

Abstract
Statistical process control (SPC) is a robust set of tools that aids in the visualization, detection, and identification of assignable causes of variation in any process that creates products, services, or information. A tool has been developed termed Statistical Process Control in Proteomics (SProCoP) which implements aspects of SPC (e.g., control charts and Pareto analysis) into the Skyline proteomics software. It monitors five quality control metrics in a shotgun or targeted proteomic workflow. None of these metrics require peptide identification. The source code, written in the R statistical language, runs directly from the Skyline interface which supports the use of raw data files from several of the mass spectrometry vendors. It provides real time evaluation of the chromatographic performance (e.g., retention time reproducibility, peak asymmetry, and resolution); and mass spectrometric performance (targeted peptide ion intensity and mass measurement accuracy for high resolving power instruments) via control charts. Thresholds are experiment- and instrument-specific and are determined empirically from user-defined quality control standards that enable the separation of random noise and systematic error. Finally, Pareto analysis provides a summary of performance metrics and guides the user to metrics with high variance. The utility of these charts to evaluate proteomic experiments is illustrated in two case studies.
Keywords: Quality Control, Statistical Process Control, Proteomics, Mass Spectrometry, Shewhart Control Charts
Introduction
The proteomics field has witnessed tremendous growth over the last decade fueled by a combination of advancements in mass spectrometry instrumentation/technology [1–4], software [5], and bioinformatics. [6–8] Instruments are even faster, more sensitive and accurate than versions introduced in the previous year. As a result, mass spectrometry has become an indispensable tool in biological studies with the number of protein identifications from a complex tissue lysate routinely in the several thousands. [9,10] When applied to complex biological problems, mass spectrometry can provide critical information at the systems level by investigating overall-protein dynamics including site-specific post translational modifications, relative quantification amongst different states, half-life, and evaluation of molecular interactions. However, this key information relies on the quality of the fundamental measurements made by LC MS/MS including retention time, chromatographic peak width, mass measurement and ion intensity of precursor and fragment ions. Data quality and robustness depends on a clear standard operating procedure and the systematic evaluation of the entire workflow. With the continued goals of biomarker discovery, clinical applications of established biomarkers, and integrating large multiomic datasets (i.e., systems biology), more accessible tools are needed to monitor data quality throughout a proteomics experiment.
An experimental design should not only be statistically sound but should also incorporate a quality control procedure that consistently evaluates appropriate performance metrics. However, descriptions of performance procedures are rarely included in the method sections of manuscripts. This omission is probably due to the lack of both tools and knowledge of suitable quality metrics. Many investigators have begun to realize the importance of assessing proteomic data quality. [11] The driving force behind these realizations is the irreproducibility of peptide and protein identifications. [12,13] While implementing robust quality control (QC) protocols will likely improve reproducibility within a laboratory, the limiting factor between laboratories will continue to be the absence of a standard operating procedure [12] and the stochastic sampling inherent of data dependent acquisition (DDA). [13–15] Regardless, for these extended biomarker discovery experiments and clinical applications it is essential that tools continue to be developed for monitoring instrument performance of a specific instrument and within a specific experiment.
National Institutes of Standards and Technology (NIST), working in collaboration with National Cancer Institute-supported Clinical Proteomic Technology Assessment for Cancer (CP-TAC) Network, developed a protocol termed MSQC for evaluating 46 system performance metrics in a shotgun proteomic experiment. [16] This list is extensive and includes everything from basic peak widths in chromatography to monitoring dynamic sampling in DDA. However, the NIST platform has not seen wide spread implementation likely due to its complexity and difficulty in incorporating different instrument and database search platforms. [17] QuaMeter performed a similar analysis as MSQC and computed 42 performance metrics for each raw data file. [17] QuaMeter did circumvent several of the original limitations of MSQC including incorporating different vendor data formats via the ProteoWizard library. [18] While both MSQC and QuaMeter are rather comprehensive, the large number of metrics reported makes it difficult for the non-expert to objectively interpret the state of the experiment.
