

Abstract

Side-chain oxysterols, such as 25-hydroxycholesterol (25-HC), are key regulators of cholesterol homeostasis. New evidence suggests that the alteration of membrane structure by 25-HC contributes to its regulatory effects. We have examined the role of oxysterol membrane effects on cholesterol accessibility within the membrane using perfringolysin O (PFO), a cholesterol-dependent cytolysin that selectively binds accessible cholesterol, as a sensor of membrane cholesterol accessibility. We show that 25-HC increases cholesterol accessibility in a manner dependent on the membrane lipid composition. Structural analysis of molecular dynamics simulations reveals that increased cholesterol accessibility is associated with membrane thinning, and that the effects of 25-HC on cholesterol accessibility are driven by these changes in membrane thickness. Further, we find that the 25-HC antagonist LY295427 (agisterol) abrogates the membrane effects of 25-HC in a nonenantioselective manner, suggesting that agisterol antagonizes the cholesterol-homeostatic effects of 25-HC indirectly through its membrane interactions. These studies demonstrate that oxysterols regulate cholesterol accessibility, and thus the availability of cholesterol to be sensed and transported throughout the cell, by modulating the membrane environment. This work provides new insights into how alterations in membrane structure can be used to relay cholesterol regulatory signals.
Side-chain oxysterols are key regulators of cellular cholesterol balance that act through activation of cholesterol-homeostatic pathways that serve to decrease the level of excess free cholesterol.1−3 Acting at a transcriptional level, side-chain oxysterols such as 25-hydroxycholesterol (25-HC) bind to the liver X receptor (LXR), activating LXR-mediated transcriptional pathways resulting in increased rates of cholesterol efflux and elimination.4 25-HC also inhibits the processing of sterol regulatory element binding proteins (SREBPs), transcription factors that stimulate de novo cholesterol synthetic pathways and enhance low-density lipoprotein receptor (LDLR) expression. 25-HC inhibits SREBP processing by promoting binding of the SREBP cleavage activating protein (SCAP) to the endoplasmic reticulum (ER) retention protein Insig, thereby retaining the SCAP–SREBP complex in the ER and preventing egress to the Golgi, the site of SREBP processing.5
In addition to these transcriptional effects, 25-HC also acutely regulates cholesterol levels through two well-characterized, nongenomic responses. First, 25-HC suppresses the activity of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), the rate-limiting enzyme in cholesterol synthesis, by promoting formation of an HMGR–Insig complex and the consequent proteosomal degradation of the enzyme.6 Second, 25-HC stimulates the esterification of cholesterol and its storage in lipid droplets. This is accomplished both by allosterically activating the ER resident cholesterol esterification enzyme acylCoA:cholesterol acyltransferase (ACAT) and by increasing the extent of transport of cholesterol to ACAT-accessible domains within ER membranes.7,8
Side-chain oxysterols are known to have strong effects on membrane structure and dynamics, expanding and disordering the membrane.9−11 Recent work with the enantiomer of 25-HC in vitro has shown that effects of side-chain oxysterols on specific physiological pathways are nonenantioselective, providing compelling evidence that side-chain oxysterols exert their cholesterol-homeostatic effects in part through nonenantioselective modulation of membrane structure rather than wholly through enantioselective protein interactions.11 Previous work on cholesterol regulatory pathways has shown that key steps in cholesterol sensing and regulation respond identically to ent-cholesterol, indicating a broad involvement for sterol–membrane interactions in cholesterol regulation.12−14
Previous attempts to link membrane structure to cholesterol regulation led to a biophysical model, termed the “cholesterol activation hypothesis”, which explains how changes in the membrane can lead to downstream effects on cholesterol levels. In this model, cholesterol is thought to exist in two distinct pools: an accessible pool that is available for cholesterol trafficking and regulation and an inaccessible pool that is complexed to the lipids around it. The membrane lipid composition determines how much cholesterol can be complexed within the inaccessible pool, and cholesterol in excess of that threshold partitions into the accessible pool.15−17
On the basis of studies of model membranes, we have recently proposed a revised model of cholesterol behavior, in which the observed increase in cholesterol accessibility at high cholesterol concentrations is not driven by saturation of cholesterol–lipid complexes but rather by cholesterol-dependent changes in membrane structure.18 In this revised model, there are no distinct pools of accessible and inaccessible cholesterol; rather, changes in membrane structure at high cholesterol concentrations lead to decreased membrane thickness and an increase in the availability of all cholesterol molecules in the membrane. As additional cholesterol is added to a membrane, all cholesterol within the membrane becomes increasingly more accessible and, at some threshold concentration, becomes available to external cholesterol sensors.
Our previous studies of the effects of 25-HC on membrane structure showed that 25-HC significantly expands the membrane area through its interfacial orientation within the bilayer.10,19 We further found that high concentrations of 25-HC in membranes significantly increase the availability of cholesterol within membranes. In the study presented here, we examine our revised model of cholesterol accessibility in the context of these membrane effects of side-chain oxysterols as well as sterol analogues known to modulate cholesterol homeostasis in vitro. We use perfringolysin O (PFO) as a sensor of physiologically available membrane cholesterol. PFO is a cholesterol-dependent cytolysin that binds to accessible membrane cholesterol, triggering PFO oligomerization, membrane insertion, and pore formation.20 PFO membrane insertion can be monitored by a change in fluorescence (as a consequence of the insertion of several tryptophan residues into the membrane). PFO insertion is dependent on the membrane cholesterol concentration and exhibits a threshold response, indicating that it acts as a sensor of available cholesterol. Using this PFO binding assay, we demonstrate that 25-HC increases the availability of membrane cholesterol, and that this cholesterol activating effect is specific to side-chain oxysterols. We also employ molecular dynamics simulations of oxysterol-containing membranes to examine the changes in cholesterol availability obtained from experimental systems, allowing us to gather specific information about changes in membrane and molecular structure caused by 25-HC and to identify the underlying structural changes associated with 25-HC-driven increases in cholesterol availability. Finally, we also analyze the effects of the molecule LY295427 (“agisterol”), a small molecule inhibitor of 25-HC-mediated cholesterol activation.21,22 We find that agisterol blocks the increase in cholesterol accessibility caused by 25-HC in a nonenantioselective manner, and that this effect of limiting cholesterol accessibility in model membranes is evident even in the absence of 25-HC. Together, our results underscore the importance of nonchiral, protein-independent membrane interactions in membrane structure and suggest how they can play an important role in the regulation of cholesterol homeostasis.
