermic, suggesting that multiple hydrogen bonds are formed when the two molecules interact.
Figure 2.

Interaction of SMARCAL1 with RPA32C. Isothermal titration calorimetry binding isotherm for titration of SMARCAL1 into RPA32C showing the raw heat release (top) and the integrated heat release (bottom). The experiment was performed at 25 °C with a concentration of 60 μM RPA32172–270 in the cell and 10 μL additions of 800 μM SMARCAL11–32.
Optimization of the RPA32C Construct by Limited Proteolysis
The solution NMR structure of RPA32C in complex with a UNG2 peptide was determined previously using a 99-residue RPA32172–270 construct.11 However, in this construct, only the 67 C-terminal residues formed the globular RPA32C domain and no residues outside of this domain were involved in binding to target proteins. Assuming that the flexible disordered N-terminal residues are not needed for binding and would inhibit crystallization, we set out to identify the minimal RPA32C fragment that retains folding and binding activity. To this end, we took advantage of the NMR assignments available for RPA32172–27011 and performed limited proteolysis in situ (in the NMR tube). This allowed us to determine how many N-terminal residues lie outside of the globular core. The NMR chemical shift is a highly sensitive, residue-specific probe of structure and binding. The use of two-dimensional 15N–1H NMR correlation spectroscopy allowed us to monitor the backbone amides of RPA32C and follow the cleavage of the disordered N-terminal residues as they were digested by Proteinase K. This protease was chosen because there are a number of consensus cleavage sites within the residues outside of the globular RPA32C core.
The series of RPA32172–270 spectra acquired prior to and after incubation with Proteinase K (Figure 3A) shows that 30 residues disappeared from the spectrum as they were cleaved by the protease. The peak for residue A202 was the most N-terminal residue that could be identified as having only very little change in peak position and intensity. Hence, this residue was chosen as the N-terminus of a new RPA32C202–270 construct. To confirm that the globular core was not affected by the truncation, we compared the two-dimensional (2D) 15N–1H correlation spectra of [15N]RPA32202–270 and RPA32172–270 (Figure 3B). The data show that there are only very minor perturbations in chemical shifts. Thus, truncation of the N-terminus to A202 did not disrupt folding of the globular RPA32C domain.
Figure 3.
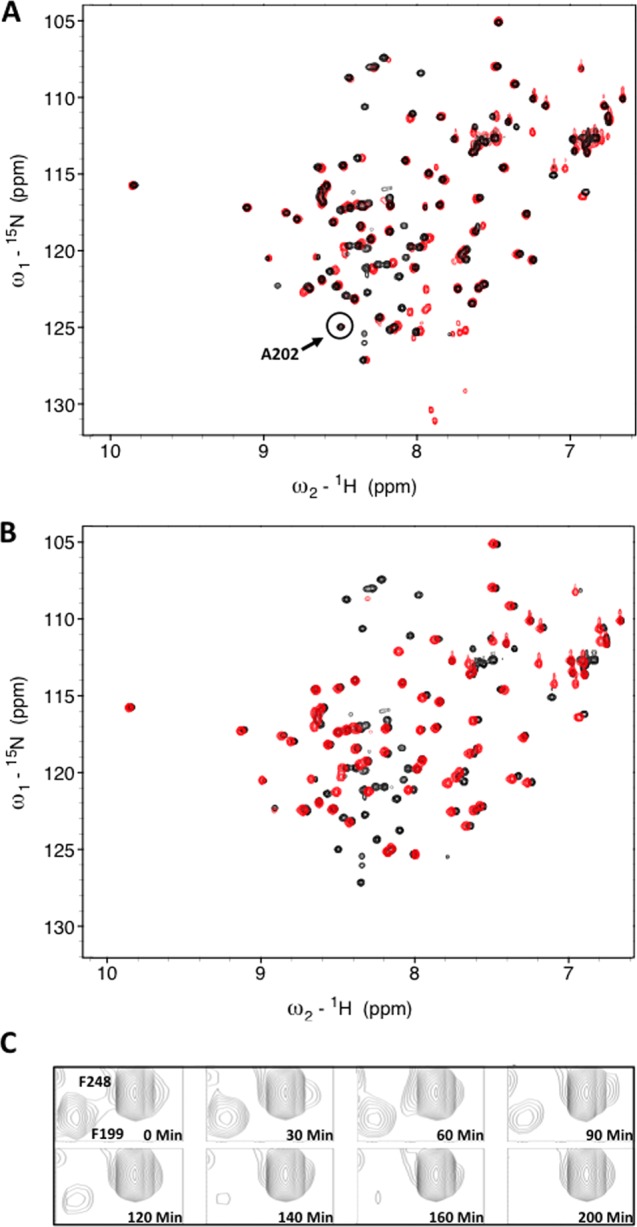
(A) 15N–1H HMQC spectra of RPA32C. 15N–1H SOFAST HMQC spectra of RPA32172–270 before (black) and after digestion with (red) Proteinase K. The most N-terminal residue not perturbed by the protease, A202, is highlighted by the circle. (B) Superposition of the 15N–1H SOFAST HMQC spectra of RPA32172–270 (black) and RPA32C (red). (C) A select region of the spectrum with multiple time points to demonstrate residues that are either protected (F248) or digested (F199).