Pichler et. al., developed a semi-automated approach for quality control termed simple automatic quality control (SIMPATICO) [19] where a designated server calculates a variety of metrics based on two types of quality control samples: 1) a sensitivity test and 2) a performance and speed test. SIMPATICO provides an interface in which one can store QC results and longitudinally track the performance of instruments; however, this software is currently limited to data acquired on Thermo Scientific instruments. Due to the dependency of the majority of existing QC metrics on database searching, most QC tools are currently used for assessment of system performance prior to and/or after an experiment, but not systematically throughout a study. Recently Abbatiello et. al. presented a system suitability protocol (SSP) for evaluating liquid chromatography stable isotope dilution selected reaction monitoring mass spectrometry (LC-SID-SRM-MS) workflows. [20] Due to the nature of targeted proteomics using SRM, this QC method does not rely on peptide identification. However, there remains a need for a simple but powerful vender neutral tool that quickly assesses system performance in either a targeted or discovery experiment that will guide the user to problematic areas, such that valuable instrument time and sample are not wasted on a sub-optimally performing system.
Statistical process control is a proven set of tools that provides an objective method to monitor the transformation of a set of inputs into desired outputs (i.e., a process). It was first routinely implemented in the United States in the automobile industry in the early 1980’s and principles from SPC are used in industries ranging from manufacturing, sales, marketing, finance, technology sectors and clinical diagnostics. [21,22] Its power as a quality control procedure lies in its primary focus on early detection of a process performing outside defined thresholds and the subsequent determination of the cause of that variation such that the process is continually improved upon. SPC aims to separate random error or error that users have little control over (referred to as common cause variation) from error that can be assigned (referred to as special cause variation). If the benefit to cost ratio is high enough, special cause variation can be examined and improved upon such that its effect on product quality is eliminated or at least mitigated. SPC has seen limited use in proteomic studies; however, a recent article by Bramwell et. al. summarizes the power of SPC as a quality control procedure and its application to a 2-D DIGE proteomics experiment. [23]
The staple in SPC is the control chart described by Shewhart [24,25] in the mid 1920’s (also referred to as the Levey-Jennings [26] chart in the clinical regime). The main purpose of the control chart is to track outputs from a process over time and to determine departure from what is considered “statistical control.” The basic principle for the control chart [24] is derived from simple statistics in which the distribution of averages from samples tends toward normal regardless of the distribution of the original population. An efficient method to summarize the data in the control chart across several metrics is via a Pareto Chart. The Pareto principle, first noted by Vilfredo Pareto and first implemented in quality management by Joseph Juran [27] in the 1940’s, states that most problems (~80%) arise from only a few (~20%) of the possible causes. This idea can be applied to proteomics in that it is unlikely that all problems (e.g., retention time drift, mass analyzer out of calibration, sensitivity issues) will occur simultaneously. The Pareto chart is a combination of a bar and line graph which displays the number of non-conformers from each metric category along with its cumulative percentage. It provides the user instant feedback on which metrics are more variable and may require immediate attention or optimization.
To help the proteomics community implement principles of SPC in their own laboratory, we developed a tool called Statistical Process Control in Proteomics (SProCoP). The LC-MS process is systematically evaluated by a QC standard and then the data is imported into the freely available Skyline software. [5] This source code is written in the R statistical language, runs directly from Skyline [28], takes user input, and produces a control chart matrix, box plots, and a Pareto chart. It monitors the performance of 5 identification-free metrics including signal intensity, mass measurement accuracy (MMA), retention time reproducibility, FWHM, and peak symmetry. The utility of this QC tool to detect systematic changes in a proteomics experiment via LC MS/MS is shown in two case studies.
Methods
Materials
Formic acid, ammonium bicarbonate, DTT, and iodoacetamide, were obtained from Sigma Aldrich (St. Louis, MO). Proteomics grade trypsin was purchased from Promega (Madison, WI). HPLC grade acetonitrile was from Burdick & Jackson (Muskegon, MI). The 6 protein digest of bovine proteins was from Bruker-Michrom (Auburn, CA) and used throughout.