Materials and Methods
Materials and Buffers
Phospholipids, sterols, and oxysterols were obtained from Steraloids [25-HC, 7α-hydroxycholesterol (7α-HC), dioleoylphosphatidylcholine (DOPC), 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), desmosterol, and 19-hydroxycholesterol (19-HC)], Sigma [7-ketocholesterol (7-KC)], Research Plus [27-hydroxycholesterol (27-HC)], and Avanti (24S-hydroxycholesterol). PFO buffer consisted of 150 mM NaCl and 50 mM Tris-HCl (pH 7.4). LY295427, which we have termed agisterol, was synthesized in two steps from cholest-4-en-3-one, as described in both its original synthesis and the synthesis of ent-agisterol from ent-cholestenone.23,24ent-Agisterol was prepared by total steroid synthesis (stepwise construction of the ring system and side chain) as described previously.23epi-Agisterol was similarly synthesized in two steps from cholestenone, but rather than reduction of the 3-ketone with K-selectride to form the 3α-alcohol, the ketone was reduced with LiAl(OtBu)3H to yield the 3β-hydroxy epimer. ent-25-Hydroxycholesterol was prepared as described previously except that one of the two methyl groups attached to C-25 was trideuterated.11
Overexpression and Purification of PFO
The His6-PFO (C459A) expression construct in the pRSETB vector (Invitrogen) was kindly provided by A. Heuck (University of Massachusetts, Amherst, MA)20 and was transformed into BL21-CodonPlus(DE3)-RIPL Escherichia coli competent cells (Stratagene). Protein expression and purification were performed according to the manufacturer’s instructions and as described previously.20 The pooled fractions containing PFO were concentrated using an Amicon Ultra 10 kDa cutoff centrifugal filter (Millipore). The PFO concentration was adjusted using PFO buffer such that, after the addition of 10% (v/v) glycerol, the final concentration was 5–9 mg/mL. The PFO was aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C.
PFO Binding Measurements
Liposomes were synthesized as described previously.25 PFO binding experiments were performed as described previously.26 Briefly, PFO binding was assessed in 96-well plates (Corning Inc.) with 200 μL reaction mixtures containing 4 μM PFO and 800 μM liposomes in PFO buffer. After incubation at 37 °C for 1 h, the tryptophan fluorescence was measured (excitation wavelength of 290 nm, emission wavelength of 340 nm, band-pass of 5 nm) using an Infinite 200 microplate reader (Tecan Group Ltd.). Fluorescence measurements were calculated as F/Fo, where F is the measured fluorescence and Fo is the fluorescence of unbound PFO (as measured in liposomes with either 0 or 20% cholesterol in DOPC), with increased fluorescence being indicative of PFO binding. Data are expressed as means ± the standard error (SE) and are the result of at least three separate experiments. Sigmoidal binding curves were fit to the data using weighted least-squares regressions, and the data are presented as fractions of maximal binding based on these fitted curves. Cholesterol activation thresholds presented in the text were calculated as the cholesterol concentrations at which the fitted curves reach between 5 and 25% of their maximal value.
Molecular Dynamics Simulations
25-HC, cholesterol, and POPC parameters and charges were used as described previously.10,19 An initial 27-HC structure and topology were prepared by modifying equilibrated 25-HC structures. 27-HC charges were parametrized as described previously for 25-HC.10 Simulations of the pure POPC bilayer, the POPC bilayer with 30% 25-HC, and POPC with cholesterol concentrations from 5 to 54% have already been described and published and are used here as controls for new simulations.10,18,19 A 30% 27-HC structure was prepared from an equilibrated 30% 25-HC structure with 25-HC molecules replaced by 27-HC molecules and relaxed as previously described to resolve steric occlusions.10,18,19 Initial bilayer structures containing cholesterol concentrations from 5 to 54% as well as an additional 5 or 10% 25-HC were prepared by taking structures from the oxysterol-free simulations with the desired cholesterol concentration and adding individual 25-HC molecules sampled from the 30% 25-HC system and similarly relaxed to resolve steric occlusions. These starting structures were then solvated with simple point charge (SPC) water molecules and K+ and Cl– ions to approximately 55 waters per lipid (both phospholipid and sterol) and a KCl concentration of 115 mM.27 These structures were then warmed to 300 K in 50 K increments and simulated for 50 ps at each temperature before being used as the starting point for the production simulations.
All molecular dynamics simulations were performed using GROMACS version 3.3.1, 4.0, or 4.528,29 and followed the same molecular dynamics protocol that was described in our previous work.10,18,19 Production simulations were run for 400 ns of total simulation time. On the basis of examination of the membrane-projected area and system energy drift, we chose to discard the first 200 ns of each simulation as equilibration time, leaving 200 ns of steady-state simulation time for analysis of each membrane composition. Statistical comparisons between simulations were conducted by treating lipids independently, with an estimated relaxation time of 5 ns between independent samples based on autocorrelation times of membrane-projected areas. Measurements of membrane and lipid properties, including lipid solvent-accessible surface area (SASA), phospholipid tail order, membrane thickness, and phospholipid interdigitation, were performed as described previously.10,18,19
Results
25-HC Increases the Availability of Cholesterol within Membranes
To determine whether oxysterols such as 25-HC increase the availability of membrane cholesterol, we used PFO binding as a sensor of physiologically accessible cholesterol. In previous studies of binary cholesterol/phospholipid liposomes, we found that the experimental cholesterol-accessibility threshold, or the threshold range at which cholesterol becomes available for PFO binding, was between 29.2 and 33.0 mol % cholesterol in liposomes composed of DOPC, establishing the baseline capacity to retain cholesterol in an inaccessible state (Figure 1A).20,26 Above this concentration threshold, cholesterol is less shielded by the membrane lipids and becomes increasingly available for PFO binding.
