

Abstract
Gastric inhibitory polypeptide (GIP) and glucagon‐like peptide‐1 (GLP‐1) are the two primary incretin hormones secreted from the intestine on ingestion of glucose or nutrients to stimulate insulin secretion from pancreatic β cells. GIP and GLP‐1 exert their effects by binding to their specific receptors, the GIP receptor (GIPR) and the GLP‐1 receptor (GLP‐1R), which belong to the G‐protein coupled receptor family. Receptor binding activates and increases the level of intracellular cyclic adenosine monophosphate in pancreatic β cells, thereby stimulating insulin secretion glucose‐dependently. In addition to their insulinotropic effects, GIP and GLP‐1 play critical roles in various biological processes in different tissues and organs that express GIPR and GLP‐1R, including the pancreas, fat, bone and the brain. Within the pancreas, GIP and GLP‐1 together promote β cell proliferation and inhibit apoptosis, thereby expanding pancreatic β cell mass, while GIP enhances postprandial glucagon response and GLP‐1 suppresses it. In adipose tissues, GIP but not GLP‐1 facilitates fat deposition. In bone, GIP promotes bone formation while GLP‐1 inhibits bone absorption. In the brain, both GIP and GLP‐1 are thought to be involved in memory formation as well as the control of appetite. In addition to these differences, secretion of GIP and GLP‐1 and their insulinotropic effects on β cells have been shown to differ in patients with type 2 diabetes compared to healthy subjects. We summarize here the similarities and differences of these two incretin hormones in secretion and metabolism, their insulinotropic action on pancreatic β cells, and their non‐insulinotropic effects, and discuss their potential in treatment of type 2 diabetes. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2010.00022.x, 2010)
Keywords: Incretin, GIP, GLP‐1
Introduction
Over 100 years have passed since the discovery of the incretin concept, which later opened up the possibility of a novel therapy in the treatment of diabetes. Inspired by Bayliss and Starling’s discovery of secretin in 19021, Moore et al. hypothesized that gut extracts contain a hormone that regulates the endocrine pancreas, and showed that administration of gut extracts reduces the amount of urine sugars in patients with diabetes, presumably through stimulation of the endocrine pancreas2. In 1929, La Barre purified the glucose‐lowering element from gut extracts, and named it incretin (INtestine seCRETtion Insulin)3. However, incretin was forgotten for three decades until radioimmunoassay to measure insulin became available in the 1960s. Oral glucose load was shown to produce a much greater insulin response than i.v. injection of glucose4,5, which now can be attributed to incretins released from the gut after ingestion of glucose or nutrients to stimulate insulin secretion from pancreatic β cells.
Merely two such gut hormones, gastric inhibitory polypeptide (GIP) and glucagon‐like peptide‐1 (GLP‐1), have been shown to act as incretins (Figure 1). GIP is a 42‐amino‐acid hormone secreted from K cells of the upper small intestine6,7. It was originally isolated from porcine intestine on the basis of its ability to inhibit gastric acid secretion8. Later, it was found that GIP administration stimulates insulin secretion in healthy volunteers9, and that GIP acts directly on pancreatic islets to stimulate insulin secretion10,11. We have also shown that endogenous GIP stimulates insulin secretion glucose‐dependently in gastrectomized patients12. These lines of evidence showed GIP to be the first incretin, which was then renamed glucose‐dependent insulinotropic polypeptide. Because immunological depletion of GIP did not abolish all insulin‐stimulating activity in gut extracts13, the existence of a second incretin was inferred. Meanwhile, a series of investigations on enteroglucagons showed that GLP‐1, a 31‐amino‐acid hormone produced from proglucagon and secreted from L cells of the lower intestine and colon14, directly acts on islets and stimulates insulin secretion in isolated islets15 as well as in healthy volunteers16. GLP‐1 was thus found to be the second incretin.
Figure 1.

The glucose‐dependent insulinotropic polypepide (GIP) gene is localized on human chromosome 17q21.3–q22 and comprises 6 exons. Proteolytic processing of preproGIP generates GIP that is secreted from K cells. The proglucagon gene is localized on human chromosome 2q36–q37 and comprises 6 exons. In the intestine, proteolytic processing of proglucagon generates glucagon‐like peptide (GLP)‐1 and GLP‐2, whereas glucagon is produced in the pancreas.
Both GIP and GLP‐1 exert their effects by binding to their specific receptors, the GIP receptor (GIPR)17–21 and the GLP‐1 receptor (GLP‐1R)22–24, which belong to the G‐protein coupled receptor family, activating adenylate cyclase and increasing levels of intracellular cyclic adenosine monophosphate (cAMP) in pancreatic β cells, thereby stimulating insulin section glucose‐dependently. Genetic ablation of GIPR and GLP‐1R separately or simultaneously in mice showed their critical roles in the entero‐insular axis and confirmed that both GIP and GLP‐1 act as incretins25–29. Furthermore, deficiency of dipeptidyl peptidase‐4 (DDP‐4), which cleaves the two NH2‐terminal amino acids of GIP and GLP‐1 in plasma and inactivates their insulinotropic activities30,31, enhances insulin secretion in response to oral glucose challenge consistently with their function as incretins32. GIP and GLP‐1 thus share common properties as incretins, but they also possess different biological characteristics (Figure 2). Here, we summarize similarities and differences in the processes of the secretion and metabolism of GIP and GLP‐1, their insulinotropic actions on pancreatic β cells, and their non‐insulinotropic effects.
Figure 2.

Pancreatic and exopancreatic function of glucose‐dependent insulinotropic polypepide (GIP) and glucagon‐like peptide (GLP)‐1. GIP acts directly on the endocrine pancreas, bone, fat, gastrointestinal (GI) tract and brain. GLP‐1 acts directly on the endocrine pancreas, gastrointestinal tract, heart and brain.
Secretion and Metabolism of GIP and GLP‐1
Because GIP and GLP‐1 rapidly undergo proteolytic degradation catalyzed by DPP‐430,31, not only intact but also total (i.e. intact plus DPP‐4‐metabolized) forms of GIP and GLP‐1 must be measured to study their secretion and processing in vivo (Figure 3). However, immunoassays for GIP and GLP‐1 levels, especially those used to measure their intact forms in plasma, require specific antibodies and are not widely available33. Furthermore, because carboxyl‐terminal arginine of GLP‐1 is susceptible to amidation, GLP‐1 occurs in both non‐amidated GLP‐1(7–37) and amidated GLP‐1(7–36)amide, both of which show similar insulinotropic effects and metabolism in humans34. Although most of the GLP‐1 secreted from the gut is amidated in humans35, careful considerations are required when measuring the levels of GLP‐1 because some antibodies only recognize amidated GLP‐1.
Figure 3.
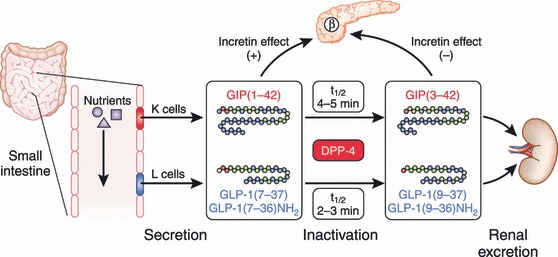
Secretion and metabolism of glucose‐dependent insulinotropic polypepide (GIP) and glucagon‐like peptide (GLP)‐1. GIP is secreted from K cells of the upper intestine; GLP‐1 is secreted from L cells of the lower intestine. Released GIP and GLP‐1 rapidly undergoes proteolytic processing by dipeptidyl peptidase‐4 (DPP‐4), and is thereby inactivated and excreted from the kidney. The intact incretins, GIP(1–42), GLP‐1(7–37), and GLP‐1(7–36)amide, have insulinotropic effects on pancreatic β cells, whereas the DPP‐4‐processed incretins, GIP(3–42), GLP‐1(9–37), and GLP‐1(9–36)amide, have lost their insulinotropic effects.
GIP secretion from K cells is enhanced in response to ingestion of meals or glucose36. A series of studies using the antibody R65, which recognizes both intact GIP(1–42) and DPP‐4‐processed GIP(3–42), shows that plasma levels of total GIP at fasting are 5–20 pM in healthy Caucasians36, indicating basal secretion in healthy Caucasians. These levels of total GIP reach 50–100 pM within 30 min in response to ingestion of 75‐gram glucose in healthy Caucasians, whereas those of total GIP reach 100–150 pM within 60 min in response to ingestion of mixed meals36,37. Although there is no direct comparison of glucose‐enhanced GIP secretion with those enhanced by proteins or fats, ingestion of proteins produces more rapid and robust GIP secretion than that of fats38. Similarly, levels of intact GIP, determined by an immunoassay using antiserum 98171, which detects GIP(1–42) but not GIP(3–42)39, increase more rapidly and robustly in response to ingestion of proteins when compared with isocaloric fat ingestion38. These results suggest that the GIP response is dependent not only on meal size but also on meal composition. Recent investigations of GIP secretion in healthy Japanese subjects using the same immunoassays showed that although peak values of total GIP levels in response to ingestion of glucose or mixed meals are higher than those of Caucasians, the peak values of intact GIP levels are similar37,40–42, suggesting enhanced processing of GIP by DPP‐4 in Japanese subjects (Figure 4). This possible racial difference in the GIP response and DPP‐4 activities needs to be studied more intensively in the future.
Figure 4.
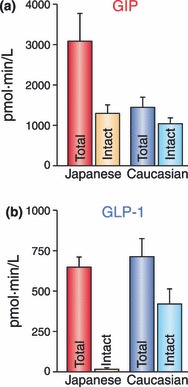
Racial differences in secretion and metabolism of (a) glucose‐dependent insulinotropic polypepide (GIP) and (b) glucagon‐like peptide (GLP)‐1. GIP secretion in healthy Japanese subjects after ingestion of glucose or mixed meals were higher than those of healthy Caucasian subjects, whereas levels of intact GIP were similar37,40–42. This suggests processing of GIP by dipeptidyl peptidase‐4 (DPP‐4) in Japanese subjects might be enhanced. Considerably low levels of intact GLP‐1 after ingestion of glucose or mixed meals are also consistent with enhanced DPP‐4 activities in Japanese subjects. To compare early phase incretin secretion and their processing by DPP‐4 between non‐obese (BMI <25) healthy Japanese and Caucasian subjects, area under the curves during 0–30 min after ingestion of mixed meals containing similar calories are plotted for total and intact forms of (a) GIP and (b) GLP‐1 from Yabe et al.42 Note that total and intact GIP and total GLP‐1 levels were measured in the same immunoassays, whereas assays for intact GLP‐1 were similar except for the presence or absence of the ethanol extraction.