SProCoP
The tool can be downloaded from the following website: (http://proteome.gs.washington.edu/software/skyline/tools/sprocop.html). This zipped file contains everything that is needed to run SProCoP from the Skyline interface. A detailed tutorial including step by step instructions is available to guide the user through the install from the website. Five quality control metrics are monitored and include: 1) Targeted peak areas (from MS1 filtering [29], selected reaction monitoring, or targeted MS/MS [30]) reported by Skyline; 2) Mass measurement accuracy for high resolving power instruments of targeted peptides; 3) full width at half maximum of targeted peptides; 4) peak asymmetry of targeted peptides; 5) retention time of targeted peptides. Further description of how each metric is calculated can be found in the supplemental information. Three different charts are used and include control charts, box plots (high resolution data), and Pareto charts.
Methods
Detection of Gross Errors in the System
Quality controls were monitored within the context of a larger study assessing the time course trypsin digestion of proteins reconstituted from dried blood spots. All experimental data was collected on a Q-Exactive tandem mass spectrometer (ThermoFisher, Bremen, Germany) coupled to a nano Acquity UPLC (Waters). The mass spectrometer settings and the targeted peptides used for these experiments are summarized in Supplemental Figure 1. Full scan MS data was collected in a cycle with eight targeted MS/MS scans. Quality controls were monitored at an interval of every 4 experimental injections. For each QC experiment, a 3μL aliquot of the diluted bovine protein digest (50 fmole/μL) was loaded from a 5 μL sample loop onto a 40 × 0.1 mm polyimide coated fused silica trap column (Polymicro Technologies) packed with Jupiter 4u Proteo 90 reverse phase beads (Phenomenex) at a flow rate of 1 μL/min for eight minutes. Peptides were then resolved on a 150 × 0.075 mm polyimide coated fused silica column (Polymicro Technologies) packed with ReproSil-Pur 3μ C18-AQ 120 reverse phase beads (Dr. Maisch GmbH) using a 30 minute linear gradient from 2% acetonitrile in 0.1 % formic acid to 40% acetonitrile in 0.1 % formic acid at a flow rate of 300 nL/min. The initial gradient was followed by steeper 5 minute linear gradient from 40% acetonitrile in 0.1% formic acid to 60% acetonitrile in 0.1% formic acid also at 300 nL/min. The column was then washed for 5 minutes at 95% acetonitrile in 0.1% formic acid and finally re-equilibrated to initial conditions at 500 nL/min.
Detection of Known Changes to the System
Data from a previous published report was used to evaluate the sensitivity of these analyses to detect different disturbances to the system (i.e., a positive control). These experiments are described in detail elsewhere. [31] In brief, the reproducibility of changing the trap cartridge was evaluated using a novel modular LC-MS source design in which the trap, column, and emitter can be replaced independent of one another. The experiment explored the reproducibility of trap cartridge replacement in which 5 QC standards were analyzed on similar length traps n=3 (4.0±0.1 cm). Thresholds for the various QC metrics were determined from the first set of 5 QC standards analyzed on the first trap. The purpose was to determine the sensitivity of these statistical analyses to display and detect a known change to the system (i.e., positive control).
Results/Discussion
Figure 1 summarizes the overall process of using SPC in a proteomics experiment. In LC-MS, as with any process, inputs will affect the measured output. As illustrated in Figure 1A, one can separate these input parameters into three categories: 1) Electrospray ionization inputs; 2) Liquid chromatography inputs; and 3) Mass spectrometry inputs. Unlike traditional industries that use SPC, these inputs are more variable and often user-, laboratory- and/or experiment-specific. However, for any one set of inputs the measured outputs throughout an experiment should be within statistical control. These data are then automatically imported into Skyline where the QC metrics for various targeted peptides are extracted. From the Skyline interface, the SProCoP tool runs and displays the metrics for each individual peptide in a control chart matrix. The control chart allows visual determination of the point where the process goes outside the empirically-defined thresholds. A detailed explanation of the control chart implemented within SProCoP is described in Supplemental Figure 2. In addition, a screen shot of the exact output from SProCoP the user can expect is available in Supplemental Figure 3.