Figure 1.

PFO binding to liposomes. (A) PFO binding to liposomes composed of DOPC (—) or 5% 25-HC in DOPC (−–−) at varying cholesterol concentrations. (B) PFO binding to liposomes composed of 25% cholesterol in DOPC at varying concentrations of 25-HC. (C) Comparison of PFO binding to liposomes composed of 25% cholesterol in DOPC, with varying concentrations of nat-25-HC (—) or ent-25-HC (−–−). (D) PFO binding to liposomes composed of DOPC (—), 5% 25-HC in DOPC (−–−), POPC (···), or 5% 25-HC in POPC (−·−).
To examine the effect of 25-HC on the cholesterol-accessibility threshold in DOPC bilayers, we synthesized liposomes composed of 5% 25-HC in DOPC and increasing amounts of cholesterol. By comparison with the previous results, 25-HC lowered the cholesterol-accessibility threshold by ∼15 mol % to 11.0–16.3 mol %, demonstrating a significant increase in the availability of cholesterol at lower cholesterol concentrations (Figure 1A). To ensure that the PFO was not binding to the 25-HC within the liposomes, and thus producing an artifactual cholesterol-accessibility threshold based upon the total sterol (cholesterol plus 25-HC) concentration, we prepared liposomes containing no cholesterol but with varying amounts of 25-HC in DOPC. PFO did not bind 25-HC at any concentration up to 60 mol %, validating PFO binding as a faithful indicator of the accessibility of cholesterol, rather than total sterol, in a ternary system (Figure S1 of the Supporting Information). In liposomes composed of 25% cholesterol in DOPC, which do not have sufficient accessible cholesterol to support PFO binding, the addition of as little as 1% 25-HC was sufficient to enhance PFO binding (Figure 1B). Experiments using the enantiomer of 25-HC (ent-25-HC) demonstrated that 25-HC-mediated cholesterol activation was largely nonenantioselective, indicating that 25-HC affects cholesterol because of its general properties in the membrane, rather than via a specific interaction with cholesterol or phospholipids in the membrane (Figure 1C).
To determine the contribution of membrane lipid saturation to the ability of 25-HC to increase cholesterol accessibility, we prepared liposomes composed of 5% 25-HC in POPC. It is known that the lipid composition affects the cholesterol-accessibility threshold within a membrane, with saturated lipids having thresholds higher than those of unsaturated lipids.30,31 Further, in previous studies, we demonstrated that acyl-chain saturation was an important determinant in mediating the membrane behavior of 25-HC.11 In agreement with previous reports, we found that the cholesterol-accessibility threshold in POPC (with one saturated and one monounsaturated acyl chain) was 41.6–45.0 mol % cholesterol, increased by ∼10 mol % compared with the DOPC cholesterol-accessibility threshold. The incorporation of 5% 25-HC increases cholesterol accessibility in POPC liposomes and lowers the cholesterol-accessibility threshold (to 36.3–39.1 mol %) (Figure 1D). However, the magnitude of the change in cholesterol accessibility was greater in unsaturated DOPC membranes than in partially saturated POPC membranes, indicating that the lipid composition not only sets the baseline cholesterol-accessibility threshold within a membrane but also affects how other compounds can alter that cholesterol-accessibility threshold.
Cholesterol Activation Is Associated with Oxysterol-Mediated Membrane Thinning
To examine the structural changes associated with 25-HC-mediated cholesterol activation, we prepared a set of 20 new molecular dynamics simulations of lipid bilayers composed of POPC with 5 or 10% 25-HC, and cholesterol concentrations from 0 to 54% to compare with previously published controls lacking 25-HC.18 As a measure of cholesterol activation, we measured the mean SASA of cholesterol molecules in each simulation (Figure 2A). At all concentrations of 25-HC, we find lower cholesterol accessibility at low cholesterol concentrations and large increases in accessibility at higher cholesterol concentrations. Moreover, membranes containing 25-HC show a concentration-dependent increase in cholesterol accessibility at cholesterol concentrations above ∼15 mol %, suggesting that the lowered PFO binding threshold for cholesterol in membranes containing 25-HC is caused by a shift in cholesterol to more accessible positions. We further measured the membrane thickness of our simulated bilayers containing 25-HC (Figure 2B) and found concentration-dependent decreases in membrane thickness in membranes containing 25-HC at cholesterol concentrations above ∼15 mol %, suggesting that increases in cholesterol accessibility are associated with reduced membrane thickness in the presence and absence of 25-HC.
Figure 2.

Analysis of membrane properties calculated from simulated membrane bilayers composed of 5% 25-HC in POPC (−–−) and 10% 25-HC in POPC (···) with varying concentrations of cholesterol, compared with data from previously published control simulations lacking 25-HC18 (solid gray lines). (A) Mean cholesterol solvent-accessible surface area. (B) Mean membrane thickness. (C) Mean phospholipid tail order. (D) Percent lipid interdigitation.
Thickness changes in membranes can be caused either by conformational changes in individual lipids tilting or folding up, reducing individual leaflet thickness, or by increased interdigitation of the lipids in each leaflet allowing closer apposition of the leaflets. In panels C and D of Figure 2, we show that 25-HC promotes changes in both tail order and leaflet interdigitation associated with decreased membrane thickness. Together, the data from these simulations indicate that 25-HC increases cholesterol accessibility through its effects on phospholipids, thereby decreasing membrane thickness and rendering cholesterol more exposed.
Cholesterol Activation Is Specific to Side-Chain Oxysterols
While 25-HC is commonly used to modulate cholesterol homeostasis in vitro, cells produce a variety of other endogenous oxysterols that similarly regulate cholesterol-homeostatic pathways (Chart 1). To determine whether the cholesterol activating effect of 25-HC was common to other side-chain oxysterols, we used the PFO binding assay to examine 27-HC, another physiologically relevant side-chain oxysterol. We found that 27-HC has effects on cholesterol accessibility nearly identical to those of 25-HC, suggesting that side-chain oxysterols share a common mechanism for disordering membrane lipids and increasing cholesterol accessibility (Figure 3A). In support of the PFO binding data, molecular dynamics simulations revealed that 27-HC has lipid disordering and membrane-expansive properties similar to those of 25-HC opposed to the normal action of cholesterol (Figure 3B).