High-Resolution X-ray Crystal Structure of RPA32C
The crystal structure of the optimized RPA32C202–270 construct (hereafter RPA32C) was determined by molecular replacement using the NMR structure of RPA32C extracted from the coordinates of the UNG2 complex (PDB entry 1DPU)11 as a search model. The crystallographic model was refined against diffraction data extending to 1.4 Å resolution (Table S1 of the Supporting Information). We note that RPA32C was soluble under all sparse matrix conditions examined at concentrations up to 40 mg/mL under standard buffer conditions, and crystals could be obtained only by drastically lowering the ionic strength of the buffer.
As anticipated, the RPA32C crystal structure reveals the same three-helix bundle capped by a β-hairpin that is observed in the NMR structure of RPA32172–270 determined in the presence of a peptide fragment of UNG2 (Figure 4A). Almost all of the noticeable differences between the structures are in loops (Figure 4C), which are presumably somewhat more flexible than the globular core. Overall, the structures are very similar; for example, the Cα atom root-mean-square deviation is only 0.57 Å. Further comparisons of the structures for the free protein and the complex reveal that the binding of targets requires only minor conformational adjustments. These comparisons also support the proposal of a significant electrostatic contribution to binding from the acidic target binding surface (Figure 4B). The similarity between the RPA32C structures in the absence and presence of the target peptide also extends to the positions of critical RPA32C side chains, which need not reorient to bind the UNG2 peptide (Figure 4C) or presumably other target proteins.
Figure 4.
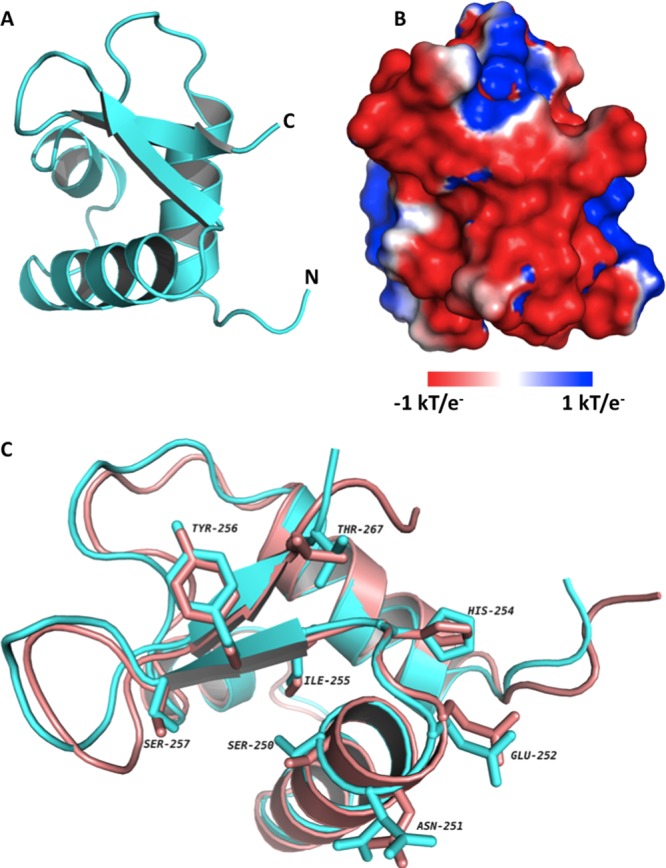
Crystal structure of RPA32C. (A) Ribbon diagram of the X-ray crystal structure of RPA32C. (B) Electrostatic surface potential of RPA32C calculated with APBS. The orientation is identical to that shown in panel A and was selected to show the highly acidic peptide binding site. (C) Superposition of the NMR structure from the RPA32C complex with the UNG2 peptide (salmon) with the crystal structure of free RPA32C (cyan). The orientation is similar to that in panels A and B, with a 45° rotation about the Z axis. A selection of side chains of RPA32C are highlighted, corresponding to those residues in the structure of the complex within 3.5 Å of the UNG2 peptide.