Figure 1.

A) A diagram summarizing the workflow for using SPC in a proteomics experiment. QC data is imported into Skyline where a control chart matrix, Pareto analysis and box plots are outputted. These charts illustrate a representative output from SProCoP when using a high resolving power mass spectrometer. The box plots of MMA would not be displayed if using a low RP mass spectrometer. B) The process is systematically evaluated approximately every 4–5 injections. C) Scan cycle consists of a MS1 full scan followed by targeted MS/MS or SRM scans.
Defining the state of statistical control (i.e., thresholds) is not straight-forward and is determined empirically as shown in Figure 1B. In practice, we will often use a reference set at the beginning of each experiment that consists of at least 3–5 standards prior and 3 or more standards after the start of an experiment to establish the thresholds. Since there is no ideal number of standards, the script allows the user to specify the number of standards (n≥3) from which to determine the thresholds. The LC-MS process is then systematically evaluated every 4th or 5th injection with the same standard used to establish the thresholds. These QC standards are then imported into Skyline. The scan cycle consists of a single MS1 scan followed by targeted MS/MS scans (or SRM scans) monitoring a user-determined number of peptides distributed across the gradient (Figure 1C). It is worth noting that peptide stability is critical for the successful implementation of this QC procedure. For this reason, targeting peptides with amino acids that are easily modified in-vitro (e.g., methionine, asparagine, glutamine) could lead to false positives and should be avoided. It is recommended to perform a thorough investigation with the user’s standard to ensure that the peptides chosen are stable across several days. A list of peptides from the Bovine protein digest standard routinely used in the laboratory for QC analysis is available in Supplemental Figure 1.
To determine a non-conformer, a threshold of ±3 standard deviations (sd) from the mean is used for all metrics except mass measurement accuracy (MMA) which is user specified. The width of these thresholds determines the run length – defined as the maximum number of runs possible without the detection of a non-conformer by chance. For example, a threshold of ± 2 standard deviations would yield an average run length of 20 (1 in 20 runs would be a false positive). Others have used a cutoff of 1 standard deviation below the mean for QC metrics (e.g., number of phosphopeptide identifications). [32] This threshold is overly conservative – considering by chance ~16% (~1 out of 6) of measurements should be below 1σ. In implementation of the SPC in the context presented here which requires systematic evaluation, a 1 or 2 standard deviation threshold would yield a large number of failures that are due simply due to random chance. It is worth noting that while we are decreasing the false positive rate (i.e., type 1 error) with a ± 3 sd threshold (~1 in 370 runs), we are also theoretically increasing the false negative rate (type 2 error).
Case Study 1 – Detecting Gross Errors
Figure 2A shows a bar graph of the integrated peaks areas from a targeted MS/MS experiment of a peptide across the study. As often the case, the start of the LC MS/MS experiment was in statistical control and any variation in the peak areas can be attributed to common causes. These subsets of runs for the different metrics are then used to establish the empirical thresholds for the remainder of the study as described vide supra. The bar graph does show a systematic shift in peak areas; however, it is difficult to quantify this shift in a statistical manner. The control chart of this data (Figure 2B) provides a nice visual determination of the systematic shift to lower peak areas and the point in time where the peptide abundance is repeatedly out of statistical control (i.e., outliers > ± 3 sd). The other targeted peptides show a similar systematic shift (Figure 2C). The first 10 quality control runs were used to establish the thresholds with the blue line representing the average and green, brown, and red representing ±1 sd, ± 2 sd, ± 3 sd from the mean, respectively. It is worth noting that unless metric(s) are considerably extreme (e.g., very low or no signal observed), less emphasis should be placed on single random occurrences for a specific peptide/metric than systematic shifts and repeatable observations of individual metrics for all or multiple peptides performing outside the empirical threshold. Supplemental Figure 4 illustrates this point further.
Figure 2.