Chart 1. Oxysterol Structures.
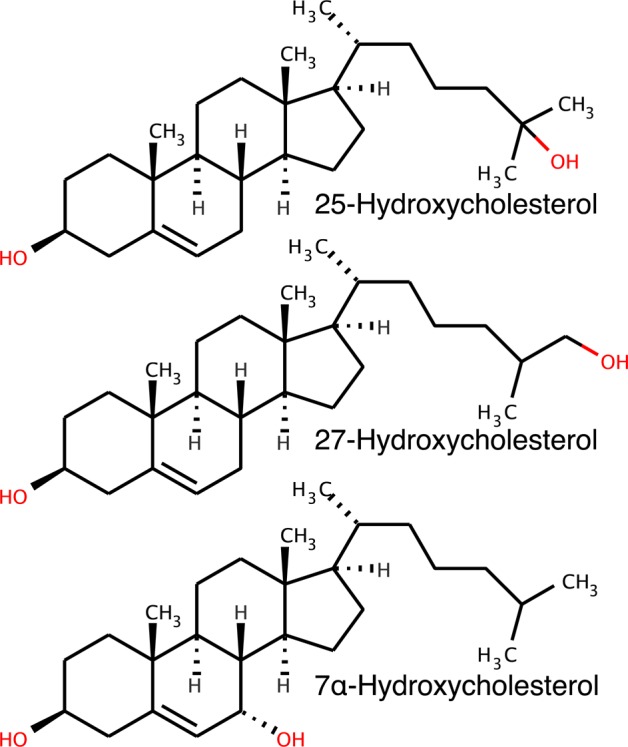
Figure 3.

(A) PFO binding to liposomes composed of POPC (—), 5% 25-HC in POPC (−–−), or 5% 27-HC in POPC (···) with varying concentrations of cholesterol. (B) Mean POPC lipid surface area in simulated membrane bilayers containing no additional sterols, an additional 30 mol % cholesterol, an additional 30 mol % 25-HC (···), or an additional 30 mol % 27-HC. Asterisks show p values of <0.01 for the difference from the control POPC simulation, as calculated with Kolmogorov–Smirnof tests. (C) PFO binding to liposomes composed of DOPC (—) or 5% 7α-HC in DOPC (−–−), with varying concentrations of cholesterol.
Side-chain oxysterols have cholesterol-homeostatic and cell signaling properties distinct from those of ring-modified oxysterols.1,2 To determine whether the homeostatic actions of oxysterols correlate with cholesterol activating properties, we synthesized liposomes composed of 5% 7α-HC, the most abundant ring-modified oxysterol in human plasma, in DOPC. We found little change in the cholesterol-accessibility threshold for the 7α-HC-containing liposomes (Figure 3C), indicating that only the side-chain oxysterols have significant effects on cholesterol accessibility. Further measurements with other sterols (desmosterol and epi-cholesterol) and oxysterols (7-ketocholesterol, 19-hydroxycholesterol, and 24S-hydroxycholesterol) showed a strong correlation between the ability to induce cholesterol activation in model membranes and reported interactions with cholesterol-homeostatic machinery, suggesting that changes in cholesterol accessibility driven by oxysterol–membrane interactions may be contributing to modulation of the sterol-sensing machinery in the ER (Figure S2 of the Supporting Information).5
Agisterol, a 25-HC Antagonist, Abrogates the Membrane Effects of 25-HC
We have amassed significant evidence that 25-HC modulates cholesterol homeostasis in part through its effects on cholesterol accessibility. As such, it is reasonable to expect that inhibitors of the effects of 25-HC on cholesterol homeostasis would also abrogate its effects on cholesterol accessibility. The hypocholesterolemic compound agisterol is well-known to antagonize the cellular effects of 25-HC, although its mechanism of action has remained elusive.21,22 To determine whether agisterol can abrogate the effects of 25-HC on cholesterol accessibility, we prepared liposomes composed of 25% cholesterol and 5% 25-HC in DOPC, with varying concentrations of agisterol. In the absence of agisterol, the 5% 25-HC in these liposomes increases cholesterol accessibility sufficiently to support partial PFO binding. The addition of agisterol prevents PFO binding in a concentration-dependent manner (Figure 4A). Experiments performed with ent-agisterol showed similar behavior, indicating that the ability of agisterol to abrogate the increase in cholesterol accessibility caused by 25-HC is nonenantioselective, and that its mechanism is based on its general membrane properties, rather than a direct stereospecific interaction between agisterol and 25-HC or cholesterol within the membrane (Figure 4A). In liposomes composed of 5% 25-HC and 10% agisterol, the cholesterol-accessibility threshold increases to 20.3–25.9 mol %, toward the cholesterol-accessibility threshold established for DOPC. Liposomes composed of 5% 25-HC and 15% agisterol completely abrogate any increase in cholesterol accessibility caused by the 5% 25-HC, further increasing the cholesterol-accessibility threshold to 27.2–31.2 mol % cholesterol (Figure 4B).
Figure 4.

(A) PFO binding to liposomes composed of 25% cholesterol and 5% 25-HC in DOPC, with varying concentrations of nat-agisterol (—) or ent-agisterol (−–−). (B) PFO binding to liposomes composed of DOPC (—), 5% 25-HC in DOPC (−–−), 10% agisterol and 5% 25-HC in DOPC (···), and 15% agisterol and 5% 25-HC (−·−), with varying concentrations of cholesterol. (C) PFO binding to liposomes composed of DOPC (—), 10% agisterol in DOPC (−–−), or 15% agisterol in DOPC (···), with varying concentrations of cholesterol.