Structural Analysis of the SMARCAL1–RPA32C Complex
Multiple-sequence alignment of SMARCAL1 suggests its RPA32C binding motif is similar to that of XPA, UNG2, Rad52, and TIPIN.11,12,45 In previous studies of the interactions of peptide fragments of these target proteins, the peptides were found to undergo a disorder-to-helix transition upon binding.11,12 To determine if this was the case for SMARCAL1, CD spectra were collected for free RPA32C, free SMARCAL11–32, and the complex (Figure 5). All three exhibited CD spectra with pronounced double minima at 208 and 222 nm indicating the presence of α-helical secondary structure. To determine if binding to RPA32C induced formation of additional helical content in SMARCAL11–32, the CD spectra of RPA32C and SMARCAL11–32 were summed and compared to the spectrum of their complex. A clear difference is seen in optical ellipticity at 222 nm between the summed spectra and the spectrum of the complex (Figure 5), suggesting a 20% increase in α-helical content upon binding of SMARCAL11–32 to RPA32C. This change in helical content is supported by the increased level of dispersion in the 1H dimension and the appearance of six new amide cross-peaks in the 2D 15N–1H NMR spectrum of [15N]SMARCAL11–32 upon addition of unlabeled RPA32C (Figure 6A). Together, these data indicate that the binding of SMARCAL11–32 to RPA32C induces a disorder-to-helix transition in SMARCAL1 similar to that induced in UNG2 and TIPIN upon binding to RPA32C.11,12
Figure 5.
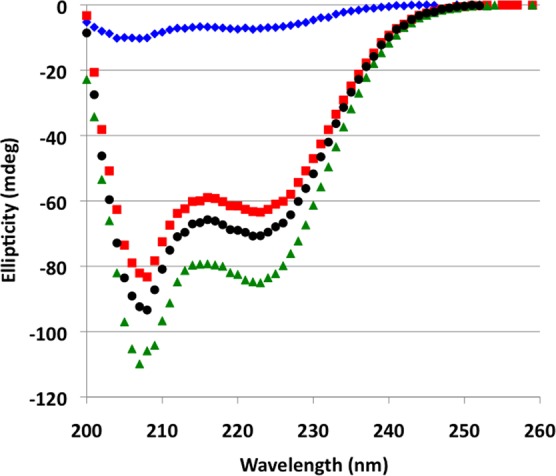
CD spectra of RPA32C, SMARCAL11–32, and the RPA32C–SMARCAL11–32 complex. CD spectra acquired at 25 °C for SMARCAL11–32 alone (blue diamonds), RPA32C alone (red squares), and the SMARCAL11–32–RPA32C complex (green triangles). The sum of the spectra for RPA32C and SMARCAL11–32 is shown with black circles.
Figure 6.

NMR analysis of [15N]SMARCAL1 with RPA32C. (A) 15N–1H SOFAST HMQC spectra of [15N]SMARCAL11–32 in the absence (black) and presence (red) of RPA32C. (B) 15N–1H SOFAST HMQC spectra of [15N]SMARCAL11–32 in complex with RPA32C obtained before (black) and after (red) a 50 min Proteinase K digestion.
Because the peptide fragments of XPA, UNG2, and Rad52 in our previous studies consisted of ∼20 residues, we hypothesized that the SMARCAL1–RPA32C binding site is similar in size and therefore not all 32 residues of SMARCAL11–32 were required for binding to RPA32C. Multiple-sequence alignment of the RPA32C binding motifs from XPA, UNG2, Rad52, and TIPIN (Figure 7) suggested SMARCAL112–27 forms the core of the binding site. To determine the minimal SMARCAL1 peptide needed to bind RPA32C, we again performed limited proteolysis in an NMR tube with the protease Proteinase K. Similar to our experiment with free RPA32C, addition of Proteinase K resulted in a progressive loss of peak intensity for a select number of [15N]SMARCAL11–32 amide cross-peaks, and a number of SMARCAL1 peaks were protected from proteolysis (Figure 6B). Even though resonance assignments for [15N]SMARCAL11–32 were not available, these data were sufficient to infer that a fragment of ∼25 residues remained bound to RPA32C, supporting our hypothesis that a shorter SMARCAL1 peptide could be designed (Figure 6B). On the basis of the data and alignments, we selected SMARCAL1 residues 7–32.
Figure 7.