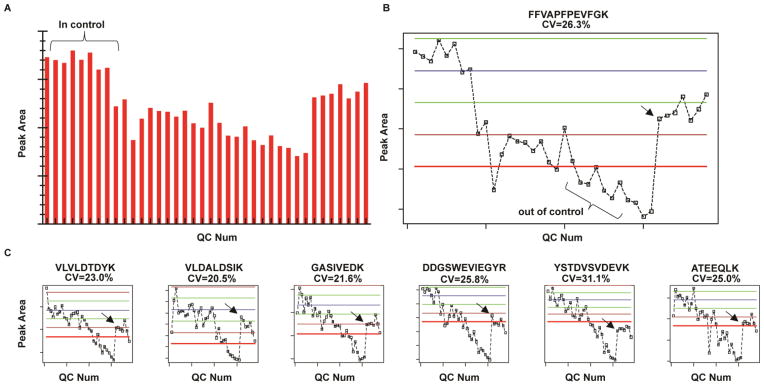
A) A bar graph of the integrated peak areas of a representative peptide (FFVAPFPEVFGK) across the study. B) Use of a control chart to visualize and quantify the systematic shift of the peptide abundance to outside the empirically defined threshold. C) Other peptides that were monitored show a similar trend. Black arrow marks the first QC that was run after the bath gas was replenished.
At QC standard #31, the operator noticed that the nitrogen C-trap bath gas on the Q-Exactive mass spectrometer was low – the gas was replenished and 7 more standards (QC# 32–38) were run to finish the experiment. The arrow in Figure 2B and Figure 2C notes the first standard that was run with the new Nitrogen supply. The signal for all peptides (Figure 2B and Figure 2C) was significantly greater QC# 32 because of the new nitrogen supply. The abundance of 6 of the 7 peptides came back within the empirically-defined thresholds (±3 sd). Interestingly, the signal from most of the peptides did not increase enough to return within 1 sd from the original mean. In fact, one peptide (YSTDVSVDEVK) did not increase enough to return within the 3 sd threshold. It is hypothesized that this failure of the signal for some peptides to return to the expected intensity is a result of other confounding factors (dirty emitter, etc) that affect peptide abundance aside from the identified principle cause of low bath gas. It is important to note that although the bath gas was low, peptide signal was still observed and data collected. This point emphasizes the importance of systematically evaluating the performance of the LC MS/MS throughout an experiment with a known concentration of standard. If performance had been based on peptide spectral matches before and after the experiment it is likely that this problem would not have been corrected as quickly.
Case Study 2 – Detecting Known Changes to the System
Figure 3 describes the effects of changing the trap cartridge on the CorConnex source [31] on the identification-free figures of merit. For simplicity only the peak areas and retention time control charts from 5 different targeted peptides are shown. Thresholds were determined from the QC #1–5 run on the first trap. The blue line represents the mean of the first 5 runs, the green line shows 1 sd, brown line 2 sd, and bold red line is 3 sd from the mean. For 4 of the 5 peptides monitored, there was negligible effect from changing the trap cartridge on peak areas. The CVs and subsequent runs (QC# 6–15) were within ± 3 sd of the mean. However, the peak areas for the peptide R.GASIVEDK.L on the third trap (QC# 11–15) were well above the 3 sd threshold. This peptide is extremely hydrophilic and the third trap was apparently more efficient at trapping the peptide. However, retention time control charts indicate a significant shift towards later elution in all peptides with the third trap. Finally the Pareto chart provides the number of non-conformers (± 3sd) and cumulative percentage as a function of the 4 different metrics. One can easily identify that the major cause of variation in the system is related to the retention time reproducibility as 79% of the non-conformers (>±3 sd) were peptides whose retention time shifted significantly from the first trap. Peak Areas, FWHM, and peak symmetry contributed 13%, 5%, and 3%, of the non-conformers, respectively.
Figure 3.
