

Abstract
Tricyclic cytosines (tC and tCO frameworks) have emerged as a unique class of fluorescent nucleobase analogues that minimally perturb the structure of B-form DNA and that are not quenched in duplex nucleic acids. Systematic derivatization of these frameworks is a likely approach to improve on and diversify photophysical properties, but has not so far been examined. Synthetic methods were refined to improve on tolerance for electron donating and electron withdrawing groups, resulting in a series of eight new, fluorescent cytidine analogues. Photophysical studies show that substitution of the framework results in a pattern of effects largely consistent across tC and tCO and provides nucleoside fluorophores that are brighter than either parent. Moreover, a range of solvent sensitivities is observed, offering promise that this family of probes can be extended to new applications that require reporting on the local environment.
Keywords: nucleosides, fluorescent probes, DNA, RNA, structure-activity relationships
Introduction
Fluorescent nucleoside analogues are used extensively as molecular probes for biophysical studies and in biotechnology.[1–6] Especially for the former, there is a great need for new, improved probes with a range of fluorescence properties and minimal structural perturbation. The vast majority of available probes either dangle freely from the major groove of the duplex (e.g. Cy5-dC)[2, 7] or are fluorescent nucleobase analogues.[8–28] Most of the latter are either incompatible with Watson-Crick hydrogen bonding (e.g. pyrene deoxyriboside)[1] or are almost completely quenched when stacked against neighboring bases (e.g. the popular, commercially available (deoxy)ribosides of 2-aminopurine and pyrrolocytosine).[29, 30] The approach of using flexible tethers (as in the Cy5-dC example) is appealing because of the predictable fluorescence properties and the possibility to maintain a useful degree of polymerase compatibility.[31] Applications of this approach are limited, however, by the large degree of structural perturbation, which can prevent normal interactions with proteins of interest. Moreover, the flexibility of the tether can lead to fluorophore intercalation and can limit the precision of distance measurements by FRET.[32, 33] Another significant, unmet need is for a way to label nucleic acids for fluorescence microscopy with sufficiently small structural perturbation so that, e.g., uptake by competent bacteria can be tracked microscopically.
In the past decade, a very promising new class of analogues has emerged, based on a tricyclic cytidine (tC) scaffold (Figure 1).[34–42] While originally developed as stabilizers for double-stranded nucleic acids (substitution of one tC for C in duplex oligonucleotides raises the Tm by an average of 3 °C),[34] tC and tCO were later found to be unique in that they retain their fluorescence (Φem ≈ 0.2) in duplex nucleic acids, varying little with the identity of neighboring bases.[37] The photophysics of these compounds, as free nucleosides and in ss- and dsDNA, are well characterized.[42] We have shown that both compounds are remarkably good substrates for A- and B-family DNA polymerases and T7 RNA polymerase, even to the point of being inserted faster than dCTP during DNA synthesis both by human pol α and Klenow fragment, albeit with approximately 10 % misincorporation of dtC(O)TP across from A (fidelity of RNA synthesis with T7 RNA polymerase is greater).[43, 44] More recently, Wilhelmsson et. al. have developed a nitro-tCO analogue that is a non-emissive FRET acceptor and shown that it can be used with tCO as a donor for inter-nucleobase FRET.[41] The G clamp molecules are also members of the tC family.[45, 46]
Figure 1.