GLP‐1 secretion from L cells, like that of GIP from K cells, is enhanced in response to ingestion of meals or glucose36. Studies using antiserum 89390 specific for GLP‐1(7–36)amide as well as DPP‐4‐processed GLP‐1(9–36)amide show that plasma levels of total GLP‐1 at fasting are 10–20 pM, indicating basal secretion in healthy Caucasians36. Levels of total GLP‐1 reach 30–60 pM within 30 min in response to ingestion of 75‐gram glucose or mixed meals in healthy Caucasians36,37. Despite the lack of direct comparison of glucose‐enhanced GLP‐1 secretion with those enhanced by proteins or fats, ingestion of proteins or isocaloric fats produces a similar GLP‐1 secretion38. The levels of intact GLP‐1 are controversial; recent studies have shown the importance of an ethanol or solid phase extraction before immunoassays for intact GLP‐1 that removes interference with substances of unknown identity and thereby reduces the large variability among individual human subjects33. Evaluation of GLP‐1 secretion in healthy Japanese subjects using the same immunoassay detecting total GLP‐1 with antiserum 89390 showed that the meal‐induced enhancement of GLP‐1 secretion is negligible, whereas GLP‐1 secretion in response to oral glucose is similar to that in healthy Caucasians36,37,40–42. The levels of intact GLP‐1 in ethanol extracted plasmas, determined by an immunoassay with the monoclonal antibodies GLP1F5 and Mab26.1, were considerably low in Japanese subjects, and showed no enhancement in response to glucose or mixed meal ingestion42. Although further studies to directly compare the GLP‐1 response of Asian and Caucasian subjects are required, the blunted meal‐induced enhancement of the GLP‐1 response and the considerably low levels of intact GLP‐1 could well account for the reduced insulin secretory capacity of Asians, including Japanese.
Secreted incretins undergo rapid degradation catalyzed by DPP‐4, which diminishes the insulinotropic effects of GIP and GLP‐143. The apparent half‐lives for intact GIP and GLP‐1 have been determined as approximately 5 and 2 min, respectively40,43,44. Studies of GLP‐1 secreted from perfused pig ileum show that approximately 75% of GLP‐1 leaving the gut is already metabolized by DPP‐445. Further degradation of GLP‐1 also occurs in the liver, which finally results in only 10–15% of newly secreted GLP‐1 in systemic circulation46. Furthermore, study of intact GLP‐1 levels in Japanese subjects shows that less than 5% of intact GLP‐1 reaches systemic circulation (Figure 4), suggesting that GLP‐1 acts on putative GLP‐1R in the portal vein and then stimulates insulin secretion through activation of the vagus nerve. Contribution of portal GLP‐1R activation in insulinotropic function of GLP‐1 could be evaluated in the future using mice lacking specifically GLP‐1R in the portal vein.
In both Caucasians and Japanese with type 2 diabetes (T2DM), the GIP response is enhanced compared with that in healthy volunteers47,48, whereas the GLP‐1 response in Caucasians with T2DM is reduced compared with that in healthy controls37. The physiological cause for the enhanced GIP response in patients with T2DM is not yet clear. Furthermore, the incretin response in T2DM patients is still controversial, because Caucasian and Japanese patients with a relatively short duration of diabetes show GIP and GLP‐1 response similar to those of healthy controls36,42. Although it is not discussed in the present review, recent advances in understanding the molecular mechanisms underlying GIP and GLP‐1 secretion from K and L cells49 should clarify the differing results on GIP and GLP‐1 responses in T2DM patients in the various studies.
Insulinotropic Actions of GIP and GLP‐1
Enhancement of glucose‐dependent insulin secretion by incretins in healthy volunteers was directly shown by i.v. infusion of GIP9 and GLP‐116; their insulinotropic effects are similar50 and additive51 in healthy humans. Critically, the effects of infused GIP at pharmacological levels are impaired in Caucasians with T2DM, although those of GLP‐1 are substantially preserved52,53. The insulinotropic effects of endogenous GIP are also diminished in Japanese T2DM patients48. Furthermore, the impaired insulinotropic action of GIP, but not that of GLP‐1, are strikingly similar in perfused islets of diabetic Goto‐Kakizaki rats54. However, the molecular mechanisms underlying the impaired insulinotropic action of GIP in T2DM patients are still largely unknown. Various studies in model animals suggest downregulation of GIPR mRNA55–58, accelerated degradation of GIPR59, and alternative splicing of GIPR mRNA60, all of which might contribute to the reduced GIP sensitivity of pancreatic β cells. Polymorphisms in the human GIPR gene have been shown to decrease GIP sensitivity in β cells, but no correlation has been observed between these polymorphisms and T2DM61. However, genetic variation in GIPR has been recently reported to be associated with the reduction in early phase insulin reaction and elevation in 2‐h glucose levels after ingestion of 75‐gram glucose, suggesting that defective GIPR signaling might play a critical role in the early pathophysiology of impaired glucose tolerance and T2DM62. It remains to be determined why GIP but not GLP‐1 signaling is selectively impaired in hyperglycemic conditions in humans.
Both GIP and GLP‐1 exert their insulinotropic effects by binding to GIP and GLP‐1 receptors expressed on pancreatic β cells. Incretin‐bound receptors increase intracellular cAMP levels63,64, thereby activating protein kinase A (PKA)65 and exchange protein activated by cAMP2 (EPAC2)/cAMP‐guanine nucleotide exchange factor (GEF) II66. PKA and EPAC2 are involved in a wide variety of intracellular events including altered ion channel activity, elevated cytosolic calcium levels and enhanced exocytosis of insulin‐containing granules, all of which contribute to stimulation of insulin section in a glucose‐dependent manner (Figure 5). Many studies have been carried out using inhibitors and activators that affect signal transduction or GLP‐1 receptor agonist exentin‐4, assuming that the molecular mechanisms downstream of both GIPR and GLP‐1R are similar. Activation of PKA results in phosphorylation of the SUR1 subunit, thereby closing the KATP channels and facilitating membrane depolarization67. PKA, together with phosphoinositide 3‐kinase (PI‐3K), also leads to inhibition of the delayed rectifying K+ (Kv) channel, which results in prolongation of action potentials68. Depolarization opens the voltage‐gated Ca2+ channels (VDCC), allowing an increase of intracellular Ca2+ concentrations, which then mobilizes Ca2+ from intracellular stores through PKA‐ and EPAC2‐dependent mechanisms69–73. The increased Ca2+ concentrations eventually trigger fusion of insulin‐containing granules with the plasma membrane and increase insulin secretion from the β cells. Mobilization of Ca2+ from intracellular stores stimulates adenosine triphosphate (ATP) synthesis in mitochondria, which further enhances membrane depolarization by KATP channel closure74. Furthermore, activation of EPAC2 has recently been shown to increase the density of insulin‐containing granules near the plasma membrane, facilitating insulin secretion from the β cells75. Taken together, these lines of evidence show the critical functions of PKA and EPAC2 pathways in the insulinotropic actions of GIP and GLP‐1.
Figure 5.
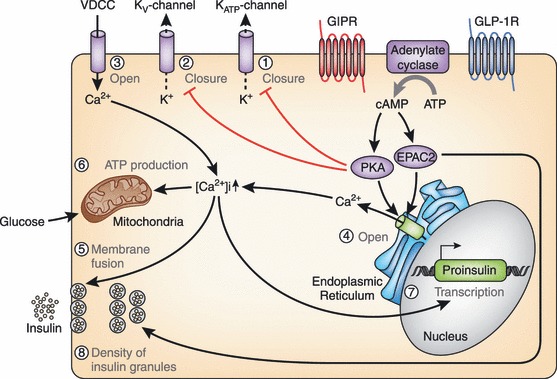
Molecular mechanisms underlying the insulinotropic effects of glucose‐dependent insulinotropic polypepide (GIP) and glucagon‐like peptide (GLP)‐1. Binding of GIP and GLP‐1 to their specific receptors, the GIP receptor (GIPR) and the GLP‐1 receptor (GLP‐1R) leads to activation of adenylate cyclase and subsequent elevation of intracellular cyclic adenosine monophosphate (cAMP) levels. Increased cAMP then activates protein kinase A (PKA) and exchange protein activated by cAMP2 (EPAC2)/cAMP‐guanine nucleotide exchange factor (GEF)II. Activation of PKA promotes closure of KATP channels and facilitates membrane depolarization. PKA also leads to inhibition of the delayed rectifying K+ (Kv) channel, a negative regulator of insulin secretion in pancreatic β cells, resulting in prolongation of action potentials. Depolarization opens the voltage‐gated Ca2+ channels (VDCC), allowing an increase of intracellular Ca2+ concentrations that mobilizes Ca2+ from intracellular stores through PKA‐ and EPAC2‐dependent mechanisms. The increased Ca2+ concentrations eventually trigger fusion of insulin‐containing granules with the plasma membrane and insulin secretion from the β cells. Increased Ca2+ levels also promote transcription of the proinsulin gene, thereby increasing the insulin content of the β cell. Activation of EPAC2 has been shown to increase the density of insulin‐containing granules near the plasma membrane to potentiate insulin secretion from the β cell. ATP, adenosine triphosphate.
To date, little is known regarding the differences in signaling events downstream of GIPR and GLP‐1R. Importantly, GLP‐1 but not GIP stimulates glucose‐dependent insulin secretion in perfused pancreatic islets from KATP channel‐deficient (Kir6.2−/−) mice76. Although the involvement of niflumic acid‐sensitive ion channels has been shown in this process77, further investigation is required to understand the potential diversity in the downstream mechanisms of GIPR and GLP‐1R. Understanding the molecular mechanism underlying the remaining GLP‐1 sensitivity in KATP channel‐deficient mice should shed light on the selective inactivation of GIPR signaling in hyperglycemic conditions.