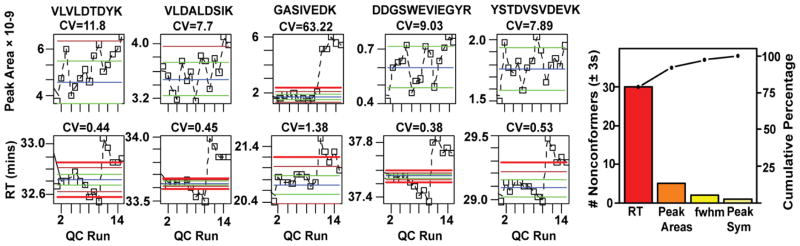
Control charts for peak areas (1st row) and retention time (2nd row) for 5 targeted peptides in an experiment where the traps (n=3) were changed every 5 QC runs. Thresholds were established on the first trap (n=5 QC standards). The majority of peak areas were within the thresholds (± 3 sd); however, the abundance of the GASIVDK peptide increased significantly using the final trap. The retention times shifted significantly for all peptides and is easily seen in the control charts and summarized in the Pareto Chart. The majority of non-conformers were related to retention time reproducibility (80%).
Conclusions
We have reported a vender neutral QC tool termed Statistical Process Control in Proteomics (SProCoP) that uses SPC techniques for monitoring the performance of an LC MS/MS experiment and allows evaluation of both targeted and discovery experiments. The tool uses freely available software and can be implemented in any lab that has access to a windows computer running Skyline and R. Future studies will focus on the addition of more identification-free metrics – for example, variations in total ion current could be used to monitor spray stability. Also we would like to implement a set of multi-rules, analogous to the Westgard rules [21,33,34] used in the clinical regime, to determine when an experiment has statistically failed. These endeavors will be met with key challenges. The huge variation between user analytical expertise, instrumentation, experimental set-ups, experimental goals, and preconceived notions on acceptable data quality will present problems. For example, more variation in metrics may be acceptable if one is performing a referenced design experiment (e.g., SILAC, PC-ID MS) than a label free spectral counting or peak intensity experiment. Regardless, as proteomics continues to find its way into the clinical regime, we believe it is important to have the capabilities to readily implement these charts and continue as a community to explore strategies for benchmarking data quality throughout an experiment, an instrument, intra-laboratory and inter-laboratory. While the emphasis of this tool has been on real time monitoring of an experiment, it can also be used retrospectively on data provided the user ran some type of peptide standard in a systematic fashion and performed MS1 profiling, targeted MS/MS, or SRM of targeted peptides. Currently, this implementation is experiment-specific but these analyses will be supported by our online repository Panorama [35] and used to track all of the QC standards from all experiments as a function of user and instrument. This combined approach will provide even further power to monitor the performance of an instrument over days, weeks, and years.
Acknowledgments
The authors acknowledge support from the National Science Foundation (grant #1109989) and National Institutes of Health grants P41 GM103533, R01 GM103551, and R01 GM107142. We also appreciate the helpful comments and discussion from Tom Corso and Colleen van Pelt from CorSolutions and Gary Valaskovic from New Objective.