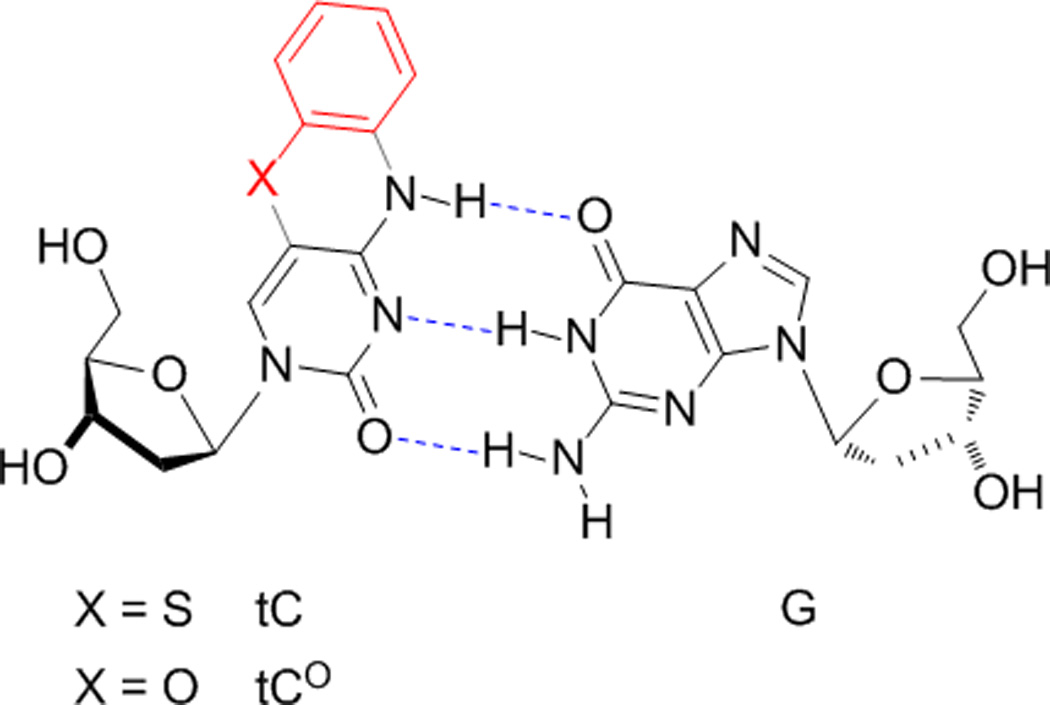
While the three nucleobases of the tC family have unique properties, they are also significantly limited in that the emissive compounds (tC and tCO) have nearly overlapping spectra and they are around 50-fold less bright than fluorescein. This brightness is inadequate for use in most single-molecule fluorescence experiments or for labelling nucleic acids for fluorescence microscopy. Nitro-tCO does not emit. Because it is difficult to predict, a priori, the photophysical properties of a molecule based on structure, we set out to design, synthesize, and characterize a new set of tC(O) analogues incorporating electron-donating and withdrawing groups at varied positions and with further extensions to conjugation (Figure 2). The results show clear patterns in the substituent effects, changes to Φem and λem, and reveal a range of solvent sensitivities. (7-Cl)tCO 1e is now the brightest known member of the tC(O) family in aqueous solution, and the strong solvent-sensitivity of the methoxy derivatives 1b, 1c, 2b, and 2c is expected make them useful as a new set of minimally perturbing and environmentally responsive fluorescence probes.
Figure 2.
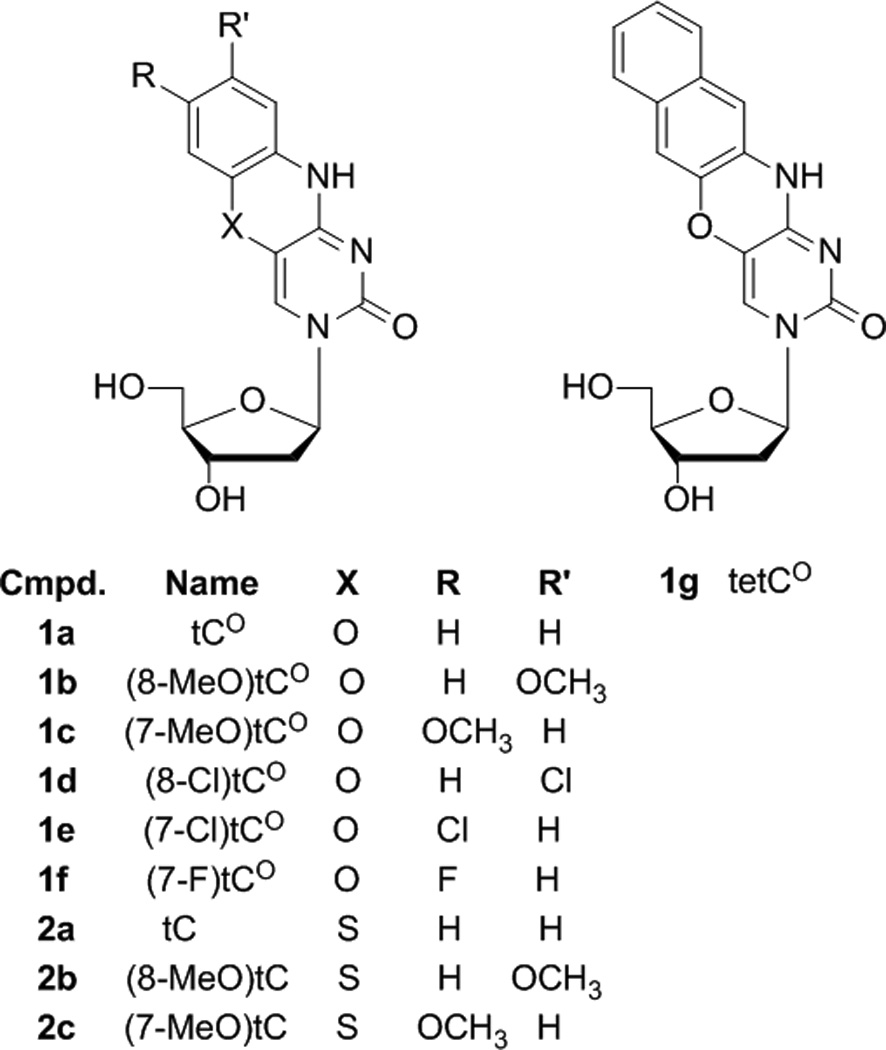
Tri- and tetracyclic cytidine analogues synthesized for this study.