Another important aspect of the insulinotropic effects of GIP and GLP‐1 is their synergy with the sulfornylurea drugs. Sulfornylureas efficiently cause mobilization of Ca2+ by closure of the KATP channels, membrane depolarization and subsequent VDCC opening, even in T2DM patients with impaired mitochondrial ATP production. As discussed earlier, GIP and GLP‐1 affects events downstream of the KATP channel closure, thereby enhancing the ability of sulfornylureas to promote insulin secretion (Figure 5). In addition, it has recently been shown that certain sulfornylureas bind to EPAC2 and activate downstream molecules78, thereby enhancing the insulinotropic effects of GIP and GLP‐1. Taken together, it is clinically important to note that sulfornylureas and incretin‐related therapies act synergistically to stimulate insulin secretion. Thus, caution is required for the combination of sulfornylureas and incretin‐related drugs that potentially cause hypoglycemia, especially among Asians in whom diabetes is characterized by reduced insulin secretory capacity and sulfornylureas would be the first choice of drugs.
Non‐insulinotropic Function of GIP and GLP‐1 on Pancreatic β Cells
Incretin was originally identified as the hormone that transmits signals from the gut to the pancreatic β cells, and the principal role of GIP and GLP‐1 has generally been thought to stimulate insulin secretion. However, it has been shown that GIP and GLP‐1 exert non‐insulinotropic actions, such as controlling pancreatic β cell proliferation and survival.
For example, GIP has been shown to have an anti‐apoptotic function in pancreatic β cells. This effect involves activation of the cAMP response element‐binding (CREB) and Akt/PKB pathways (Figure 6). In INS‐1 cells, binding of GIP to GIPR leads to the elevation of intracellular cAMP levels and activation of PKA, which then migrates into the nucleus and directly phosphorylates nuclear CREB79,80. In addition, activated PKA inhibits AMPK, which then results in de‐phosphorylation and nuclear import of the transducer of regulated CREB activity 2 (TORC2)79. In the nucleus, phosphorylated CREB and TORC2 form a complex in the promoter of the anti‐apoptotic gene bcl2, thereby promoting its gene transcription79. Binding of GIP to GIPR also results in the activation of Akt/PKB, promoting phosphorylation of the nuclear transcription factor Foxo1 in INS‐1 cells (Figure 6)81. Phosphorylated Foxo1 is exported from the nucleus, leading to downregulation of the pro‐apoptotic gene bax, one of the Foxo1 direct target genes, which promotes β cell apoptosis in response to glucolipotoxicity81. Downregulation of Bax and upregulation of Bcl‐2 is observed not only in INS‐1 cells, but also in islets of the Vancouver diabetic fatty (VDF) Zucker rats receiving 2‐week continuous infusion of GIP81. While the mechanisms underlying activation of Akt/PKB by GIP are yet unclear, it has been shown recently that activation of EPAC2, but not PI3‐K, Erk1/2 or PKA, is responsible for Akt/PKB activation and the anti‐apoptotic function of GIP82. Furthermore, activation of Akt/PKB by GIP has been shown to suppress mitochondrial translocation of Bad and BimEL and the subsequent activation of caspase‐3 by inhibiting p38 MAPK and JNK in INS‐1 cells exposed to staurosporin, a rapid activator of the mitochondria‐mediated apoptotic pathway83,84. Suppression of p38 MAPK and JNK has also been shown to be critical for the anti‐apoptotic actions of GIP in INS‐1 cells exposed to endoplasmic reticulum (ER) stress and genotoxic stress84.
Figure 6.
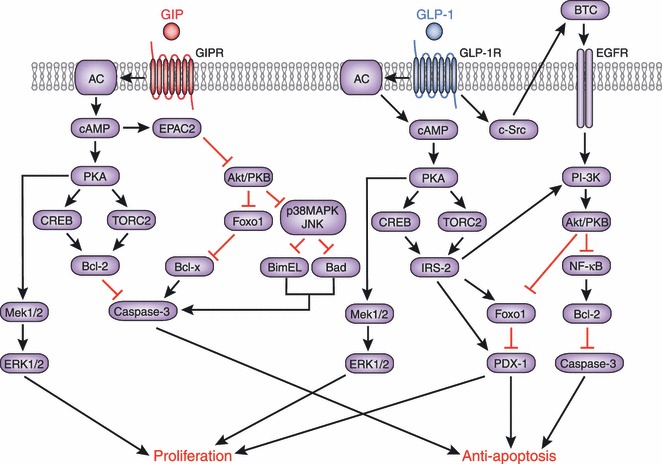
Molecular mechanisms underlying the anti‐apoptotic and proliferative effects of glucose‐dependent insulinotropic polypepide (GIP) and glucagon‐like peptide (GLP)‐1. Signaling cascades linking the GIP receptor (GIPR) and the GLP‐1 receptor (GLP‐1R) with anti‐apoptotic and proliferative effects share similarities and differences as shown. Involvement of epidermal growth factor (EGFR) and phosphoinositide 3‐kinase (PI‐3K) has been shown to be a critical difference between the GIPR‐ and GLP‐1R‐signaling pathways. AC, adenylate cyclase; Akt, v‐akt murine thymoma viral oncogene homolog; Bad, Bcl‐2 antagonist of cell death; Bcl, B‐cell CLL/lymphoma; BimEL, Bcl‐2 interacting mediator of cell death EL; BTC, betacellulin; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element‐binding; c‐Src, proto‐oncogene tyrosine‐protein kinase Src; EPAC2, exchange protein directly activated by cAMP2; ERK, extracellular signal‐regulated kinase; Foxo1, forkhead box protein O1; IRS‐2, insulin receptor substrate 2; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinase; Mek, mitogen‐activated protein kinase kinase; NFκB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; PDX‐1, pancreas/duodenum homeobox protein 1; PKA, protein kinase A; PKB, protein kinase B; TORC2, transducer of regulated CREB activity 2.
The anti‐apoptotic function of GLP‐1 on pancreatic β cells was suggested by studies of the pancreas of exendin‐4‐treated db/db mice or GLP‐1‐infused diabetic VDF Zucker rats, which showed an increase in β cell mass and a decrease in apoptotic β cells85,86. Subsequently, it was shown that activation of GLP‐1R by exendin‐4 inhibits apoptosis of MIN6 cells exposed to hydrogen peroxide in a cAMP‐ and PI3K‐dependent manner, in association with upregulation of Bcl‐2 and Bcl‐xL, and reduced poly‐(ADP‐ribose)‐polymerase87. Activation of PKA and EPAC2 by elevated cAMP levels was also shown to inhibit activation of caspase‐3 and subsequent apoptosis of palmitate‐treated RINm5F cells88 (Figure 6). Activation of Akt/PKB, a downstream effecter of PI3K, by GLP‐1 was also shown to prevent apoptosis of INS‐1 cells in response to glucolipotoxicity or staurosporin89,90, possibly through the activation of nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NFκB) and the upregulation of the anti‐apoptotic NFκB target genes bcl2 and IAP289. GLP‐1R activation also reduces the ER stress response, thereby promoting β cell survival in INS‐1 cells as well as in human islets91–93. A critical difference in the anti‐apoptotic function of GLP‐1 is the requirement for PI3K, which is not required for the anti‐apoptotic action of GIP. While activation of Akt/PKB by GIP is mediated by EPAC282, activation of Akt/PKB by GLP‐1 requires CREB‐mediated induction of IRS‐2, which might also potentiate PI3K and Akt/PKB94 (Figure 6). The physiological impact of differences in PI3K requirement and other yet to be identified differences between GIP and GLP‐1 have recently been investigated in mice treated with GLP‐1R agonist, GIPR agonist or DPP‐4 inhibitor95. This study showed that GLP‐1R agonist exerts the most robust effect on the survival of β cells compared with that of GIPR agonist or DPP‐4 inhibitor in vivo in mice95. Further investigation of the mechanisms underlying the effects of GIP and GLP‐1 could show potential therapeutic targets to increase β cell mass by inhibiting apoptosis.
Another important aspect of GIP and GLP‐1 action on β cells is the stimulation of the proliferation of β cells and/or progenitor cells. An early investigation evaluating the effects of exendin‐4 in rats showed that activation of GLP‐1R promotes proliferation and neogenesis of pancreatic β cells96. Later, proliferative effects were also shown of GIPR activation in INS‐1 cells97,98. It has been shown that GIP activates the Raf‐Mek1/2‐ERK1/2 signaling module through cAMP/PKA signaling in GIPR‐overexpressing Chinese hamster ovary cells98–100 (Figure 6). Consistent with these observations, PKA and MEK inhibitors have been shown to prevent GIP‐induced proliferation of cultured islet cells101. The same study further showed that GIP, as well as GLP‐1, induces transcription of cyclin D1 that is critical for G1 phase progression and S‐phase entry in most cell types101. Interestingly, PI3K inhibitor also prevents GIP and GLP‐1‐dependent proliferation of cultured islet cells and INS‐1 cells, suggesting involvement of the PI3K pathway in the proliferation of pancreatic β cells induced not only by GLP‐1, but also by GIP. Delineation of the proliferative action of GLP‐1 further showed that it involves activation of PI3K and upregulation of PDX‐1 transcription through transactivation of epidermal growth factor receptor (EGFR)102,103. Although these findings could explain the involvement of EGFR transactivation and subsequent PI3K activation in the proliferative action of GLP‐1, whether or not the same mechanism might be involved in the proliferative action of GIP must be investigated. Importantly, the proliferative and anti‐apoptotic effects of GIP and GLP‐1 on pancreatic β cells could be achieved by pharmacological levels of incretins rather than physiological levels, because no reduction of β cell mass has been reported in animals lacking both GIPR and GLP‐1R25,27,29. Nevertheless, the proliferative effects of GIP and GLP‐1 on pancreatic β cells are clinically relevant for treatment of diabetes, and remain to be tested in patients with diabetes.
Effects on Glucagon Secretion from Pancreatic α Cells
The effects of GLP‐1 and GIP on glucagon secretion from pancreatic α cells are opposing. As early as the 1970s, infusion of GIP was shown to counteract suppression of glucagon secretion by glucose in rats or isolated rat islets11,104. Later, this was confirmed in healthy humans during euglycemic, but not during hyperglycemic, clamp studies105, as well as in T2DM patients during meal‐tolerance tests106. Furthermore, it has been shown that GIPR is expressed in human and mouse pancreatic α cells and that GIP stimulates glucagon secretion from cultured α cell‐line αTC‐1 cells with a concomitant increase in intracellular cAMP levels106. Although its physiological importance remains unknown, enhancement of glucagon secretion by GIP hinders clinical usage of GIP as a treatment for diabetes.