References
- 1.Hu QZ, Noll RJ, Li HY, Makarov A, Hardman M, Cooks RG. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 2.Andrews GL, Simons BL, Young JB, Hawkridge AM, Muddiman DC. Performance Characteristics of a New Hybrid Quadrupole Time-of-Flight Tandem Mass Spectrometer (TripleTOF 5600) Anal Chem. 2011;83:5442–5446. doi: 10.1021/ac200812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol Cell Proteomics. 2011;10:M111 011015. doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bereman MS, Canterbury JD, Egertson JD, Horner J, Remes PM, Schwartz J, Zabrouskov V, MacCoss MJ. Evaluation of front-end higher energy collision-induced dissociation on a benchtop dual-pressure linear ion trap mass spectrometer for shotgun proteomics. Anal Chem. 2012;84:1533–1539. doi: 10.1021/ac203210a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and Sensitive Peptide Identification with Mascot Percolator. J Proteome Res. 2009;8:3176–3181. doi: 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 8.Spivak M, Weston J, Bottou Lo, Kall L, Noble WS. Improvements to the Percolator Algorithm for Peptide Identification from Shotgun Proteomics Data Sets. J Proteome Res. 2009;8:3737–3745. doi: 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thakur SS, Geiger T, Chatterjee B, Bandilla P, Frohlich F, Cox J, Mann M. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol Cell Proteomics. 2011;10:M110 003699. doi: 10.1074/mcp.M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunner E, Ahrens CH, Mohanty S, Baetschmann H, Loevenich S, Potthast F, Deutsch EW, Panse C, de Lichtenberg U, Rinner O, Lee H, Pedrioli PGA, Malmstrom J, Koehler K, Schrimpf S, Krijgsveld J, Kregenow F, Heck AJR, Hafen E, Schlapbach R, Aebersold R. A high-quality catalog of the Drosophila melanogaster proteome. Nature Biotechnology. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- 11.Mann M. Comparative analysis to guide quality improvements in proteomics. Nat Methods. 2009;6:717–719. doi: 10.1038/nmeth1009-717. [DOI] [PubMed] [Google Scholar]
- 12.Bell AW, Deutsch EW, Au CE, Kearney RE, Beavis R, Sechi S, Nilsson T, Bergeron JJM, Grp HTSW. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat Methods. 2009;6:423–U440. doi: 10.1038/nmeth.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabb DL, Vega-Montoto L, Rudnick PA, Variyath AM, Ham AJ, Bunk DM, Kilpatrick LE, Billheimer DD, Blackman RK, Cardasis HL, Carr SA, Clauser KR, Jaffe JD, Kowalski KA, Neubert TA, Regnier FE, Schilling B, Tegeler TJ, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Fisher SJ, Gibson BW, Kinsinger CR, Mesri M, Rodriguez H, Stein SE, Tempst P, Paulovich AG, Liebler DC, Spiegelman C. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J Proteome Res. 2010;9:761–776. doi: 10.1021/pr9006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoopmann MR, Merrihew GE, von Haller PD, MacCoss MJ. Post analysis data acquisition for the iterative MS/MS sampling of proteomics mixtures. J Proteome Res. 2009;8:1870–1875. doi: 10.1021/pr800828p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michalski A, Cox J, Mann M. More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC MS/MS. J Proteome Res. 2011;10:1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 16.Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, Neta P, Blonder N, Billheimer DD, Blackman RK, Bunk DM, Cardasis HL, Ham AJL, Jaffe JD, Kinsinger CR, Mesri M, Neubert TA, Schilling B, Tabb DL, Tegeler TJ, Vega-Montoto L, Variyath AM, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Carr SA, Fisher SJ, Gibson BW, Paulovich AG, Regnier FE, Rodriguez H, Spiegelman C, Tempst P, Liebler DC, Stein SE. Performance Metrics for Liquid Chromatography-Tandem Mass Spectrometry Systems in Proteomics Analyses. Mol Cell Proteomics. 