Results and Discussion
Synthesis of Fluorescent Nucleoside Analogues
Synthetic methods for the parent compounds 1a (tC) and 2a (tCO) are established, but the substituent effects of the EDGs and EWGs necessitated re-optimization to improve yields and functional group tolerance. In our hands, the original route to tCO worked poorly or not at all when applied to the synthesis of compounds 1b–g, but our modified route provided adequate yields.[34] Moreover, our re-optimization of the route to tC substantially improves the overall yield (from 10–15% to 43% for 10a to 2a), facilitating the synthesis of 2b,c.[47]
The original synthesis of tCO (1a) entails the activation of the 4-OH tautomer of 3′,5′-di-O-acetyl-5-bromo-2′-deoxyuridine as a mesitylsulfonyl ester, formation of a secondary amine by SNAr of this mesylate, removal of the acetate protective groups, and ring closure by SNAr of the aryl bromide, promoted by KF in ethanol (c.f. Figure 3).[34] Two problems limited the application of this route to derivative synthesis. First, chloro and methoxy groups meta to N of 2-aminophenols attenuate the nucleophilicity, which has a detrimental effect on secondary amine formation. Second, chloro and methoxy groups meta to O attenuate the SNAr reactivity with the aryl bromide, inhibiting ring closure.
Figure 3.
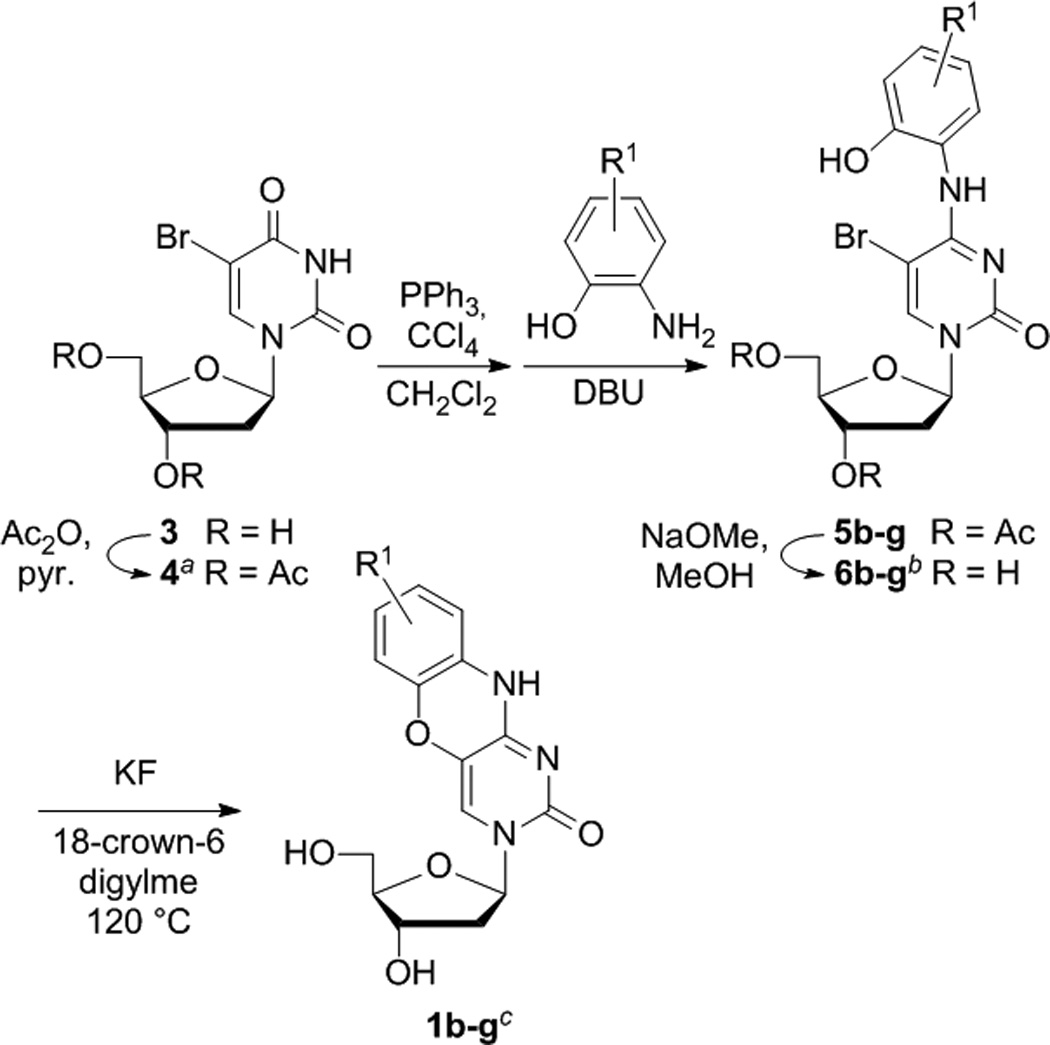
Synthesis of tCO analogues. Yields are (a) 85%, (b) over two steps, 6b 40%, 6c 48%, 6d 41%, 6e 20%, 6f 61%, 6g 44%, (c) 1b 20%, 6c 11%, 1d 24%, 1e 3%, 1f 11%, 1g 53%. The parent tCO 1a was synthesized using the original procedure.[34]