In contrast, GLP‐1 has been shown to suppress glucagon secretion when plasma glucose levels are above fasting level107. This is clinically important because GLP‐1 loses its inhibitory effect on glucagon secretion at hypoglycemic levels and does not attenuate the counter‐regulatory responses to hypoglycemia. Furthermore, it has recently reported that insulin stimulation and glucagon inhibition contribute equally to the effect of GLP‐1 on glucose turnover in T2DM patients108. Despite of its clinical importance, the mechanism underlying the suppression of glucagon secretion by GLP‐1 remains unclear. While GLP‐1R mRNA is detected in approximately 20% of cultured α cell‐lines109, expression of the GLP‐1 receptor in pancreatic α cells in vivo is controversial, and there is no evidence that GLP‐1 directly acts on α cells. How then does GLP‐1 inhibit glucagon secretion? Glucagon secretion is strongly inhibited by GLP‐1 in type 1 diabetes patients with no remaining insulin secretory capacity110, suggesting that insulin, which is generally thought to suppress glucagon secretion, is not required in this process. Importantly, it has been shown that GLP‐1 stimulates pancreatic somatostatin secretion111 and that the inhibitory effect of GLP‐1 on glucagon secretion is abolished by somatostatin antibodies and a somatostatin receptor 2 antagonist in isolated rat pancreas112. Although expression of the GLP‐1R in δ cells also remains controversial, these studies suggest that the suppression of glucagon secretion by GLP‐1 is mediated by somatostatin.
Physiological Function of GIP in Fat Accumulation
Fats strongly enhance GIP secretion38,113, and GIP levels are high in obese T2DM patients114. GIP has been proposed to have a physiological role on nutrient uptake into adipose tissues, thereby linking overnutrition to obesity. An initial clue came in the early 1980s from an experiment showing that GIP, in the presence of insulin, induces fatty acid incorporation into rat epididymal fat pads115. Later, GIPR was shown to be expressed in adipose tissues116, and genetic ablation of GIPR further shows the critical role of GIP in fat accumulation26.
High‐fat diets are one of the well‐known environmental determinants of obesity. Although control mice on a high‐fat diet for 50 weeks show weight gain and a marked increase in visceral and subcutaneous fat mass and liver steatosis, such weight gain and adiposity was not observed in GIPR‐deficient mice on the same high‐fat diets26. GIPR‐deficient mice on high‐fat diets showed energy intake similar to that of control mice, but showed higher energy expenditure as revealed by a reduction of oxygen consumption and respiratory quotient during the light phase, the latter indicating that fat is utilized as the preferred energy substrate in GIPR‐deficient mice. Furthermore, GIPR‐deficient mice show increased adiponectin secretion, which promotes fat oxidation in muscle and increases the respiratory quotient117,118. In addition, genetic ablation of GIPR in obese ob/ob mice, in which a defect in the leptin gene results in hyperphagia and subsequent obesity119, ameliorates not only obesity by increasing energy expenditure26,53, but also insulin insensitivity and glucose tolerance without seriously affecting insulin secretion54. These observations were confirmed in high‐fat fed mice and obese ob/ob mice treated with a GIPR antagonist, (Pro3)GIP120–122 and in mice lacking GIP‐secreting K cells123, establishing the critical role of GIP in fat accumulation.
Although GIP was shown to increase the activity of lipoprotein lipase (LPL), an enzyme that is bound to the cell surface of adipocytes and hydrolyzes lipoprotein‐associated triglycerides to produce free fatty acids available for local uptake26, the molecular mechanism by which GIP acts on adipocytes is largely unknown. It was recently shown that binding of GIP to GIPR in 3T3‐L1 cells and rat epididymal fat results in enhanced secretion of resistin through a pathway involving p38 MAPK and the stress‐activated protein kinase/Jun amino‐terminal kinase (SAPK/JNK)124. GIP activates PI3K and Akt/PKB through secreted resistin, thereby suppressing AMPK and increasing LPL activity in adipocytes124,125. Interestingly, another GIPR agonist, D‐Ala2GIP(1–30), shows a potency equivalent to GIP(1–42) on β cell function and survival, but greatly reduced action on lipoprotein lipase activity in 3T3‐L1 cells126. Further investigation on the differential effects of D‐Ala2GIP(1–30) in β cell and adipocytes might shed light on the molecular mechanisms underlying GIP action in fat accumulation as well as might open up a possibility of GIP‐based anti‐diabetic therapy that does not promote obesity. Importantly, GLP‐1 does not show any role in fat accumulation. While GLP‐1R is expressed in adipocytes127, activation of GLP‐1R affects none of the aforementioned signaling molecules and does not increase LPL activity in adipocytes124,125.
Endogenous GIP and GLP‐1 in Bone Metabolism
Regulation of bone metabolism is another important physiological function of GIP and GLP‐1. A role of GIP in bone metabolism was first suggested by the presence of GIPR in bone and the suppression of ovariectomy‐induced bone loss by GIP administration128. Later, the role of endogenous GIP in bone formation became evident in GIPR‐deficient mice, which show thinner bone trabeculae reminiscent of osteoporosis129. Bone histomorphometrical analyses showed that bone formation parameters are significantly lower and the number of osteoclasts is significantly increased in GIPR‐deficient mice, indicating high‐turnover osteoporosis129. In addition, GIP suppresses apoptosis of osteoblasts in vitro, suggesting that GIP stimulates bone formation by inhibiting apoptosis of osteoblasts129. Enhancement of bone formation by GIP, through suppression of osteoclasts and prevention of osteoclast apoptosis, has also been reproduced in GIP transgenic mice130–132. Furthermore, GIP might facilitate calcium deposition in bone in response to meal ingestion because postprandial plasma calcium levels are enhanced in GIPR‐deficient mice128. Although these findings imply that postmenopausal women might be osteoporosis‐prone partly as a result of reduced GIP response133, it remains to be determined whether GIP exerts an osteogenic function in humans.
A role of endogenous GLP‐1 in bone metabolism was shown by detailed analyses of GLP‐1R‐deficient mice that showed cortical osteopenia and bone fragility in addition to increased osteoclastic numbers and bone resorption activity134. Unlike GIP, GLP‐1 has no direct effect on osteoblasts and osteoclasts, and GLP‐1 inhibits bone resorption indirectly through upregulation of calcitonin134,135. Although exendin‐4 administered at pharmacological levels has been shown to promote bone formation in rats136, whether GLP‐1‐based therapies show any effects on bone metabolism in human remains to be addressed in the future.
Physiological Functions of Endogenous GIP and GLP‐1 in Other Organs
Receptors for GIP and GLP‐1 are expressed in a wide variety of organs in addition to the pancreas, fats and bones. Within the brain, GIP is strongly expressed in neurons of the hippocampus, and the olfactory bulb and Purkinje cells of the cerebellum137,138, and GIPR is expressed in several brain regions including the cerebral cortex, hippocampus and olfactory bulb139,140. Notably, expression of GIPR in neuronal progenitors of the dentate gyrus in the hippocampus suggests involvement of GIP in regulation of neurogenesis and memory formation137. Indeed, proliferation of neuronal progenitors is enhanced by infusion of GIP, and is decreased in the dentate gyrus of GIPR‐deficient mice137. Consistent with the proliferative effects of GIP on neuronal progenitors, activation of GIPR by GIP analogue enhances LTP formation in hippocampal slice culture, whereas inhibition of GIPR by the GIP antagonist (Pro3)GIP reduces LTP141. GIP‐transgenic (Tg) mice show improved performance in a memory‐related behavioral task142. Similarly, GLP‐1 enhances proliferation of neuronal progenitors143,144 and has been shown to enhance LTP145–147, and GLP‐1R‐deficient mice show impaired performance in memory‐related behavioral tasks145. In addition, GLP‐1 is protective against neuronal apoptosis in the Alzheimer’s disease model148,149. Taken together, both GIP and GLP‐1 could proliferate neuronal progenitors, thereby enhancing memory formation.
Another function of GIP in the brain is regulation of appetite and satiety. Ovariectomy‐induced obesity has been prevented by GIPR deficiency, which could be explained partly by reduced expression of orexigenic neuropeptide Y (NPY) in the hypothalamus and subsequent reduction of food intake in the absence of GIPR150. Cerebral infusion of NPY stimulates neuronal secretion of GIP, implying that GIP acts as a negative regulator of NPY and can thereby control food intake151. However, careful consideration is required for anti‐obesity function of GIP in the brain because GIP has its direct effect on adipose tissues. Future studies using brain‐specific GIPR‐deficient mice could clarify roles of GIP in the central nervous system. Regarding the regulation of appetite and satiety, both intracerebroventricular and peripheral infusion of GLP‐1R agonists also inhibit food intake152,153. Further examination using the GLP‐1 and GLP‐1R antagonist exendin(9–39) confirms the inhibitory actions of endogenous GLP‐1 on food intake154,155. GLP‐1R is expressed in the arcuate nucleus and other hypothalamic regions involved in regulation of food intake156, and destruction of the arcuate nucleus abolishes the inhibitory effect of GLP‐1 on food intake157. These lines of evidence imply that not only GIP, but also GLP‐1 controls food intake and satiety in humans.
In addition to these functions, GLP‐1 is involved in gastrointestinal motility as well as in the reduction of cardiac contractility. Although GLP‐1 inhibits gastric emptying158,159, GIP has been shown to have little effect on gastric emptying in humans and mice106,160. GLP‐1 action on the heart was also studied in GLP‐1R‐deficient mice that display increased left ventricle thickness, impaired left ventricle contractility and diastolic dysfunction161. In addition, GLP‐1 administration in animal models or humans with cardiac injuries (e.g. acute myocardial infarction and dilated cardiomyopathy) significantly improves cardiac perormance162–166, suggesting not only beneficial effects of GLP‐1 in heart diseases but also supporting the existence of physiological roles of GLP‐1 on the heart. GIP action on the heart remains to be examined in the future.