To identify whether agisterol affects the cholesterol-accessibility threshold in membranes independent of oxysterols, we prepared liposomes composed of 10 and 15% agisterol in DOPC. We found that agisterol increases the cholesterol-accessibility threshold even in the absence of 25-HC and thus decreases cholesterol availability in a manner independent of any inhibitory effects on 25-HC-mediated cholesterol activation (Figure 4C).
epi-Agisterol, the 3β-Hydroxy Epimer of Agisterol, Does Not Oppose the Cholesterol Activating Effects of 25-HC
Although agisterol does not compete with 25-HC at any known binding sites, it is generally thought that agisterol inhibits 25-HC via a specific binding interaction. This is principally based on finding that epi-agisterol, the 3β-hydroxy epimer of agisterol, lacks 25-HC-inhibitory activity and the premise that substituting an α-hydroxyl for a β-hydroxyl at the 3-position in the steroid ring would be expected to disrupt potential protein-binding interactions (Chart 2).22,32 This inference assumes that the membrane properties of these epimers are similar, yet it is known that changing a hydroxyl from an axial to an equatorial position in a ring system can have considerable effects on a molecule’s membrane properties.33−35 Using liposomes composed of 25% cholesterol and 5% 25-HC, which supports partial PFO binding, we examined the effect of epi-agisterol on the increase in cholesterol accessibility caused by 25-HC. Notably, we found that epi-agisterol exerted the opposite effect of agisterol and further increased cholesterol accessibility in a concentration-dependent manner (Figure 5A). Experiments using liposomes composed of 5% 25-HC and 10% epi-agisterol demonstrated that epi-agisterol lowered the cholesterol-accessibility threshold to 4.6–12.1 mol % cholesterol, effectively enhancing the ability of 25-HC to increase cholesterol accessibility (Figure 5B). We further examined the effects of epi-agisterol on cholesterol accessibility independent of its effects on 25-HC. Using liposomes composed of 10% epi-agisterol in DOPC, we found that epi-agisterol lowers the accessibility threshold to 22.0–25.7 mol % cholesterol in the absence of 25-HC (Figure 5C).
Chart 2. Agisterol Structures.
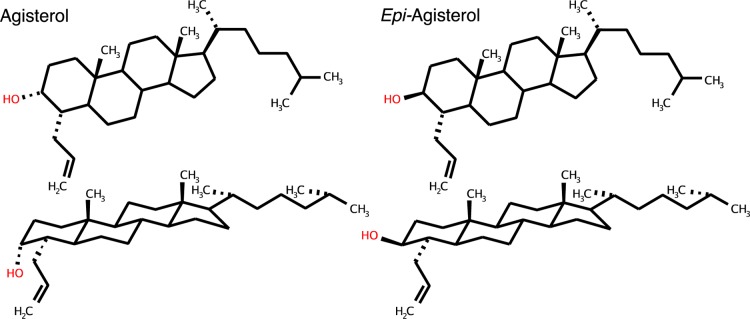
Figure 5.

(A) PFO binding to liposomes composed of 25% cholesterol and 5% 25-HC in DOPC, with varying concentrations of agisterol (—) and epi-agisterol (−–−). (B) PFO binding to liposomes composed of 5% 25-HC in DOPC (—), 10% agisterol and 5% 25-HC in DOPC (−–−), and 10% epi-agisterol and 5% 25-HC in DOPC (···), with varying concentrations of cholesterol. (C) PFO binding to liposomes composed of DOPC (—), 10% agisterol in DOPC (−–−), and 10% epi-agisterol in DOPC (···), with varying concentrations of cholesterol.
Discussion
In this study, we used PFO binding as a sensor of cholesterol availability in a membrane bilayer and demonstrated that the side-chain oxysterol 25-HC increases the cholesterol accessibility within membranes. We found that 25-HC-mediated cholesterol activation was nonenantioselective, indicating that increased cholesterol accessibility is due to the general lipid disordering and membrane expansive properties of 25-HC, rather than direct stereospecific interactions with cholesterol in the membrane. We further show that 25-HC-driven cholesterol activation is caused by effects on lipid order and leaflet interdigitation that, in turn, significantly reduce the membrane thickness to leave cholesterol in a more accessible state. Taken together, we conclude from these findings that a specific cholesterol:oxysterol ratio does not determine cholesterol accessibility, but rather that low concentrations of oxysterols can activate a relatively larger percentage of membrane cholesterol. In our assay, a detectable increase in cholesterol accessibility was seen with as little as 1% 25-HC. While bulk oxysterol concentrations in the ER, where the cholesterol regulatory machinery largely resides, has been measured to be several orders of magnitude lower than those examined in these model membranes, these data suggest that, if oxysterols are locally concentrated within specialized domains, their membrane effects could result in measurable changes in cholesterol accessibility and sterol-homeostatic responses.36
It is known that the lipid composition determines the cholesterol-accessibility threshold of a membrane, and previous studies have shown that the lipid composition influences the magnitude of any oxysterol membrane disordering effects (i.e., 25-HC has stronger membrane disordering effects in unsaturated vs saturated lipid environments).10,11,37 We found that lipid composition also affects the magnitude of oxysterol-mediated changes in cholesterol accessibility. 25-HC causes a larger increase in cholesterol accessibility in unsaturated membranes, which is of particular interest as the ER membrane—the site of cholesterol synthesis, esterification, and sterol sensing—is highly enriched in unsaturated lipids.38 Thus, the different lipid composition of the ER that results in a much lower basal cholesterol activation threshold would also be expected to amplify the oxysterol-induced membrane effects, allowing cholesterol-homeostatic machinery to be more sensitive to small changes in cellular cholesterol levels. Recent results showing increased cholesterol accessibility in ER membranes and deactivation of the SREBP transcription factor occurring concurrently with changes in ER cholesterol concentrations support this model of cholesterol homeostasis dependence on membrane structural changes.26
The strikingly different effects of side-chain oxysterols versus ring-modified oxysterols on cellular cholesterol homeostasis are well-established. In previous studies, we showed that side-chain oxysterols such as 25-HC and 27-HC disorder membranes whereas ring-modified oxysterols such as 7α-HC and 7-KC condense membranes.11 In this study, we show that the oxysterol-induced increase in cholesterol accessibility is specific to side-chain oxysterols, as 25-HC, 27-HC, and 24S-HC all significantly increase cholesterol accessibility, while ring-modified sterols such as 7α-HC, 7-KC, and 19-HC have little effect. This distinction between side-chain and ring-modified oxysterols is in agreement with the observed functional differences between these oxysterol classes. Side-chain oxysterols regulate physiologically relevant cholesterol-homeostatic pathways: LXR activation, inhibition of SREBP processing and stimulation of HMGR degradation through interaction with Insig-2, and stimulation of cholesterol esterification. Ring-modified oxysterols, on the other hand, while able to influence several cellular processes, are not physiological effectors, and have little or no effect on these specific cholesterol-homeostatic pathways.1,5,31,39 The finding that side-chain and ring-modified oxysterols do not share the same ability to increase cholesterol-accessibility membrane properties further suggests that the unique membrane behavior of the side-chain oxysterols, specifically increasing the accessibility of membrane cholesterol, is a key determinant in regulating cholesterol regulatory pathways.