An improved synthesis begins with the same 3′,5′-di-O-acetyl-5-bromo-2′-deoxyuridine as starting material, but activates the O4 using Appel chemistry (PPh3, CCl4) for the conversion to a 4-Cl (Figure 3).[48] The 4-Cl intermediate was used in situ for SNAr with substituted 2-aminophenols in the presence of DBU, resulting in good yields of the corresponding secondary amines for all derivatives tested. Removal of the acetyl protective groups is required for ring closure, as previously reported by Matteucci,[34] likely owing to the intermediacy of a 5′,6-ether, itself resulting from reversible 1,4-addition of the 5′-OH to the uracil moiety. The attenuated reactivity of several of the targeted derivatives necessitated more forceful conditions for this step and the removal of competing nucleophiles. Nucleophilic deamination of the secondary amine (either by solvent or H2O) is likely the main competing side reaction. The efficacy of ring closure could be improved significantly by switching from KF to CsF and changing solvents to N-methylpyrrolidinone, but the hygroscopicity of CsF made it difficult to handle without introducing water into the reaction mixture. Fortunately, a further change to KF, activated by 18-crown-6, in anhydrous diglyme at 120°C proved to offer a significant improvement, resulting in substantially improved yields of all derivatives in a reaction time of approximately 20 minutes. The formation of what are likely deaminated side products (as evidenced by 1H NMR) was greatly minimized under these conditions.