Incretin‐Based Therapies for T2DM
Although endogenous GIP exerts strong insulinotropic effects in healthy subjects, the severe reduction in the insulinotropic effect of GIP52,53 and the GIP‐dependent enhancement of postprandial glucagon response106 have discouraged development of GIP‐based therapies for T2DM. In contrast, the insulinotropic effect of GLP‐1 is substantially preserved in T2DM53,167. Long‐term intravenous infusion of GLP‐1 has been shown to improve glycemic control168, establishing GLP‐1 and GLP‐1R signaling as attractive therapeutic targets for T2DM. Indeed, GLP‐1R agonists (e.g. liraglutide and exenatide) and DPP‐4 inhibitors (e.g. sitagliptin and vildagliptin) have been widely and successfully used. Furthermore, recent clinical data suggest that incretin‐based therapies are more effective in Japanese patients compared with Caucasian patients169–175. The effectiveness of incretin‐based therapies is consistent with the reduced early insulin secretory capacity in T2DM patients in Asia countries including Japan176, and further suggests that such reduced early insulin secretory capacity could be partly due to their considerably lower levels of intact GLP‐1, which has been recently revealed in Japanese subjects42. Incretin‐based therapies have recently become widely available in Asian countries. However, their effectiveness in the regulation of long‐term glycemic control, preservation of β cell mass and function, and the prevention of macro and microcomplications are not known and must be carefully followed for years. Nevertheless, given the pathophysiology of Asian T2DM (insulin deficiency rather than insulin resistance), incretin‐based therapies that primarily correct impaired early insulin secretion might well be highly suitable in the treatment of Asian T2DM and have the potential to be a first choice therapy as is presently the case for metformin in Caucasian T2DM177,178.
Acknowledgement
The authors deeply thank current and former colleagues in the laboratory of Seino, and apologize for citing only part of the relevant work in this field (due to limited space) and are indebted to many authors for their contributions. The authors have no conflict of interest.
References
- 1.Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol 1902; 28: 325–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore B. On the treatment of diabetus mellitus by acid extract of duodenal mucous membrane. Biochem J 1906; 1: 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zunz E, La Barre J. Contributiona a l’etude des variations physiologiques de la secretion interne du pancreas: Relations entre les secretions externe et interne du pancreas. Arch Int Physiol Biochim 1929; 31: 20–44 [Google Scholar]
- 4.Elrick H, Stimmler L, Hlad CJ Jr, et al. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 1964; 24: 1076–1082 [DOI] [PubMed] [Google Scholar]
- 5.McIntyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet 1964; 2: 20–21 [DOI] [PubMed] [Google Scholar]
- 6.Inagaki N, Seino Y, Takeda J, et al. Gastric inhibitory polypeptide: Structure and chromosomal localization of the human gene. Mol Endocrinol 1989; 3: 1014–1021 [DOI] [PubMed] [Google Scholar]
- 7.Takeda J, Seino Y, Tanaka K, et al. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc Natl Acad Sci USA 1987; 84: 7005–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown JC, Mutt V, Pederson RA. Further purification of a polypeptide demonstrating enterogastrone activity. J Physiol 1970; 209: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupre J, Ross SA, Watson D, et al. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 1973; 37: 826–828 [DOI] [PubMed] [Google Scholar]
- 10.Adrian TE, Bloom SR, Hermansen K, et al. Pancreatic polypeptide, glucagon and insulin secretion from the isolated perfused canine pancreas. Diabetologia 1978; 14: 413–417 [DOI] [PubMed] [Google Scholar]
- 11.Taminato T, Seino Y, Goto Y, et al. Synthetic gastric inhibitory polypeptide. Stimulatory effect on insulin and glucagon secretion in the rat. Diabetes 1977; 26: 480–484 [DOI] [PubMed] [Google Scholar]
- 12.Takemura J, Seino Y, Yamamura T, et al. The role of endogenous gastric inhibitory polypeptide in the enteroinsular axis. J Clin Endocrinol Metab 1982; 54: 909–913 [DOI] [PubMed] [Google Scholar]
- 13.Ebert R, Unger H, Creutzfeldt W. Preservation of incretin activity after removal of gastric inhibitory polypeptide (GIP) from rat gut extracts by immunoadsorption. Diabetologia 1983; 24: 449–454 [DOI] [PubMed] [Google Scholar]
- 14.Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 1983; 302: 716–718 [DOI] [PubMed] [Google Scholar]
- 15.Schmidt WE, Siegel EG, Creutzfeldt W. Glucagon‐like peptide‐1 but not glucagon‐like peptide‐2 stimulates insulin release from isolated rat pancreatic islets. Diabetologia 1985; 28: 704–707 [DOI] [PubMed] [Google Scholar]
- 16.Kreymann B, Williams G, Ghatei MA, et al. Glucagon‐like peptide‐1 7‐36: A physiological incretin in man. Lancet 1987; 2: 1300–1304 [DOI] [PubMed] [Google Scholar]
- 17.Gremlich S, Porret A, Hani EH, et al. Cloning, functional expression, and chromosomal localization of the human pancreatic islet glucose‐dependent insulinotropic polypeptide receptor. Diabetes 1995; 44: 1202–1208 [DOI] [PubMed] [Google Scholar]
- 18.Volz A, Goke R, Lankat‐Buttgereit B, et al. Molecular cloning, functional expression, and signal transduction of the GIP‐receptor cloned from a human insulinoma. FEBS Lett 1995; 373: 23–29 [DOI] [PubMed] [Google Scholar]
- 19.Wheeler MB, Gelling RW, McIntosh CH, et al. Functional expression of the rat pancreatic islet glucose‐dependent insulinotropic polypeptide receptor: Ligand binding and intracellular signaling properties. Endocrinology 1995; 136: 4629–4639 [DOI] [PubMed] [Google Scholar]
- 20.Yamada Y, Hayami T, Nakamura K, et al. Human gastric inhibitory polypeptide receptor: Cloning of the gene (GIPR) and cDNA. Genomics 1995; 29: 773–776 [DOI] [PubMed] [Google Scholar]
- 21.Yasuda K, Inagaki N, Yamada Y, et al. Hamster gastric inhibitory polypeptide receptor expressed in pancreatic islets and clonal insulin‐secreting cells: Its structure and functional properties. Biochem Biophys Res Commun 1994; 205: 1556–1562 [DOI] [PubMed] [Google Scholar]
- 22.Dillon JS, Tanizawa Y, Wheeler MB, et al. Cloning and functional expression of the human glucagon‐like peptide‐1 (GLP‐1) receptor. Endocrinology 1993; 133: 1907–1910 [DOI] [PubMed] [Google Scholar]
- 23.Stoffel M, Espinosa R 3rd, Le Beau MM, et al. Human glucagon‐like peptide‐1 receptor gene. Localization to chromosome band 6p21 by fluorescence in situ hybridization and linkage of a highly polymorphic simple tandem repeat DNA polymorphism to other markers on chromosome 6. Diabetes 1993; 42: 1215–1218 [DOI] [PubMed] [Google Scholar]
- 24.Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco‐incretin hormone glucagon‐like peptide 1. Proc Natl Acad Sci USA 1992; 89: 8641–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansotia T, Baggio LL, Delmeire D, et al. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP‐IV inhibitors. Diabetes 2004; 53: 1326–1335 [DOI] [PubMed] [Google Scholar]
- 26.Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742 [DOI] [PubMed] [Google Scholar]
- 27.Preitner F, Ibberson M, Franklin I, et al. Gluco‐incretins control insulin secretion at multiple levels as revealed in mice lacking GLP‐1 and GIP receptors. J Clin Invest 2004; 113: 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scrocchi LA, Brown TJ, MaClusky N, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon‐like peptide 1 receptor gene. Nat Med 1996; 2: 1254–1258 [DOI] [PubMed] [Google Scholar]
- 29.Miyawaki K, Yamada Y, Yano H, et al. Glucose intolerance caused by a defect in the entero‐insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA 1999; 96: 14843–14847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose‐dependent insulinotropic polypeptide and truncated glucagon‐like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995; 136: 3585–3596 [DOI] [PubMed] [Google Scholar]
- 31.Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl‐peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon‐like peptide‐1(7‐36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 1993; 214: 829–835 [DOI] [PubMed] [Google Scholar]
- 32.Marguet D, Baggio L, Kobayashi T, et al. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc Natl Acad Sci USA 2000; 97: 6874–6879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deacon CF, Holst JJ. Immunoassays for the incretin hormones GIP and GLP‐1. Best Pract Res Clin Endocrinol Metab 2009; 23: 425–432 [DOI] [PubMed] [Google Scholar]
- 34.Orskov C, Wettergren A, Holst JJ. Biological effects and metabolic rates of glucagonlike peptide‐1 7‐36 amide and glucagonlike peptide‐1 7‐37 in healthy subjects are indistinguishable. Diabetes 1993; 42: 658–661 [DOI] [PubMed] [Google Scholar]
- 35.Orskov C, Rabenhoj L, Wettergren A, et al. Tissue and plasma concentrations of amidated and glycine‐extended glucagon‐like peptide I in humans. Diabetes 1994; 43: 535–539 [DOI] [PubMed] [Google Scholar]
- 36.Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008; 57: 678–687 [DOI] [PubMed] [Google Scholar]
- 37.Vilsboll T, Krarup T, Deacon CF, et al. Reduced postprandial concentrations of intact biologically active glucagon‐like peptide 1 in type 2 diabetic patients. Diabetes 2001; 50: 609–613 [DOI] [PubMed] [Google Scholar]
- 38.Carr RD, Larsen MO, Winzell MS, et al. Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiol Endocrinol Metab 2008; 295: E779–E784 [DOI] [PubMed] [Google Scholar]
- 39.Deacon CF, Nauck MA, Meier J, et al. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab 2000; 85: 3575–3581 [DOI] [PubMed] [Google Scholar]
- 40.Meier JJ, Nauck MA, Kranz D, et al. Secretion, degradation, and elimination of glucagon‐like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 2004; 53: 654–662 [DOI] [PubMed] [Google Scholar]