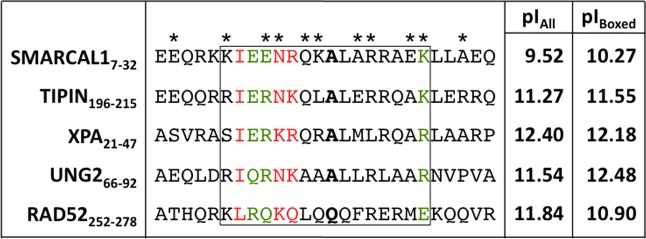
Sequence alignment of RPA32C target interaction motifs. The asterisks above the SMARCAL1-RMB sequence identify the SMARCAL1-RBM residues in contact with RPA32C in our RosettaDock model. Residues colored green and red represent those that are conserved and highly conserved, respectively. The residues corresponding to the critical alanine residue at position 14 in SMARCAL1 are highlighted in bold. The box is drawn to show the residues that correspond to the RPA32C binding region in the NMR structure of the complex with UNG2. The two columns at right list the pI values of all residues in the motif (pIall) and of only residues in the box (pIbox). The alignment was generated using ClustalW.45
To determine if this smaller fragment (hereafter SMARCAL1-RBM) bound to RPA32C in the same manner as SMARCAL11–32, a progressive series of complexes were analyzed by 2D 15N–1H NMR using [15N]RPA32172–270 and [15N]RPA32C titrated with SMARCAL11–32 and SMARCAL1-RBM (Figure 8A–C). The first step was to determine chemical shift perturbations of RPA32172–270 induced by the binding of SMARCAL11–32 (Figure 8A). To compare the effect of reducing the flexible N-terminal linker of RPA32C, the experiment was repeated with RPA32C (Figure 8B). The comparison of the two sets of spectra shows that SMARCAL11–32 induces similar chemical shift perturbations, as reflected in the overlay of the spectra obtained in the presence of the peptide (Figure 8D). Thus, truncation of the linker does not interfere with the binding of SMARCAL11–32. The third step of this analysis was to examine the chemical shift perturbations induced in RPA32C by the truncated SMARCAL1-RBM identified from our proteolytic digest (Figure 8C). To compare the effect of removing the six N-terminal residues, the spectrum of RPA32C bound to SMARCAL1-RBM was overlaid on the spectrum with SMARCAL11–32 (Figure 8E). The observation of similar chemical shift perturbations shows that the smaller peptide binds at the same site. A small number of minor differences are evident in Figure 8E, which we attribute to the different lengths of the two SMARCAL1 peptides. Together, these data show that structural analysis of the complex can be performed with RPA32C and SMARCAL1-RBM.
Figure 8.
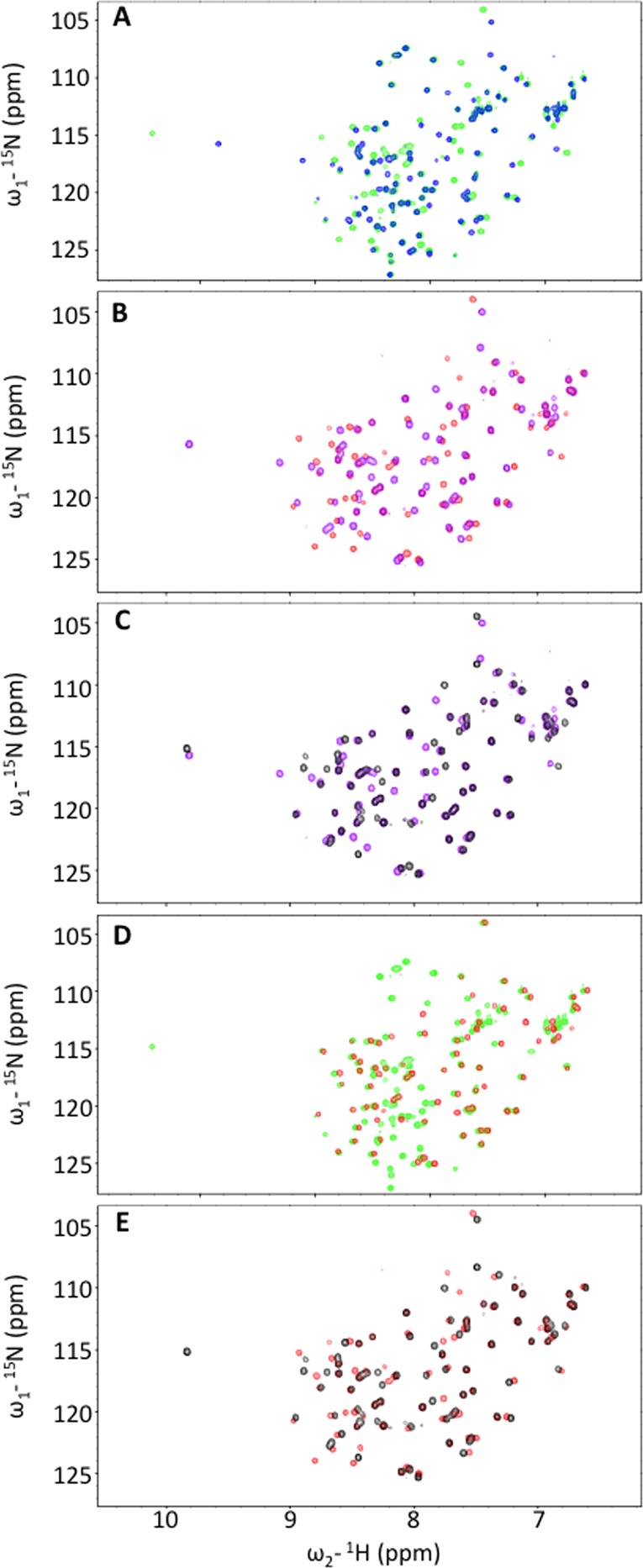
NMR analysis to define the minimal regions required for formation of the RPA32C–SMARCAL1 complex. Superposition of pairs of 15N–1H SOFAST HMQC spectra. (A) RPA32172–270 in the absence (blue) and presence of SMARCAL11–32 (green). (B) RPA32C in the absence (purple) and presence of SMARCAL11–32 (red). (C) RPA32C in the absence (purple) and presence of SMARCAL1-RBM (black). (D) RPA32172–270 (green) and RPA32C (red) in the presence of SMARCAL11–32. (E) RPA32C in the presence of SMARCAL11–32 (red) and SMARCAL1-RBM (black).
Deeper insight into how SMARCAL1-RBM interacts with RPA32C was obtained from an examination of the magnitude of the NMR chemical shift perturbations in RPA32C induced by the binding of the peptide. Using a cutoff of 0.24 ppm, a total of 15 residues were identified as being significantly perturbed (Figure 9A). The RPA32C residues exhibiting the greatest chemical shift change upon binding of SMARCAL17–32 were T267, Y256, D268, and T258. The chemical shift perturbations are mapped on the crystal structure in Figure 9B. Notably, these RPA32C residues correspond well to the residues closest to the UNG2 peptide in the RPA32C complex (Figure 9C). This observation provides additional support for the generation of structural models of the RPA32C–SMARCAL1-RBM complex based on the homology to the structure of the UNG2 complex and NMR chemical shift perturbation data.