Novel insight into the molecular basis of the cholesterol regulatory effects of side-chain oxysterols has come from study of the agisterol compound. Agisterol (LY295427) was discovered to antagonize the cholesterol regulatory effects of 25-HC nearly two decades ago, yet its mechanism of action has remained elusive.22 Others have proposed that agisterol competes with 25-HC at a specific binding site. Evidence that was suggested to support a specific-binding mechanism is the fact that epi-agisterol, the 3β-hydroxy epimer of agisterol, has no inhibitory effect on 25-HC-mediated cholesterol-homeostatic responses.21,32,40 However, subsequent studies found that agisterol did not compete with 25-HC for Insig binding.5 While studies examining the effect of agisterol on inhibiting oxysterol-mediated activation of LXR-regulated targets found that agisterol antagonized the LXR activating effects of several side-chain oxysterols, agisterol did not inhibit LXR activation by TO-901317, a small molecule activator of LXR.41 Taken together, it seems unlikely that agisterol directly competes with oxysterols for binding to Insig or the LXR binding site. Rather, agisterol appears to work through a mechanism that directly interferes with oxysterol effects on membrane cholesterol accessibility.
Our results show that agisterol abrogates the effects of 25-HC on cholesterol accessibility in a nonenantioselective manner, suggesting that its in vivo effects on 25-HC-mediated cholesterol-homeostatic responses are due to these membrane effects rather than any stereospecific protein interactions. Further, this suppression of cholesterol accessibility by agisterol is not dependent on the presence of 25-HC and also occurs in 25-HC-free membranes. Thus, it appears that agisterol and 25-HC both directly modify membrane structure to modulate cholesterol accessibility, but in opposing ways, and that these opposing effects are responsible for the observed antagonism of agisterol and 25-HC.
Our data also suggest an alternative explanation for the regulatory inactivity of epi-agisterol. We found that epi-agisterol, unlike ent-agisterol, was unable to suppress cholesterol accessibility in model membranes. The observed inability of epi-agisterol to inhibit the cholesterol regulatory effects of 25-HC is thus likely due not to impaired protein interactions but rather to its altered membrane interactions, providing further evidence of the importance of membrane structure in regulating cholesterol homeostasis.
Our previous work has shown that changes in cholesterol accessibility as a function of cholesterol concentration are driven largely by decreases in membrane thickness at high cholesterol concentrations.18 In this setting, the phospholipids and the water interface move inward while the cholesterol remains fixed in position, leaving it more exposed to solvent and extramembrane acceptors. This membrane thinning observed at high cholesterol concentrations is consistent with both other simulated models and nuclear magnetic resonance and X-ray studies.42−45 We had independently shown that high concentrations of oxysterols can significantly thin the membrane, because of their strong disordering effects on lipid acyl chains and their adoption of interfacial orientations within the membrane.10,19 In this study, these observations are extended to understanding the behavior of cholesterol in oxysterol-containing membranes. We find that oxysterols thin the membrane in a manner that is independent of cholesterol, in turn partially exposing cholesterol, and thus increasing cholesterol accessibility indirectly through changes to membrane structure.
In this study, we add to the mounting evidence the fact that side-chain oxysterols increase the accessibility of membrane cholesterol via their membrane properties, and that this mechanism contributes to 25-HC-mediated cholesterol regulation. As demonstrated in earlier work, increases in cholesterol accessibility expose cholesterol to cytosolic acceptors such as cyclodextrin or apolipoproteins, providing a possible mechanism for how 25-HC could enhance intracellular cholesterol trafficking or elimination of cholesterol from the cell.17,46,47 It seems plausible that 25-HC-mediated increases in cholesterol accessibility could allow other cholesterol-sensing molecules to bind the newly available cholesterol. It is intriguing that SREBP processing is inhibited once the cholesterol content within the ER rises above 5 mol %, the same threshold at which cholesterol becomes available for PFO binding.26 The implication is that membrane cholesterol must be in an accessible conformation to be sensed and bound by SCAP, ultimately leading to Insig binding and the retention of the Insig–SCAP–SREBP complex in the ER. If cholesterol-sensing proteins bind only accessible cholesterol, then by lowering the cholesterol-accessibility threshold, 25-HC can increase the sensitivity of these cholesterol-sensing proteins by increasing the fraction of cholesterol available to bind to these proteins.
Maintaining a narrow cholesterol range within cell membranes is required for cell viability. While a number of transcriptional mechanisms are known to be essential for the maintenance of cholesterol homeostasis, these mechanisms necessarily act on a comparatively slow time scale. It is apparent that a membrane-based biophysical mechanism for the control of cholesterol homeostasis offers significant advantages for rapid responses to abrupt changes in cellular cholesterol levels, as would be encountered in vivo. Our previous work shows that 27-HC is synthesized within minutes of cholesterol loading, indicating their ability to rapidly respond to changes and mediate sterol-homeostatic responses.48 While these oxysterols act on a number of transcriptional cholesterol regulatory pathways, we propose that the oxysterols initially act in a feed-forward pathway to further activate cholesterol by altering the membrane environment, resulting in an increased level of plasma membrane to ER cholesterol trafficking for esterification, as well as enhanced sensitivity of cholesterol-homeostatic machinery to cholesterol. In this work, we show that the lipid environment and cholesterol accessibility are poised to respond to the presence of side-chain oxysterols, contributing to downstream cholesterol regulation. Further investigation of the molecular basis of oxysterol-mediated activation of cholesterol will improve our understanding of the mechanisms through which cholesterol is transported and sensed.