2010;9:225–241. doi: 10.1074/mcp.M900223-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma ZQ, Polzin KO, Dasari S, Charnbers MC, Schilling B, Gibson BW, Tran BQ, Vega-Montoto L, Liebler DC, Tabb DL. QuaMeter: Multivendor Performance Metrics for LC-MS/MS Proteomics Instrumentation. Anal Chem. 2012;84:5845–5850. doi: 10.1021/ac300629p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kessner D, Chambers M, Burke R, Agusand D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pichler P, Mazanek M, Dusberger F, Weilnbock L, Huber CG, Stingl C, Luider TM, Straube WL, Kocher T, Mechtler K. SIMPATIQCO: A Server-Based Software Suite Which Facilitates Monitoring the Time Course of LC-MS Performance Metrics on Orbitrap Instruments. J Proteome Res. 2012;11:5540–5547. doi: 10.1021/pr300163u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abbatiello SE, Mani DR, Schilling B, MacLean B, Zimmerman LJ, Feng X, Cusack MP, Sedransk N, Hall SC, Addona T, Allen S, Dodder NG, Ghosh M, Held JM, Hedrick V, Inerowicz HD, Jackson A, Keshishian H, Kim JW, Lyssand JS, Riley CP, Rudnick P, Sadowski P, Shaddox K, Smith D, Tomazela D, Wahlander A, Waldemarson S, Whitwell CA, You J, Zhang S, Kinsinger CR, Mesri M, Rodriguez H, Borchers CH, Buck C, Fisher SJ, Gibson BW, Liebler D, MacCoss M, Neubert TA, Paulovich A, Regnier F, Skates SJ, Tempst P, Wang M, Carr SA. Design, Implementation and Multisite Evaluation of a System Suitability Protocol for the Quantitative Assessment of Instrument Performance in Liquid Chromatography-Multiple Reaction Monitoring-MS (LC-MRM-MS) Mol Cell Proteomics. 2013;12:2623–2639. doi: 10.1074/mcp.M112.027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westgard JO, Groth T. Design and Evaluation of Statistical Control Procedures - Applications of a Computer-Quality Control Simulator Program. Clinical Chemistry. 1981;27:1536–1545. [PubMed] [Google Scholar]
- 22.Westgard JO. Statistical Quality Control Procedures. Clinics in Laboratory Medicine. 2013;33:111–124. doi: 10.1016/j.cll.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 23.Bramwell D. An introduction to statistical process control in research proteomics. Journal of Proteomics. doi: 10.1016/j.jprot.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Shewhart WA. Application of statistical methods to manufacturing problems. J Frankl Inst. 1938;226:163–186. [Google Scholar]
- 25.Shewhart WA. Some applications of statistical methods to the analysis of physical and engineering data. Bell Syst Tech J. 1924;3:43–87. [Google Scholar]
- 26.Levey S, Jennings ER. The Use of Control Charts in the Clinical Laboratory. Am J Clin Pathol. 1950;20:1059–1066. doi: 10.1093/ajcp/20.11_ts.1059. [DOI] [PubMed] [Google Scholar]
- 27.Bunkley N. New York Times. 2008. [Google Scholar]
- 28.Broudy D, Killeen K, Choi M, Shulman N, Mani DR, Abbatiello SE, Mani D, Ahmad R, Sahu AK, Schilling B, Gibson BW, Carr SA, Vitek O, MacCoss MJ, MacLean B. A Framework for Installable External Tools in Skyline. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btu148. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, Sorensen DJ, Bereman MS, Jing E, Wu CC, Verdin E, Kahn CR, Maccoss MJ, Gibson BW. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol Cell Proteomics. 2012;11:202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherrod SD, Myers MV, Li M, Myers JS, Carpenter KL, MacLean B, MacCoss MJ, Liebler DC, Ham AJL. Label-Free Quantitation of Protein Modifications by Pseudo Selected Reaction Monitoring with Internal Reference Peptides. J Proteome Res. 2012;11:3467–3479. doi: 10.1021/pr201240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bereman MS, Hsieh EJ, Corso TN, Van Pelt CK, Maccoss MJ. Development and characterization of a novel plug and play liquid chromatography-mass spectrometry (LC-MS) source that automates connections between the capillary trap, column, and emitter. Mol Cell Proteomics. 2013;12:1701–1708. doi: 10.1074/mcp.O112.024893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocher T, Pichler P, Swart R, Mechtler K. Quality control in LC-MS/MS. Proteomics. 2011;11:1026–1030. doi: 10.1002/pmic.201000578. [DOI] [PubMed] [Google Scholar]
- 33.Eggert AA, Westgard JO, Barry PL, Emmerich KA. Implementation of a multirule, multistage quality control program in a clinical laboratory computer system. Journal of medical systems. 1987;11:391–411. doi: 10.1007/BF00993007. [DOI] [PubMed] [Google Scholar]
- 34.Westgard JO, Barry PL, Hunt MR, Groth T. A Multi-Rule Shewhart Chart for Quality-Control in Clinical-Chemistry. Clinical Chemistry. 1981;27:493–501. [PubMed] [Google Scholar]
- 35. [Acessed December 22, 2013]; http://panoramaweb.org.