Figure 9.

NMR chemical shift perturbations in RPA32C induced by the binding of SMARCAL1-RBM. (A) Chemical shift perturbations of [15N]RPA32C induced by the binding of the SMARCAL1 peptide. (B) Surface representation of RPA32C with the significant chemical shift perturbations from panel A colored yellow. (C) Surface representation of RPA32C from the UNG2 peptide complex with RPA32C residues within 3.5 Å of the peptide colored green.
Three different approaches were utilized to independently model the RPA32C–SMARCAL1-RBM complex. First, a homology model of the complex was directly generated using Modeler with the structure of the RPA32C–UNG2 peptide complex as a template. The model showed that SMARCAL1-RBM could be readily accommodated at the same binding site in RPA32C as the UNG2 peptide, in particular, the formation of an α-helix by the SMARCAL1 residues (14–27) directly in the binding site. Notably, the remaining SMARCAL1 residues, 7–13 and 28–32, were disordered in this model because the algorithm does not generate structural models for residues lacking the template and only 17 residues in the UNG2 peptide are ordered. Next, the HEX rigid-body docking server in conjunction with RPA32C NMR chemical shift perturbations defining the SMARCAL1-RBM binding site were used to perform a series of docking calculations using our RPA32C crystal structure and a computationally generated α-helical model of SMARCAL17–32. The critical finding from this docking calculation is that SMARCAL1-RBM bound not only to the same surface of RPA32C as the Modeler model but also with the same polarity and register.
To enhance the quality of the model, we turned to RosettaDock, a Monte Carlo-based algorithm, to calculate the lowest-energy conformation of the RPA32C–SMARCAL1-RBM complex.42−44 Docking of the peptide to the protein using cycles of random rigid-body perturbation was performed followed by optimization of side chain conformations using the complex of RPA32C with the UNG2 peptide (PDB entry 1DPU) as a template. A total of 1000 independent models of the RPA32C–SMARCAL1-RBM complex were generated and scored on the basis of van der Waals bonding, hydrogen bonding, implicit Gaussian solvation, side chain rotamer probabilities, and electrostatics.42−44 Figure 10 shows the lowest-energy conformer from the ensemble of best scoring RosettaDock models. Notably, the SMARCAL17–32 peptide adopted an α-helical conformation, through residues not only in the RPA32C binding site but also throughout the entire peptide. While our CD and NMR analyses support the disorder-to-helix transition in the peptide, there is no direct evidence that the peptide forms a stable helix throughout its whole length as suggested by this model. Contacts of Structural Units analysis46 was used to assign contacts between RPA32C and the SMARCAL1-RBM peptide (Figure 10). The complex is seen to have a number of complementary electrostatic interactions between acidic residues of RPA32C (e.g., E252 and D258) and basic residues in SMARCAL1-RBM (e.g., R17 and K19). Hydrophobic interactions are also observed, including A20 in SMARCAL1-RBM, which contacts S250, G253, I255, Y256, and T267 in RPA32C. These RPA32C residues also play a key role in the hydrophobic interactions with XPA, UNG2, RAD52, and TIPIN peptides.11
Figure 10.