Acknowledgments
We thank Alejandro Heuck for generously providing the PFO expression construct.
Glossary
Abbreviations
- 25-HC
25-hydroxycholesterol
- PFO
perfringolysin O
- agisterol
- LXR
liver X receptor
- SREBPs
sterol regulatory element binding proteins
- LDLR
low-density lipoprotein receptor
- SCAP
SREBP cleavage activating protein
- ER
endoplasmic reticulum
- HMGR
3-hydroxy-3-methylglutaryl-CoA reductase
- ACAT
acylCoA:cholesterol acyltransferase
- DOPC
dioleoylphosphatidylcholine
- ent-25-HC
enantiomer of 25-HC
- POPC
1-palmitoyl-2-oleoylphosphatidylcholine
- 27-HC
27-hydroxycholesterol
- 7-αHC
7-α-hydroxycholesterol
- 7-KC
7-ketocholesterol
- epi-agisterol
3β-hydroxy epimer of agisterol.
Supporting Information Available
PFO binding measurements with 25-HC, agisterol, and ent-agisterol in membranes as well as measurements of the effects of desmosterol, 24S-hydroxycholesterol, 7-ketocholesterol, 19-hydroxycholesterol, and epicholesterol on the binding of PFO to cholesterol. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.A.B. and B.N.O. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grant F30 HL97563 to A.A.B., Cardiovascular Biology Training Grant T32HL007275 to A.A.B. and B.N.O., and NIH Grant HL067773 to D.S.O., D.F.C., P.H.S., and N.A.B. Computational resources were provided by the Texas Advanced Computing Center through Teragrid Grants TG-MCB060053 and TG-MCA08X003 as well as the National Biomedical Computation Resource (NIH Grant P41 RR0860516). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant OCI-1053575.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gill S.; Chow R.; Brown A. J. (2008) Sterol regulators of cholesterol homeostasis and beyond: The oxysterol hypothesis revisited and revised. Prog. Lipid Res. 47, 391–404. [DOI] [PubMed] [Google Scholar]
- Olkkonen V. M.; Hynynen R. (2009) Interactions of oxysterols with membranes and proteins. Mol. Aspects Med. 30, 123–133. [DOI] [PubMed] [Google Scholar]
- Brown M. S.; Goldstein J. L. (2008) Cholesterol feedback: From Schoenheimer’s bottle to Scap’s MELADL. J. Lipid Res. 50, S15–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski B. A.; Willy P. J.; Devi T. R.; Falck J. R.; Mangelsdorf D. J. (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 383, 728–731. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A.; Ikeda Y.; Kwon H. J.; Brown M. S.; Goldstein J. L. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. U.S.A. 104, 6511–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBose-Boyd R. A. (2008) Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 18, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D.; Chang C. C.; Qu X.; Chang T. Y. (1995) Activation of acyl-coenzyme A:cholesterol acyltransferase by cholesterol or by oxysterol in a cell-free system. J. Biol. Chem. 270, 685–695. [DOI] [PubMed] [Google Scholar]
- Lange Y.; Ye J.; Rigney M.; Steck T. L. L. (1999) Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J. Lipid Res. 40, 2264–2270. [PubMed] [Google Scholar]
- Ohvo-Rekilä H.; Ramstedt B.; Leppimäki P.; Slotte J. P. (2002) Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 41, 66–97. [DOI] [PubMed] [Google Scholar]
- Olsen B. N.; Schlesinger P. H.; Baker N. A. (2009) Perturbations of membrane structure by cholesterol and cholesterol derivatives are determined by sterol orientation. J. Am. Chem. Soc. 131, 4854–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale S. E.; Westover E. J.; Dudley N.; Krishnan K.; Merlin S.; Scherrer D. E.; Han X.; Zhai X.; Brockman H. L.; Brown R. E.; Covey D. F.; Schaffer J. E.; Schlesinger P.; Ory D. S. (2009) Side chain oxygenated cholesterol regulates cellular cholesterol homeostasis through direct sterol-membrane interactions. J. Biol. Chem. 284, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder C. M.; Westover E. J.; Kumar A. S.; Ostlund R. E.; Covey D. F. (2001) Enantiospecificity of Cholesterol Function in Vivo. J. Biol. Chem. 276, 44369–44372. [DOI] [PubMed] [Google Scholar]
- Covey D. F. (2009) ent-Steroids: Novel tools for studies of signaling pathways. Steroids 74, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Chang C. C. Y.; Westover E. J.; Covey D. F.; Chang T.-Y. (2005) Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: In vitro and intact cell studies. Biochem. J. 391, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A.; McConnell H. (2000) Chemical activity of cholesterol in membranes. Biochemistry 39, 8119–8124. [DOI] [PubMed] [Google Scholar]
- Lange Y.; Steck T. L. L. (2008) Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 47, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck T. L.; Lange Y. (2010) Cell cholesterol homeostasis: Mediation by active cholesterol. Trends Cell Biol. 20, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B. N.; Bielska A. A.; Lee T.; Daily M. D.; Covey D. F.; Schlesinger P. H.; Baker N. A.; Ory D. S. (2013) The Structural Basis of Cholesterol Accessibility in Membranes. Biophys. J. 105, 1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B. N.; Schlesinger P. H.; Ory D. S.; Baker N. A. (2011) 25-Hydroxycholesterol Increases the Availability of Cholesterol in Phospholipid Membranes. Biophys. J. 100, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan J. J.; Tweten R. K.; Johnson A. E.; Heuck A. P. (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski B. A. A.; Shan B.; Russell D. W. W. (2001) The hypocholesterolemic agent LY295427 reverses suppression of sterol regulatory element-binding protein processing mediated by oxysterols. J. Biol. Chem. 276, 45408–45416. [DOI] [PubMed] [Google Scholar]