The very different reactivity of aryl alcohols and aryl thiols necessitates a different route to X = S (tC) derivatives (Figures 1 and 4). Matteucci’s and Wilhelmsson’s syntheses of tC 2a begins with 2-aminothiophenol,[34, 47] but derivatives of this compound have very limited commercial availability and are highly sensitive to oxidative degradation. 4- and 5-Methoxy-2-aminothiophenol are accessible through ring opening of more readily available benzothiazole precursors, a reaction previously carried out using concentrated hydroxide. We found this procedure to be unreliable for methoxy starting materials 7b,c, and a change to hydrazine provided significant improvement. Because of the tendency of the resulting 2-amino-(7/8)-methoxythiophenols to undergo oxidative degradation, we performed a controlled, in situ oxidation to the disulfides 8b,c using hydrogen peroxide, giving a product that could be purified chromatographically. A one-pot reduction of this disulfide using triethylphosphine and nucleophilic substitution with 5-bromouracil gave thioether compounds 9b,c. The methoxy group meta to N attenuates its nucleophilicity, but condensation conditions more forcing than those of the original procedure nonetheless produced tricyclic nucleobases 10b,c. The original ribosylation procedure uses the sodium salt of the nucleobase (from NaH) in a reaction with 3,5-di-O-(p-toluoyl)-2′-deoxyribofuranosyl chloride (Hoffer’s chlorosugar)[49] and yields only 10–15%, but we found that substantially better yields of the 2′-deoxyriboside could be obtained by instead activating the nucleobases 10a–c as the TMS ether using BSA, followed by ribosylation in situ, Vorbrüggen’s Silyl-Hilbert-Johnson method.[50, 51] Yields were typically around 80% and consisted of a ~ 1:1 mixture of anomers. The pure β-anomers were separable in around 40% yield from 10. Removal of the toluoyl groups using methoxide completed the synthesis of the fluorescent nucleoside analogues 2. Yields were unfortunately significantly lower for compound 2c because of an apparent high sensitivity to oxidative degradation of 8c and 9c and preferential formation of the α-anomer during 2′-deoxyribosylation.
Figure 4.
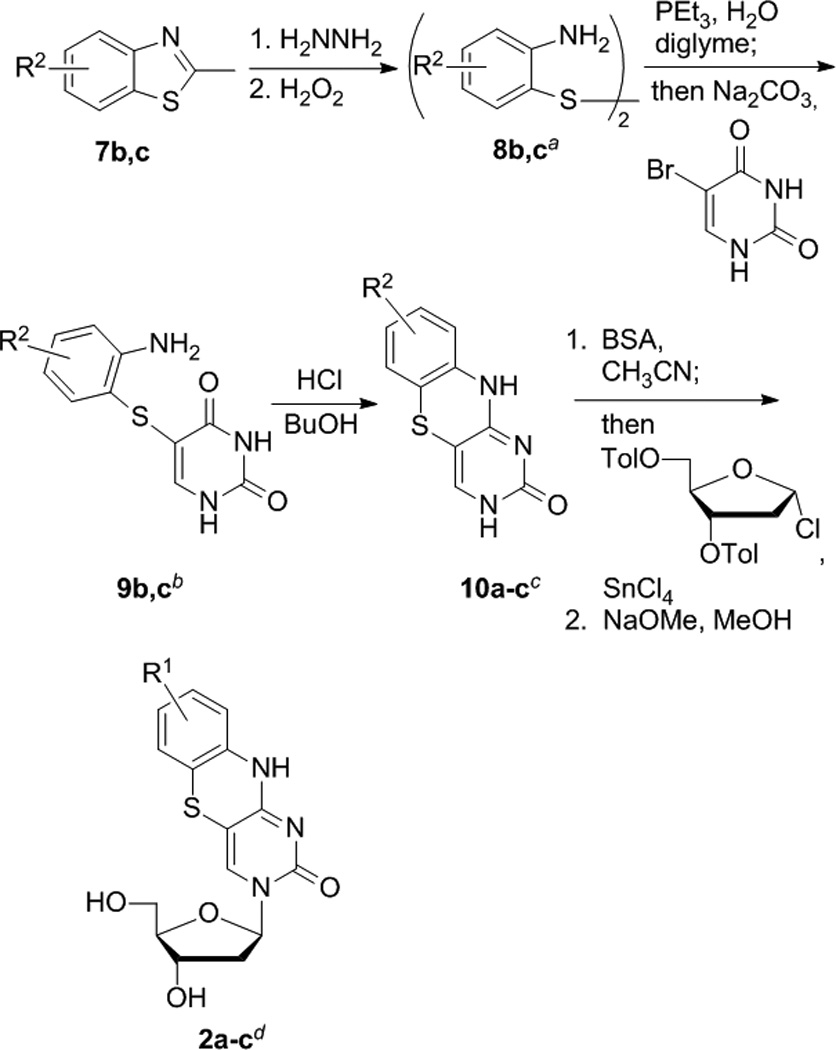
Synthesis of tC analogues. Yields are (a) 8b > 98%, 8c 60%, (b) 9b 86%, 9c 24%, (c) 10b 86%, 10c, 27% (d) 2a 43%, 2b 39%, 2c 8% over two steps.