Model of the RPA32C–SMARCAL1-RBM complex. The top left panel shows a ribbon representation of the RosettaDock model of RPA32C (cyan) in complex with the SMARCAL1-RMB peptide (yellow). The top right panel shows an overlay of the structure shown in the top left panel with the complex of RPA32C (salmon) in complex with the UNG2 peptide (green). The orientation is shifted by 90° with respect to the top[ left panel. The bottom panel lists contacts between SMARCAL1-RBM residues (yellow) and residues in RPA32C (cyan). Contacts were assigned using Contacts of Structural Units analysis.46
Discussion
The in situ proteolysis NMR method used here to identify the optimal RPA32C construct length should be applicable to other proteins with well-folded domains connected via flexible linkers and provides an additional example of the complementarity of NMR and X-ray crystallography. The crystal structure of free RPA32C allowed the first detailed analysis of the conformation changes required for target binding. Comparisons with the NMR structures of RPA32C in complex with UNG2 and TIPIN reveal that in fact, target binding requires only modest changes in the structure of RPA32C. The chemical shift perturbation data reported here confirm that like other RPA32C interaction partners, SMARCAL1 binds to the common RPA32C interface. Our structural model is consistent with these data, suggesting that there is little change in RPA32C structure outside of select side chain rearrangements.
The structural model of SMARCAL1-RBM bound to RPA32C is similar to the experimentally determined structures of UNG2 and TIPIN bound to RPA32C.11,12 As noted in our previous study, the RPA32C binding interface is devoid of hydrophobic pockets typically found at protein binding interfaces and, rather, is relatively flat.11 The importance of this characteristic is underscored by the strong conservation of alanine at the position in the binding motifs that contacts Y256 of RPA32C (Figure 7). The flatness of the binding site combined with a modest burial of hydrophobic surface (613.7 Å2 for the SMARCAL1-RBM) suggests that additional molecular forces have significant roles in molecular recognition of targets by RPA32C.
The contribution of electrostatic interactions to the binding of RPA32C targets is evident from the charge complementarity of the acidic RPA32C binding surface and the presence of multiple basic residues in the target binding motifs.11 Sequence alignment with UNG2, XPA, RAD52, and TIPIN shows that SMARCAL1-RBM is considerably less basic than other RPA32C binding partners because of the incorporation of a number of glutamates between residues 7 and 15 (Figure 7). Hence, we did not anticipate the SMARCAL1-RBM to bind to RPA32C with an affinity as strong as that of the other targets and were initially surprised to observe that in contrast, it bound more tightly than XPA, RAD52, and UNG2. However, further consideration of the sequences suggests an explanation: stabilization of the helical conformation in the RBM by key side chain residue pairs. In this model, α-helical side chain i + 4 interactions (E7–K11, E8–K12, R10–E14, K11–E15, E15–K19, and K27–E31) in SMARCAL1-RBM serve to lower the energetic cost of the disorder-to-helix transition required for binding of SMARCAL1-RBM to RPA32C and offset the absence of favorable intermolecular electrostatic interactions for other RPA32C targets such as XPA, UNG2, and RAD52. A similar line of reasoning would also explain the stronger binding of TIPIN to RPA32C, as it has the same potential helix-stabilizing i + 4 interactions as SMARCAL1-RBM (Figure 7).
In order to interact with its numerous targets involved in DNA replication, damage response and repair, RPA32C must be able to orchestrate the recruitment of other proteins in the DNA processing machinery to the DNA substrate. To perform this function efficiently, RPA32C is tethered via a flexible linker to the DNA binding apparatus of RPA, allowing it to adopt a wide range of orientations to facilitate recruitment and remodeling of the multi-protein machinery. In response to replication stalling caused by genotoxic stress, SMARCAL1 regresses replication forks to generate 4-way junctions important for fork restart, thereby preventing the accumulation of excess ssDNA when helicase and polymerase activity are uncoupled. Thus, SMARCAL1’s function is vital to efficiently reset the DNA replication fork to enable repair of fork-stalling lesions.