- Bensch W. R.; Gadski R. A.; Bean J. S.; Beavers L. S.; Schmidt R. J.; Perry D. N.; Murphy A. T.; McClure D. B.; Eacho P. I.; Breau A. P.; Archer R. A.; Kauffman R. F. (1999) Effects of LY295427, a low-density lipoprotein (LDL) receptor up-regulator, on LDL receptor gene transcription and cholesterol metabolism in normal and hypercholesterolemic hamsters. J. Pharmacol. Exp. Ther. 289, 85–92. [PubMed] [Google Scholar]
- Bielska A. A.; Ory D. S.; Covey D. F. (2011) Synthesis of the enantiomer of the oxysterol-antagonist LY295427. Steroids 76, 986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.-S.; Rampersaud A. A.; Archer R. A.; Pawlak J. M.; Beavers L. S.; Schmidt R. J.; Kauffman R. F.; Bensch W. R.; Bumol T. F. (1995) Synthesis and Biological Evaluation of a New Series of Sterols as Potential Hypocholesterolemic Agents. J. Med. Chem. 38, 277–288. [DOI] [PubMed] [Google Scholar]
- Pan H.; Marsh J. N.; Christenson E. T.; Soman N. R.; Ivashyna O.; Lanza G. M.; Schlesinger P. H.; Wickline S. A. (2012) Postformulation peptide drug loading of nanostructures. Methods Enzymol. 508, 17–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov A.; Radhakrishnan A. (2010) Accessibility of cholesterol in endoplasmic reticulum (ER) membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 285, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. (1987) The Missing Term in Effective Pair Potentials. J. Phys. Chem. 91, 6269–6271. [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56. [Google Scholar]
- Lindahl E.; Hess B.; van der Spoel D. (2001) GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7, 306–317. [Google Scholar]
- Huang J.; Buboltz J. T.; Feigenson G. W. (1999) Maximum solubility of cholesterol in phosphatidylcholine and phosphatidylethanolamine bilayers. Biochim. Biophys. Acta 1417, 89–100. [DOI] [PubMed] [Google Scholar]
- Wang J.; Megha; London E. (2004) Relationship between sterol/steroid structure and participation in ordered lipid domains (lipid rafts): Implications for lipid raft structure and function. Biochemistry 43, 1010–1018. [DOI] [PubMed] [Google Scholar]
- Du X.; Pham Y. H.; Brown A. J. (2004) Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: Implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J. Biol. Chem. 279, 47010–47016. [DOI] [PubMed] [Google Scholar]
- Dufourc E. J. (2008) Sterols and membrane dynamics. J. Chem. Biol. 1, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey J. B.; Pownall H. J. (2006) Structures of biologically active oxysterols determine their differential effects on phospholipid membranes. Biochemistry 45, 10747–10758. [DOI] [PubMed] [Google Scholar]
- Rog T.; Pasenkiewiczgierula M. (2003) Effects of Epicholesterol on the Phosphatidylcholine Bilayer: A Molecular Simulation Study. Biophys. J. 84, 1818–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A.; Goldstein J. L.; McDonald J. G.; Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 8, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell H. (2003) Condensed complexes of cholesterol and phospholipids. Biochim. Biophys. Acta 1610, 159–173. [DOI] [PubMed] [Google Scholar]
- Borradaile N. M.; Han X.; Harp J. D.; Gale S. E.; Ory D. S.; Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737. [DOI] [PubMed] [Google Scholar]
- Janowski B. A.; Grogan M. J.; Jones S. A.; Wisely G. B.; Kliewer S. A.; Corey E. J.; Mangelsdorf D. J. (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. U.S.A. 96, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. S.; Rampersaud A. A.; Beavers L. S.; McClure D. B.; Gardner A. J.; Eacho P. I.; Foxworthy P. S.; Gadski R. A. (1999) Synthesis and in vitro biological activity of 4α-(2-propenyl)-5α-cholest-24-en-3α,12α-diol, a 12α-hydroxyl analog of 4α-(2-propenyl)-5α-cholest-24-en-3α-ol: The latter is a potent activator of the low-density lipoprotein R. Steroids 64, 735–741. [DOI] [PubMed] [Google Scholar]
- Janowski B. A. (2002) The hypocholesterolemic agent LY295427 up-regulates INSIG-1, identifying the INSIG-1 protein as a mediator of cholesterol homeostasis through SREBP. Proc. Natl. Acad. Sci. U.S.A. 99, 12675–12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; MacCallum J. L.; Tieleman D. P. (2009) Thermodynamic analysis of the effect of cholesterol on dipalmitoylphosphatidylcholine lipid membranes. J. Am. Chem. Soc. 131, 1972–1978. [DOI] [PubMed] [Google Scholar]
- Ivankin A.; Kuzmenko I.; Gidalevitz D. (2010) Cholesterol-Phospholipid Interactions: New Insights from Surface X-ray Scattering Data. Phys. Rev. Lett. 104, 108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaram M. B.; Thompson T. E. (1990) Modulation of phospholipid acyl chain order by cholesterol. A solid-state deuterium nuclear magnetic resonance study. Biochemistry 29, 10676–10684. [DOI] [PubMed] [Google Scholar]
- Pencer J.; Nieh M.-P.; Harroun T. A.; Krueger S.; Adams C.; Katsaras J. (2005) Bilayer thickness and thermal response of dimyristoylphosphatidylcholine unilamellar vesicles containing cholesterol, ergosterol and lanosterol: A small-angle neutron scattering study. Biochim. Biophys. Acta 1720, 84–91. [DOI] [PubMed] [Google Scholar]
- Haynes M.; Phillips M.; Rothblat G. (2000) Efflux of cholesterol from different cellular pools. Biochemistry 39, 4508–4517. [DOI] [PubMed] [Google Scholar]
- Slotte J.; Bierman E. (1988) Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem. J. 250, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange Y.; Steck T. L.; Ye J.; Lanier M. H.; Molugu V.; Ory D. (2009) Regulation of fibroblast mitochondrial 27-hydroxycholesterol production by active plasma membrane cholesterol. J. Lipid Res. 50, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.