Photophysical Measurements
In order to compare the effects of substituents and to determine how the new analogues compare with tC and tCO, we carried out photophysical measurements in 1X PBS buffer, solvent mixtures, and 1,4-dioxane (Table 1 and Figure 5). The results show substituent effects that are consistent across the tC and tCO derivatives. (7-Cl)tCO (1e) is now the brightest known member of this family in an aqueous environment, and the solvent effects point to highly desirable properties for biophysical probe development.
Table 1.
Photophysical properties of analogues 1–2 dissolved in 1X PBS buffer and in 1,4-dioxane.
| compound | solvent | λmax, abs / nm | λmax, em / nm | Stokes shift / nm |
ε / L mol−1 cm−1 | Φem | brightness (ε·Φem) | polarity sensitivitya |
|---|---|---|---|---|---|---|---|---|
| 1a tCO | dioxane | 357 | 442 | 85 | 7400 | 0.46 | 3400 | 6 |
| 1X PBS | 355 | 460 | 105 | 6800 | 0.24 | 1600 | ||
| 1b (8-MeO)tCO | dioxane | 360 | 449 | 89 | 4800 | 0.38 | 1800 | 5 |
| 1X PBS | 361 | 486 | 125 | 4000 | 0.05 | 200 | ||
| 1c (7-MeO)tCO | dioxane | 358 | 449 | 91 | 2700 | 0.37 | 1000 | 2 |
| 1X PBS | 360 | 487 | 127 | 3000 | 0.05 | 150 | ||
| 1d (8-Cl)tCO | dioxane | 366 | 417 | 51 | 8500 | 0.49 | 4200 | 2 |
| 1X PBS | 361 | 455 | 94 | 5600 | 0.35 | 2000 | ||
| 1e (7-Cl)tCO | dioxane | 363 | 446 | 83 | 8700 | 0.46 | 4000 | 4 |
| 1X PBS | 357 | 456 | 99 | 6700 | 0.35 | 2300 | ||
| 1f (7-F)tCO | dioxane | 366 | 452 | 86 | 2700 | 0.41 | 1100 | 5 |
| 1X PBS | 360 | 462 | 102 | 1800 | 0.17 | 300 | ||
| 1g tetCO | dioxane | 362 | 438 | 76 | 6700 | 0.43 | 2900 | 7 |
| 1X PBS | 361 | 434 | 73 | 5300 | 0.27 | 1400 | ||
| 2a tC | dioxane | 369 | 472 | 103 | 4300 | 0.34 | 1500 | 3 |
| 1X PBS | 377 | 503 | 126 | 4700 | 0.09 | 400 | ||
| 2b (8-MeO)tC | dioxane | 372 | 490 | 118 | 3800 | 0.28 | 1100 | 7 |
| 1X PBS | 379 | 538 | 159 | 3800 | 0.01 | 40 | ||
| 2c (7-MeO)tC | dioxane | 372 | 477 | 105 | 2200 | 0.27 | 600 | 7 |
| 1X PBS | 379 | 508 | 129 | 2000 | 0.05 | 100 |
Expressed as the slope of Stokes shift plotted against solvent polarity as measured using the Dimroth-Reichardt ET(30) scale. Units are 101 cm−1/(kcal·mol−1).
Figure 5.