Upon encountering DNA damage, proteins involved in halting the cell cycle (e.g. TIPIN) and in resetting the replication fork (e.g. SMARCAL1) are required prior to recruitment of DNA repair proteins (e.g. UNG2, XPA, RPA52) to mend the damaged DNA. The higher affinity of SMARCAL1 and TIPIN for RPA32C compared to the DNA repair proteins may play a role in the sequence of DNA processing events at DNA damage sites where RPA orchestrates the recruitment of multiple proteins. Although this speculation is intriguing, the mechanisms for this cascade of hand-offs remain obscure and are currently under investigation in our laboratory.
Acknowledgments
We thank Dr. David Cortez for his intellectual contributions in the conception of this project and the Life Sciences Collaborative Access Team (LS-CAT) staff at the Advanced Photon Source for help with data collection.
Supporting Information Available
One table showing the data collection and refinement statistics for the X-ray crystal structure of RPA32C. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Atomic coordinates and structure factors for the crystal structure of RPA32C have been deposited as PDB entry 4OU0.
The authors declare no competing financial interest.
This research was supported in part by National Institutes of Health (NIH) Grants R01 GM65484 and P01 CA092584 to W.J.C., P30 ES00267 to the Vanderbilt Center in Molecular Toxicology, and P30 CA068485 to the Vanderbilt Ingram Cancer Center. A.C.M. was funded by an American Cancer Society postdoctoral fellowship (PF-12-220-01) and the Vanderbilt Training Program in Environmental Toxicology (T32 ES07028). M.D.F. was supported by National Research Service Award Postdoctoral Fellowship F32 ES021690. Support for NMR instrumentation was provided in part by grants from the National Science Foundation (0922862), the NIH (S10 RR025677), and Vanderbilt University matching funds. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zegerman P.; Diffley J. F. (2009) DNA replication as a target of the DNA damage checkpoint. DNA Repair 8, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Bansbach C. E.; Betous R.; Lovejoy C. A.; Glick G. G.; Cortez D. (2009) The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich K. A.; Cortez D. (2008) ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. A.; Eisen J. A.; Mohrenweiser H. W. (2000) Cloning and characterization of HARP/SMARCAL1: A prokaryotic HepA-related SNF2 helicase protein from human and mouse. Genomics 65, 274–282. [DOI] [PubMed] [Google Scholar]
- Yusufzai T.; Kadonaga J. T. (2008) HARP is an ATP-driven annealing helicase. Science 322, 748–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A.; Bredemeyer A. L.; Sowa M. E.; Terret M. E.; Jallepalli P. V.; Harper J. W.; Elledge S. J. (2009) The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 23, 2415–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L.; Woo E. M.; Chait B. T.; Funabiki H. (2009) Identification of SMARCAL1 as a component of the DNA damage response. J. Biol. Chem. 284, 35951–35961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C.; Bansbach C. E.; Zhao R.; Jung S. Y.; Qin J.; Cortez D. (2014) Phosphorylation of a C-terminal auto-inhibitory domain increases SMARCAL1 activity. Nucleic Acids Res. 42, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch F. B.; Bansbach C. E.; Driscoll R.; Luzwick J. W.; Glick G. G.; Betous R.; Carroll C. M.; Jung S. Y.; Qin J.; Cimprich K. A.; Cortez D. (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerkoel C. F.; Takashima H.; John J.; Yan J.; Stankiewicz P.; Rosenbarker L.; Andre J. L.; Bogdanovic R.; Burguet A.; Cockfield S.; Cordeiro I.; Frund S.; Illies F.; Joseph M.; Kaitila I.; Lama G.; Loirat C.; McLeod D. R.; Milford D. V.; Petty E. M.; Rodrigo F.; Saraiva J. M.; Schmidt B.; Smith G. C.; Spranger J.; Stein A.; Thiele H.; Tizard J.; Weksberg R.; Lupski J. R.; Stockton D. W. (2002) Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 30, 215–220. [DOI] [PubMed] [Google Scholar]
- Mer G.; Bochkarev A.; Gupta R.; Bochkareva E.; Frappier L.; Ingles C. J.; Edwards A. M.; Chazin W. J. (2000) Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell 103, 449–456. [DOI] [PubMed] [Google Scholar]
- Ali S. I.; Shin J. S.; Bae S. H.; Kim B.; Choi B. S. (2010) Replication protein A 32 interacts through a similar binding interface with TIPIN, XPA, and UNG2. Int. J. Biochem. Cell Biol. 42, 1210–1215. [DOI] [PubMed] [Google Scholar]
- Betous R.; Couch F. B.; Mason A. C.; Eichman B. F.; Manosas M.; Cortez D. (2013) Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 3, 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold M. S.; Kelly T. (1988) Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. U.S.A. 85, 2523–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold M. S. (1997) Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92. [DOI] [PubMed] [Google Scholar]