- 41.Vilsboll T, Krarup T, Sonne J, et al. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003; 88: 2706–2713 [DOI] [PubMed] [Google Scholar]
- 42.Yabe D, Kuroe A, Lee S, et al. Little enhancement of meal‐induced GLP‐1 secretion in Japanese: Comparison of type 2 diabetes and healthy controls. J Diabetes Invest 2010; 1: 56–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon‐like peptide‐1 by human plasma in vitro yields an N‐terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 1995; 80: 952–957 [DOI] [PubMed] [Google Scholar]
- 44.Vilsboll T, Agerso H, Lauritsen T, et al. The elimination rates of intact GIP as well as its primary metabolite, GIP 3‐42, are similar in type 2 diabetic patients and healthy subjects. Regul Pept 2006; 137: 168–172 [DOI] [PubMed] [Google Scholar]
- 45.Hansen L, Deacon CF, Orskov C, et al. Glucagon‐like peptide‐1‐(7‐36)amide is transformed to glucagon‐like peptide‐1‐(9‐36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140: 5356–5363 [DOI] [PubMed] [Google Scholar]
- 46.Deacon CF, Pridal L, Klarskov L, et al. Glucagon‐like peptide 1 undergoes differential tissue‐specific metabolism in the anesthetized pig. Am J Physiol 1996; 271: E458–E464 [DOI] [PubMed] [Google Scholar]
- 47.Ross SA, Brown JC, Dupre J. Hypersecretion of gastric inhibitory polypeptide following oral glucose in diabetes mellitus. Diabetes 1977; 26: 525–529 [DOI] [PubMed] [Google Scholar]
- 48.Takemura J, Seino Y, Tsuda K, et al. Hypersecretion of gastric inhibitory polypeptide induced by glucose ingestion in diabetes mellitus. Endocrinol Jpn 1981; 28: 17–21 [DOI] [PubMed] [Google Scholar]
- 49.Parker HE, Habib AM, Rogers GJ, et al. Nutrient‐dependent secretion of glucose‐dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009; 52: 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vilsboll T, Krarup T, Madsbad S, et al. Both GLP‐1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003; 114: 115–121 [DOI] [PubMed] [Google Scholar]
- 51.Nauck MA, Bartels E, Orskov C, et al. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon‐like peptide‐1‐(7‐36) amide infused at near‐physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab 1993; 76: 912–917 [DOI] [PubMed] [Google Scholar]
- 52.Nauck MA, Heimesaat MM, Orskov C, et al. Preserved incretin activity of glucagon‐like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type‐2 diabetes mellitus. J Clin Invest 1993; 91: 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilsboll T, Krarup T, Madsbad S, et al. Defective amplification of the late phase insulin response to glucose by GIP in obese type II diabetic patients. Diabetologia 2002; 45: 1111–1119 [DOI] [PubMed] [Google Scholar]
- 54.Yamada Y, Miyawaki K, Tsukiyama K, et al. Pancreatic and extrapancreatic effects of gastric inhibitory polypeptide. Diabetes 2006; 55: S88–S91 [Google Scholar]
- 55.Lynn FC, Pamir N, Ng EH, et al. Defective glucose‐dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes 2001; 50: 1004–1011 [DOI] [PubMed] [Google Scholar]
- 56.Lynn FC, Thompson SA, Pospisilik JA, et al. A novel pathway for regulation of glucose‐dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 2003; 17: 91–93 [DOI] [PubMed] [Google Scholar]
- 57.Piteau S, Olver A, Kim SJ, et al. Reversal of islet GIP receptor down‐regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun 2007; 362: 1007–1012 [DOI] [PubMed] [Google Scholar]
- 58.Younan SM, Rashed LA. Impairment of the insulinotropic effect of gastric inhibitory polypeptide (GIP) in obese and diabetic rats is related to the down‐regulation of its pancreatic receptors. Gen Physiol Biophys 2007; 26: 181–193 [PubMed] [Google Scholar]
- 59.Zhou J, Livak MF, Bernier M, et al. Ubiquitination is involved in glucose‐mediated downregulation of GIP receptors in islets. Am J Physiol Endocrinol Metab 2007; 293: E538–E547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harada N, Yamada Y, Tsukiyama K, et al. A novel GIP receptor splice variant influences GIP sensitivity of pancreatic beta‐cells in obese mice. Am J Physiol Endocrinol Metab 2008; 294: E61–E68 [DOI] [PubMed] [Google Scholar]
- 61.Kubota A, Yamada Y, Hayami T, et al. Identification of two missense mutations in the GIP receptor gene: A functional study and association analysis with NIDDM: No evidence of association with Japanese NIDDM subjects. Diabetes 1996; 45: 1701–1705 [DOI] [PubMed] [Google Scholar]
- 62.Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010; 42: 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drucker DJ, Philippe J, Mojsov S, et al. Glucagon‐like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA 1987; 84: 3434–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szecowka J, Grill V, Sandberg E, et al. Effect of GIP on the secretion of insulin and somatostatin and the accumulation of cyclic AMP in vitro in the rat. Acta Endocrinol (Copenh) 1982; 99: 416–421 [DOI] [PubMed] [Google Scholar]
- 65.Fehmann HC, Goke R, Goke B. Cell and molecular biology of the incretin hormones glucagon‐like peptide‐I and glucose‐dependent insulin releasing polypeptide. Endocr Rev 1995; 16: 390–410 [DOI] [PubMed] [Google Scholar]
- 66.Holz GG. Epac: A new cAMP‐binding protein in support of glucagon‐like peptide‐1 receptor‐mediated signal transduction in the pancreatic beta‐cell. Diabetes 2004; 53: 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Light PE, Manning Fox JE, Riedel MJ, et al. Glucagon‐like peptide‐1 inhibits pancreatic ATP‐sensitive potassium channels via a protein kinase A‐ and ADP‐dependent mechanism. Mol Endocrinol 2002; 16: 2135–2144 [DOI] [PubMed] [Google Scholar]
- 68.MacDonald PE, Wang X, Xia F, et al. Antagonism of rat beta‐cell voltage‐dependent K+ currents by exendin 4 requires dual activation of the cAMP/protein kinase A and phosphatidylinositol 3‐kinase signaling pathways. J Biol Chem 2003; 278: 52446–52453 [DOI] [PubMed] [Google Scholar]
- 69.Ding WG, Gromada J. Protein kinase A‐dependent stimulation of exocytosis in mouse pancreatic beta‐cells by glucose‐dependent insulinotropic polypeptide. Diabetes 1997; 46: 615–621 [DOI] [PubMed] [Google Scholar]
- 70.Kang G, Chepurny OG, Holz GG. cAMP‐regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+‐induced Ca2+ release in INS‐1 pancreatic beta‐cells. J Physiol 2001;536:375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang G, Joseph JW, Chepurny OG, et al. Epac‐selective cAMP analog 8‐pCPT‐2′‐O‐Me‐cAMP as a stimulus for Ca2+‐induced Ca2+ release and exocytosis in pancreatic beta‐cells. J Biol Chem 2003; 278: 8279–8285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Renstrom E, Eliasson L, Rorsman P. Protein kinase A‐dependent and ‐independent stimulation of exocytosis by cAMP in mouse pancreatic B‐cells. J Physiol 1997; 1: 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yaekura K, Kakei M, Yada T. cAMP‐signaling pathway acts in selective synergism with glucose or tolbutamide to increase cytosolic Ca2+ in rat pancreatic beta‐cells. Diabetes 1996;45:295–301 [DOI] [PubMed] [Google Scholar]
- 74.Tsuboi T, da Silva Xavier G, Holz GG, et al. Glucagon‐like peptide‐1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 beta‐cells. Biochem J 2003; 369: 287–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shibasaki T, Takahashi H, Miki T, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333–19338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miki T, Minami K, Shinozaki H, et al. Distinct effects of glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1 on insulin secretion and gut motility. Diabetes 2005; 54: 1056–1063 [DOI] [PubMed] [Google Scholar]
- 77.Fujimoto W, Miki T, Ogura T, et al. Niflumic acid‐sensitive ion channels play an important role in the induction of glucose‐stimulated insulin secretion by cyclic AMP in mice. Diabetologia 2009; 52: 863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang CL, Katoh M, Shibasaki T, et al. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 2009; 325: 607–610 [DOI] [PubMed] [Google Scholar]
- 79.Kim SJ, Nian C, Widenmaier S, et al. Glucose‐dependent insulinotropic polypeptide‐mediated up‐regulation of beta‐cell antiapoptotic Bcl‐2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP‐responsive CREB coactivator 2. Mol Cell Biol 2008; 28: 1644–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trumper A, Trumper K, Horsch D. Mechanisms of mitogenic and anti‐apoptotic signaling by glucose‐dependent insulinotropic polypeptide in beta(INS‐1)‐cells. J Endocrinol 2002; 174: 233–246 [DOI] [PubMed] [Google Scholar]
- 81.Kim SJ, Winter K, Nian C, et al. Glucose‐dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta‐cell survival is dependent upon phosphatidylinositol 3‐kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down‐regulation of bax expression. J Biol Chem 2005; 280: 22297–22307 [DOI] [PubMed] [Google Scholar]
- 82.Widenmaier SB, Sampaio AV, Underhill TM, et al. Noncanonical activation of Akt/protein kinase B in {beta}‐cells by the incretin hormone glucose‐dependent insulinotropic polypeptide. J Biol Chem 2009; 284: 10764–10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ehses JA, Casilla VR, Doty T, et al. Glucose‐dependent insulinotropic polypeptide promotes beta‐(INS‐1) cell survival via cyclic adenosine monophosphate‐mediated caspase‐3 inhibition and regulation of p38 mitogen‐activated protein kinase. Endocrinology 2003; 144: 4433–4445 [DOI] [PubMed] [Google Scholar]
- 84.Widenmaier SB, Ao Z, Kim SJ, et al. Suppression of p38 MAPK and JNK via Akt‐mediated inhibition of apoptosis signal‐regulating kinase 1 constitutes a core component of the beta‐cell pro‐survival effects of glucose‐dependent insulinotropic polypeptide. J Biol Chem 2009; 284: 30372–30382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farilla L, Hui H, Bertolotto C, et al. Glucagon‐like peptide‐1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 2002; 143: 4397–4408 [DOI] [PubMed] [Google Scholar]