- Bochkarev A.; Pfuetzner R. A.; Edwards A. M.; Frappier L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 385, 176–181. [DOI] [PubMed] [Google Scholar]
- Lao Y.; Gomes X. V.; Ren Y.; Taylor J. S.; Wold M. S. (2000) Replication protein A interactions with DNA. III. Molecular basis of recognition of damaged DNA. Biochemistry 39, 850–859. [DOI] [PubMed] [Google Scholar]
- Treuner K.; Ramsperger U.; Knippers R. (1996) Replication protein A induces the unwinding of long double-stranded DNA regions. J. Mol. Biol. 259, 104–112. [DOI] [PubMed] [Google Scholar]
- Zou L.; Liu D.; Elledge S. J. (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Vaithiyalingam S.; Glick G. G.; Mordes D. A.; Chazin W. J.; Cortez D. (2008) The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell. Biol. 28, 7345–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva E.; Kaustov L.; Ayed A.; Yi G. S.; Lu Y.; Pineda-Lucena A.; Liao J. C.; Okorokov A. L.; Milner J.; Arrowsmith C. H.; Bochkarev A. (2005) Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U.S.A. 102, 15412–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R. M. Jr.; Orren D. K.; Nehlin J. O.; Ravn P. H.; Kenny M. K.; Machwe A.; Bohr V. A. (1999) Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- Fanning E.; Klimovich V.; Nager A. R. (2006) A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34, 4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughdrill G. W.; Buchko G. W.; Botuyan M. V.; Arrowsmith C.; Wold M. S.; Kennedy M. A.; Lowry D. F. (2003) Chemical shift changes provide evidence for overlapping single-stranded DNA- and XPA-binding sites on the 70 kDa subunit of human replication protein A. Nucleic Acids Res. 31, 4176–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki Y.; Zou L. (2006) ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc. Natl. Acad. Sci. U.S.A. 103, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T.; Saijo M.; Kuraoka I.; Kobayashi T.; Nakatsu Y.; Nagai A.; Enjoji T.; Masutani C.; Sugasawa K.; Hanaoka F.; et al. (1995) DNA repair protein XPA binds replication protein A (RPA). J. Biol. Chem. 270, 4152–4157. [DOI] [PubMed] [Google Scholar]
- Nagelhus T. A.; Haug T.; Singh K. K.; Keshav K. F.; Skorpen F.; Otterlei M.; Bharati S.; Lindmo T.; Benichou S.; Benarous R.; Krokan H. E. (1997) A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 272, 6561–6566. [DOI] [PubMed] [Google Scholar]
- Park M. S.; Ludwig D. L.; Stigger E.; Lee S. H. (1996) Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J. Biol. Chem. 271, 18996–19000. [DOI] [PubMed] [Google Scholar]
- Yoshizawa-Sugata N.; Masai H. (2007) Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 282, 2729–2740. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P.; Kupce E.; Brutscher B. (2005) SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 33, 199–211. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Hoffman R. M.; Li M. X.; Sykes B. D. (2005) The binding of W7, an inhibitor of striated muscle contraction, to cardiac troponin C. Biochemistry 44, 15750–15759. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M. A.; Stuart A. C.; Fiser A.; Sanchez R.; Melo F.; Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Fiser A.; Do R. K.; Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2006) Comparative protein structure modeling using Modeller. Current Protocols in Bioinformatics, Chapter 5, Unit 5, 6, Wiley, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macindoe G.; Mavridis L.; Venkatraman V.; Devignes M. D.; Ritchie D. W. (2010) HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 38, W445–W449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S.; Gray J. J. (2008) The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 36, W233–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S.; Chou F. C.; Conchuir S. O.; Der B. S.; Drew K.; Kuroda D.; Xu J.; Weitzner B. D.; Renfrew P. D.; Sripakdeevong P.; Borgo B.; Havranek J. J.; Kuhlman B.; Kortemme T.; Bonneau R.; Gray J. J.; Das R. (2013) Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S.; Berrondo M.; Weitzner B. D.; Muthu P.; Bergman H.; Gray J. J. (2011) Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6, e22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A.; Blackshields G.; Brown N. P.; Chenna R.; McGettigan P. A.; McWilliam H.; Valentin F.; Wallace I. M.; Wilm A.; Lopez R.; Thompson J. D.; Gibson T. J.; Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Sobolev V.; Sorokine A.; Prilusky J.; Abola E. E.; Edelman M. (1999) Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327–332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.