- 86.Wang Q, Brubaker PL. Glucagon‐like peptide‐1 treatment delays the onset of diabetes in 8 week‐old db/db mice. Diabetologia 2002; 45: 1263–1273 [DOI] [PubMed] [Google Scholar]
- 87.Hui H, Nourparvar A, Zhao X, et al. Glucagon‐like peptide‐1 inhibits apoptosis of insulin‐secreting cells via a cyclic 5′‐adenosine monophosphate‐dependent protein kinase A‐ and a phosphatidylinositol 3‐kinase‐dependent pathway. Endocrinology 2003; 144: 1444–1455 [DOI] [PubMed] [Google Scholar]
- 88.Kwon G, Pappan KL, Marshall CA, et al. cAMP Dose‐dependently prevents palmitate‐induced apoptosis by both protein kinase A‐ and cAMP‐guanine nucleotide exchange factor‐dependent pathways in beta‐cells. J Biol Chem 2004;279:8938–8945 [DOI] [PubMed] [Google Scholar]
- 89.Buteau J, El‐Assaad W, Rhodes CJ, et al. Glucagon‐like peptide‐1 prevents beta cell glucolipotoxicity. Diabetologia 2004; 47: 806–815 [DOI] [PubMed] [Google Scholar]
- 90.Wang Q, Li L, Xu E, et al. Glucagon‐like peptide‐1 regulates proliferation and apoptosis via activation of protein kinase B in pancreatic INS‐1 beta cells. Diabetologia 2004; 47: 478–487 [DOI] [PubMed] [Google Scholar]
- 91.Cunha DA, Ladriere L, Ortis F, et al. Glucagon‐like peptide‐1 agonists protect pancreatic beta‐cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009; 58: 2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsunekawa S, Yamamoto N, Tsukamoto K, et al. Protection of pancreatic beta‐cells by exendin‐4 may involve the reduction of endoplasmic reticulum stress; in vivo and in vitro studies. J Endocrinol 2007; 193: 65–74 [DOI] [PubMed] [Google Scholar]
- 93.Yusta B, Baggio LL, Estall JL, et al. GLP‐1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 2006; 4: 391–406 [DOI] [PubMed] [Google Scholar]
- 94.Jhala US, Canettieri G, Screaton RA, et al. cAMP promotes pancreatic beta‐cell survival via CREB‐mediated induction of IRS2. Genes Dev 2003;17:1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maida A, Hansotia T, Longuet C, et al. Differential importance of glucose‐dependent insulinotropic polypeptide vs glucagon‐like peptide 1 receptor signaling for beta cell survival in mice. Gastroenterology 2009; 137: 2146–2157 [DOI] [PubMed] [Google Scholar]
- 96.Xu G, Stoffers DA, Habener JF, et al. Exendin‐4 stimulates both beta‐cell replication and neogenesis, resulting in increased beta‐cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999; 48: 2270–2276 [DOI] [PubMed] [Google Scholar]
- 97.Bocker D, Verspohl EJ. Role of protein kinase C, PI3‐kinase and tyrosine kinase in activation of MAP kinase by glucose and agonists of G‐protein coupled receptors in INS‐1 cells. Int J Exp Diabetes Res 2001; 2: 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trumper A, Trumper K, Trusheim H, et al. Glucose‐dependent insulinotropic polypeptide is a growth factor for beta (INS‐1) cells by pleiotropic signaling. Mol Endocrinol 2001; 15: 1559–1570 [DOI] [PubMed] [Google Scholar]
- 99.Ehses JA, Pelech SL, Pederson RA, et al. Glucose‐dependent insulinotropic polypeptide activates the Raf‐Mek1/2‐ERK1/2 module via a cyclic AMP/cAMP‐dependent protein kinase/Rap1‐mediated pathway. J Biol Chem 2002; 277: 37088–37097 [DOI] [PubMed] [Google Scholar]
- 100.Kubota A, Yamada Y, Yasuda K, et al. Gastric inhibitory polypeptide activates MAP kinase through the wortmannin‐sensitive and ‐insensitive pathways. Biochem Biophys Res Commun 1997; 235: 171–175 [DOI] [PubMed] [Google Scholar]
- 101.Friedrichsen BN, Neubauer N, Lee YC, et al. Stimulation of pancreatic beta‐cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J Endocrinol 2006; 188: 481–492 [DOI] [PubMed] [Google Scholar]
- 102.Buteau J, Foisy S, Joly E, et al. Glucagon‐like peptide 1 induces pancreatic beta‐cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003; 52: 124–132 [DOI] [PubMed] [Google Scholar]
- 103.Perfetti R, Zhou J, Doyle ME, et al. Glucagon‐like peptide‐1 induces cell proliferation and pancreatic‐duodenum homeobox‐1 expression and increases endocrine cell mass in the pancreas of old, glucose‐intolerant rats. Endocrinology 2000; 141: 4600–4605 [DOI] [PubMed] [Google Scholar]
- 104.Pederson RA, Brown JC. Interaction of gastric inhibitory polypeptide, glucose, and arginine on insulin and glucagon secretion from the perfused rat pancreas. Endocrinology 1978; 103: 610–615 [DOI] [PubMed] [Google Scholar]
- 105.Meier JJ, Gallwitz B, Siepmann N, et al. Gastric inhibitory polypeptide (GIP) dose‐dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003; 46: 798–801 [DOI] [PubMed] [Google Scholar]
- 106.Chia CW, Carlson OD, Kim W, et al. Exogenous glucose‐dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes 2009; 58: 1342–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nauck MA, Heimesaat MM, Behle K, et al. Effects of glucagon‐like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab 2002; 87: 1239–1246 [DOI] [PubMed] [Google Scholar]
- 108.Hare KJ, Vilsboll T, Asmar M, et al. The glucagononostatic and insulinotropic effects of glucagon‐like peptide‐1 contribute equally to its glucose‐lowering action. Diabetes 2010; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Heller RS, Kieffer TJ, Habener JF. Insulinotropic glucagon‐like peptide I receptor expression in glucagon‐producing alpha‐cells of the rat endocrine pancreas. Diabetes 1997; 46: 785–791 [DOI] [PubMed] [Google Scholar]
- 110.Creutzfeldt WO, Kleine N, Willms B, et al. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon‐like peptide I(7–36) amide in type I diabetic patients. Diabetes Care 1996; 19: 580–586 [DOI] [PubMed] [Google Scholar]
- 111.Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon‐like peptide‐1 [proglucagon‐(78‐107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology 1988; 123: 2009–2013 [DOI] [PubMed] [Google Scholar]
- 112.de Heer J, Rasmussen C, Coy DH, et al. Glucagon‐like peptide‐1, but not glucose‐dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008; 51: 2263–2270 [DOI] [PubMed] [Google Scholar]
- 113.Thomsen C, Rasmussen O, Lousen T, et al. Differential effects of saturated and monounsaturated fatty acids on postprandial lipemia and incretin responses in healthy subjects. Am J Clin Nutr 1999; 69: 1135–1143 [DOI] [PubMed] [Google Scholar]
- 114.Creutzfeldt W, Ebert R, Willms B, et al. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia 1978; 14: 15–24 [DOI] [PubMed] [Google Scholar]
- 115.Beck B, Max JP. Gastric inhibitory polypeptide enhancement of the insulin effect on fatty acid incorporation into adipose tissue in the rat. Regul Pept 1983; 7: 3–8 [DOI] [PubMed] [Google Scholar]
- 116.Yip RG, Boylan MO, Kieffer TJ, et al. Functional GIP receptors are present on adipocytes. Endocrinology 1998; 139: 4004–4007 [DOI] [PubMed] [Google Scholar]
- 117.Naitoh R, Miyawaki K, Harada N, et al. Inhibition of GIP signaling modulates adiponectin levels under high‐fat diet in mice. Biochem Biophys Res Commun 2008; 376: 21–25 [DOI] [PubMed] [Google Scholar]
- 118.Zhou H, Yamada Y, Tsukiyama K, et al. Gastric inhibitory polypeptide modulates adiposity and fat oxidation under diminished insulin action. Biochem Biophys Res Commun 2005; 335: 937–942 [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432 [DOI] [PubMed] [Google Scholar]
- 120.Gault VA, McClean PL, Cassidy RS, et al. Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high‐fat and cafeteria diets. Diabetologia 2007; 50: 1752–1762 [DOI] [PubMed] [Google Scholar]
- 121.Irwin N, McClean PL, O’Harte FP, et al. Early administration of the glucose‐dependent insulinotropic polypeptide receptor antagonist (Pro3)GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia 2007; 50: 1532–1540 [DOI] [PubMed] [Google Scholar]
- 122.McClean PL, Irwin N, Cassidy RS, et al. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high‐fat diet. Am J Physiol Endocrinol Metab 2007; 293: E1746–E1755 [DOI] [PubMed] [Google Scholar]
- 123.Althage MC, Ford EL, Wang S, et al. Targeted ablation of glucose‐dependent insulinotropic polypeptide‐producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J Biol Chem 2008; 283: 18365–18376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim SJ, Nian C, McIntosh CH. Resistin is a key mediator of glucose‐dependent insulinotropic polypeptide (GIP) stimulation of lipoprotein lipase (LPL) activity in adipocytes. J Biol Chem 2007; 282: 34139–34147 [DOI] [PubMed] [Google Scholar]
- 125.Kim SJ, Nian C, McIntosh CH. Activation of lipoprotein lipase by glucose‐dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP‐activated protein kinase cascade. J Biol Chem 2007; 282: 8557–8567 [DOI] [PubMed] [Google Scholar]
- 126.Widenmaiser SB, Kim SJ, Yang GK, et al. A GIP receptor agnoist exhibits β‐cell anti‐apoptotic actions in rat models of diabetes resulting in improved β‐cell function and glycemic control. PLoS ONE 2010; 5: e9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Montrose‐Rafizadeh C, Yang H, Wang Y, et al. Novel signal transduction and peptide specificity of glucagon‐like peptide receptor in 3T3‐L1 adipocytes. J Cell Physiol 1997; 172: 275–283 [DOI] [PubMed] [Google Scholar]
- 128.Bollag RJ, Zhong Q, Ding KH, et al. Glucose‐dependent insulinotropic peptide is an integrative hormone with osteotropic effects. Mol Cell Endocrinol 2001; 177: 35–41 [DOI] [PubMed] [Google Scholar]
- 129.Tsukiyama K, Yamada Y, Yamada C, et al. Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol 2006; 20: 1644–1651 [DOI] [PubMed] [Google Scholar]
- 130.Ding KH, Shi XM, Zhong Q, et al. Impact of glucose‐dependent insulinotropic peptide on age‐induced bone loss. J Bone Miner Res 2008; 23: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xie D, Zhong Q, Ding KH, et al. Glucose‐dependent insulinotropic peptide‐overexpressing transgenic mice have increased bone mass. Bone 2007; 40: 1352–1360 [DOI] [PubMed] [Google Scholar]
- 132.Zhong Q, Itokawa T, Sridhar S, et al. Effects of glucose‐dependent insulinotropic peptide on osteoclast function. Am J Physiol Endocrinol Metab 2007; 292: E543–E548 [DOI] [PubMed] [Google Scholar]
- 133.Ahren B, Larsson H, Holst JJ. Reduced gastric inhibitory polypeptide but normal glucagon‐like peptide 1 response to oral glucose in postmenopausal women with impaired glucose tolerance. Eur J Endocrinol 1997; 137: 127–131 [DOI] [PubMed] [Google Scholar]
- 134.Yamada C, Yamada Y, Tsukiyama K, et al. The murine glucagon‐like peptide‐1 receptor is essential for control of bone resorption. Endocrinology 2008; 149: 574–579 [DOI] [PubMed] [Google Scholar]
- 135.Lamari Y, Boissard C, Moukhtar MS, et al. Expression of glucagon‐like peptide 1 receptor in a murine C cell line: Regulation of calcitonin gene by glucagon‐like peptide 1. FEBS Lett 1996; 393: 248–252 [DOI] [PubMed] [Google Scholar]
- 136.Nuche‐Berenguer B, Moreno P, Portal‐Nunez S, et al. Exendin‐4 exerts osteogenic actions in insulin‐resistant and type 2 diabetic states. Regul Pept 2010; 159: 61–66 [DOI] [PubMed] [Google Scholar]
- 137.Nyberg J, Anderson MF, Meister B, et al. Glucose‐dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J Neurosci 2005; 25: 1816–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nyberg J, Jacobsson C, Anderson MF, et al. Immunohistochemical distribution of glucose‐dependent insulinotropic polypeptide in the adult rat brain. J Neurosci Res 2007; 85: 2099–2119 [DOI] [PubMed] [Google Scholar]
- 139.Kaplan AM, Vigna SR. Gastric inhibitory polypeptide (GIP) binding sites in rat brain. Peptides 1994; 15: 297–302 [DOI] [PubMed] [Google Scholar]
- 140.Usdin TB, Mezey E, Button DC, et al. Gastric inhibitory polypeptide receptor, a member of the secretin‐vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993; 133: 2861–2870 [DOI] [PubMed] [Google Scholar]
- 141.Gault VA, Holscher C. Protease‐resistant glucose‐dependent insulinotropic polypeptide agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta‐amyloid. J Neurophysiol 2008; 99: 1590–1595 [DOI] [PubMed] [Google Scholar]
- 142.Ding KH, Zhong Q, Xie D, et al. Effects of glucose‐dependent insulinotropic peptide on behavior. Peptides 2006; 27: 2750–2755 [DOI] [PubMed] [Google Scholar]
- 143.Belsham DD, Fick LJ, Dalvi PS, et al. Ciliary neurotrophic factor recruitment of glucagon‐like peptide‐1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J 2009; 23: 4256–4265 [DOI] [PubMed] [Google Scholar]
- 144.Bertilsson G, Patrone C, Zachrisson O, et al. Peptide hormone exendin‐4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res 2008; 86: 326–338 [DOI] [PubMed] [Google Scholar]
- 145.Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP‐1 receptor KO mice: Interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res 2009; 205: 265–271 [DOI] [PubMed] [Google Scholar]
- 146.Gault VA, Holscher C. GLP‐1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta‐amyloid. Eur J Pharmacol 2008; 587: 112–117 [DOI] [PubMed] [Google Scholar]
- 147.McClean PL, Gault VA, Harriott P, et al. Glucagon‐like peptide‐1 analogues enhance synaptic plasticity in the brain: A link between diabetes and Alzheimer’s disease. Eur J Pharmacol 2010; 630: 158–162 [DOI] [PubMed] [Google Scholar]
- 148.Perry T, Lahiri DK, Sambamurti K, et al. Glucagon‐like peptide‐1 decreases endogenous amyloid‐beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res 2003; 72: 603–612 [DOI] [PubMed] [Google Scholar]
- 149.Qin Z, Sun Z, Huang J, et al. Mutated recombinant human glucagon‐like peptide‐1 protects SH‐SY5Y cells from apoptosis induced by amyloid‐beta peptide (1–42). Neurosci Lett 2008; 444: 217–221 [DOI] [PubMed] [Google Scholar]
- 150.Isken F, Pfeiffer AF, Nogueiras R, et al. Deficiency of glucose‐dependent insulinotropic polypeptide receptor prevents ovariectomy‐induced obesity in mice. Am J Physiol Endocrinol Metab 2008; 295: E350–E355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yavropoulou MP, Kotsa K, Kesisoglou I, et al. Intracerebroventricular infusion of neuropeptide Y increases glucose dependent‐insulinotropic peptide secretion in the fasting conscious dog. Peptides 2008; 29: 2281–2285 [DOI] [PubMed] [Google Scholar]
- 152.Tang‐Christensen M, Larsen PJ, Goke R, et al. Central administration of GLP‐1‐(7–36) amide inhibits food and water intake in rats. Am J Physiol 1996; 271: R848–R856 [DOI] [PubMed] [Google Scholar]
- 153.Turton MD, O’Shea D, Gunn I, et al. A role for glucagon‐like peptide‐1 in the central regulation of feeding. Nature 1996; 379: 69–72 [DOI] [PubMed] [Google Scholar]
- 154.Kinzig KP, D’Alessio DA, Seeley RJ. The diverse roles of specific GLP‐1 receptors in the control of food intake and the response to visceral illness. J Neurosci 2002; 22: 10470–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schick RR, Zimmermann JP, vorm Walde T, et al. Peptides that regulate food intake: Glucagon‐like peptide 1‐(7–36) amide acts at lateral and medial hypothalamic sites to suppress feeding in rats. Am J Physiol Regul Integr Comp Physiol 2003; 284: R1427–R1435 [DOI] [PubMed] [Google Scholar]
- 156.Goke R, Larsen PJ, Mikkelsen JD, et al. Distribution of GLP‐1 binding sites in the rat brain: Evidence that exendin‐4 is a ligand of brain GLP‐1 binding sites. Eur J Neurosci 1995; 7: 2294–2300 [DOI] [PubMed] [Google Scholar]
- 157.Tang‐Christensen M, Vrang N, Larsen PJ. Glucagon‐like peptide 1(7–36) amide’s central inhibition of feeding and peripheral inhibition of drinking are abolished by neonatal monosodium glutamate treatment. Diabetes 1998; 47: 530–537 [DOI] [PubMed] [Google Scholar]
- 158.Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon‐like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol 1997; 273: E981–E988 [DOI] [PubMed] [Google Scholar]
- 159.Wettergren A, Schjoldager B, Mortensen PE, et al. Truncated GLP‐1 (proglucagon 78‐107‐amide) inhibits gastric and pancreatic functions in man. Dig Dis Sci 1993; 38: 665–673 [DOI] [PubMed] [Google Scholar]
- 160.Meier JJ, Goetze O, Anstipp J, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab 2004; 286: E621–E625 [DOI] [PubMed] [Google Scholar]
- 161.Gros R, You X, Baggio LL, et al. Cardiac function in mice lacking the glucagon‐like peptide‐1 receptor. Endocrinology 2003; 144: 2242–2252 [DOI] [PubMed] [Google Scholar]
- 162.Ban K, Noyan‐Ashraf MH, Hoefer J, et al. Cardioprotective and vasodilatory actions of glucagon‐like peptide 1 receptor are mediated through both glucagon‐like peptide 1 receptor‐dependent and ‐independent pathways. Circulation 2008; 117: 2340–2350 [DOI] [PubMed] [Google Scholar]
- 163.Bose AK, Mocanu MM, Carr RD, et al. Glucagon‐like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2005; 54: 146–151 [DOI] [PubMed] [Google Scholar]
- 164.Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon‐like peptide‐1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004; 109: 962–965 [DOI] [PubMed] [Google Scholar]
- 165.Noyan‐Ashraf MH, Momen MA, Ban K, et al. GLP‐1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes 2009; 58: 975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nystrom T, Gutniak MK, Zhang Q, et al. Effects of glucagon‐like peptide‐1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab 2004; 287: E1209–E1215 [DOI] [PubMed] [Google Scholar]
- 167.Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab 1986; 63: 492–498 [DOI] [PubMed] [Google Scholar]
- 168.Zander M, Madsbad S, Madsen JL, et al. Effect of 6‐week course of glucagon‐like peptide 1 on glycaemic control, insulin sensitivity, and beta‐cell function in type 2 diabetes: A parallel‐group study. Lancet 2002; 359: 824–830 [DOI] [PubMed] [Google Scholar]
- 169.Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes: Systematic review and meta‐analysis. JAMA 2007; 298: 194–206 [DOI] [PubMed] [Google Scholar]
- 170.Kikuchi M, Abe N, Kato M, et al. Vildagliptin dose‐dependently improves glycemic control in Japanese patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 2009; 83: 233–240 [DOI] [PubMed] [Google Scholar]
- 171.Nonaka K, Kakikawa T, Sato A, et al. Efficacy and safety of sitagliptin monotherapy in Japanese patients with type 2 diabetes. Diabetes Res Clin Pract 2008; 79: 291–298 [DOI] [PubMed] [Google Scholar]
- 172.Seino Y, Nakajima H, Miyahara H, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of albiglutide, a long‐acting GLP‐1‐receptor agonist, in Japanese subjects with type 2 diabetes mellitus. Curr Med Res Opin 2009; 25: 3049–3057 [DOI] [PubMed] [Google Scholar]
- 173.Seino Y, Rasmussen MF, Zdravkovic M, et al. Dose‐dependent improvement in glycemia with once‐daily liraglutide without hypoglycemia or weight gain: A double‐blind, randomized, controlled trial in Japanese patients with type 2 diabetes. Diabetes Res Clin Pract 2008; 81: 161–168 [DOI] [PubMed] [Google Scholar]
- 174.Seino Y, Rasmussen MF, Nishida T, et al. Efficacy and safety of the once‐daily human GLP‐1 analogue, liraglutide, vs glibenclamide monotherapy in Japanese patients with type 2 diabetes. Curr Med Res Opin 2010; in press. [DOI] [PubMed] [Google Scholar]
- 175.Kaku K, Rasmussen MF, Clauson P, et al. Improved glycaemic control with minimal hypoglycaemia and no weight change with the once‐daily human GLP‐1 analogue liraglutide as add‐on to sulfonylurea in Japanese patients with type 2 diabetes. Diabetes Obes Metab 2010; in press. [DOI] [PubMed] [Google Scholar]
- 176.Fukushima M, Suzuki H, Seino Y. Insulin secretion capacity in the development from normal glucose tolerance to type 2 diabetes. Diabetes Res Clin Pract 2004; 66S: S37–S44 [DOI] [PubMed] [Google Scholar]
- 177.Nathan DM, Buse JB, Davidson MB, et al. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nathan DM, Buse JB, Davidson MB, et al. Medical management of hyperglycaemia in type 2 diabetes mellitus: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2009; 52: 17–30 [DOI] [PubMed] [Google Scholar]