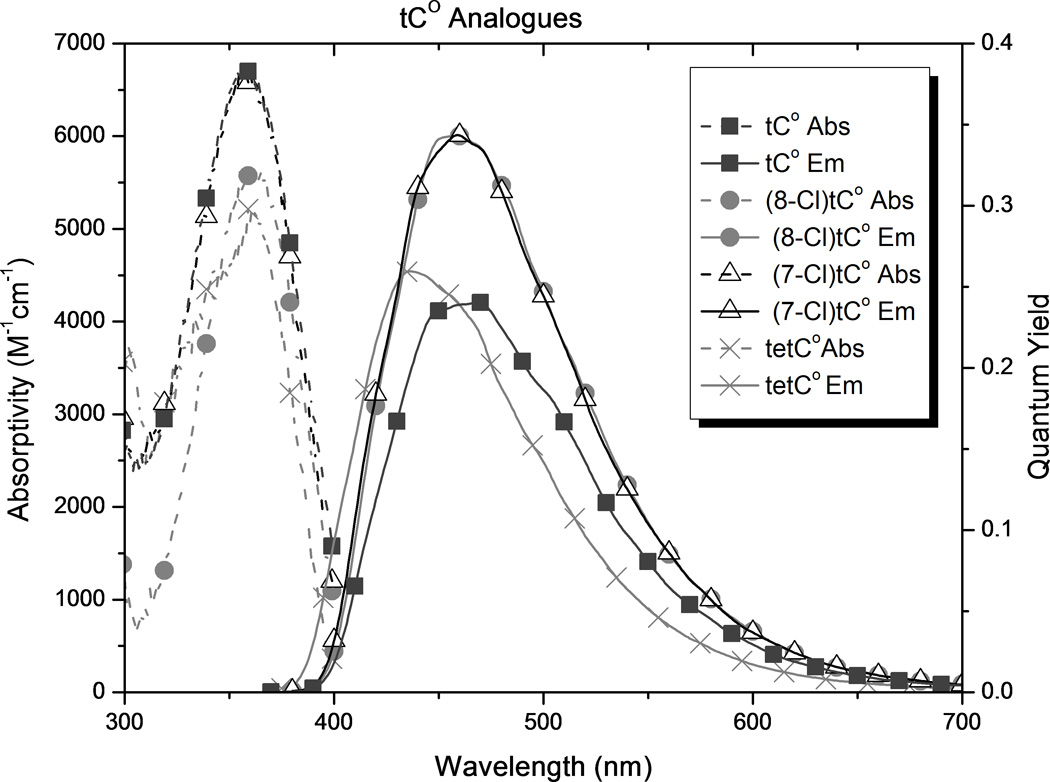
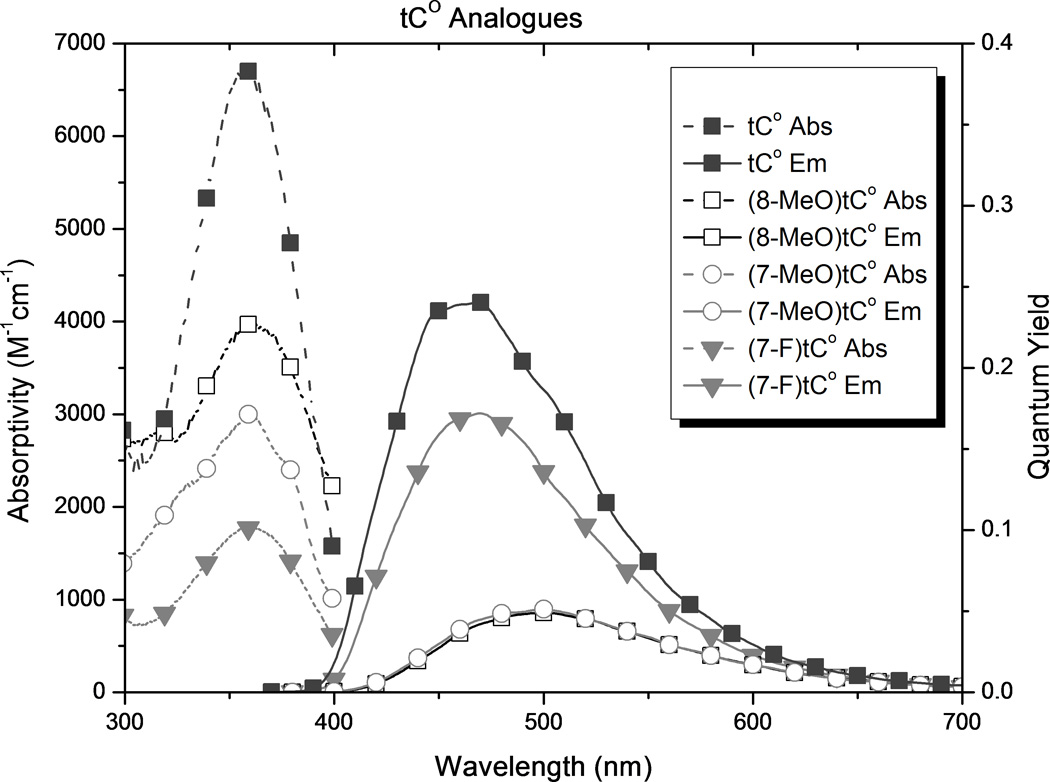
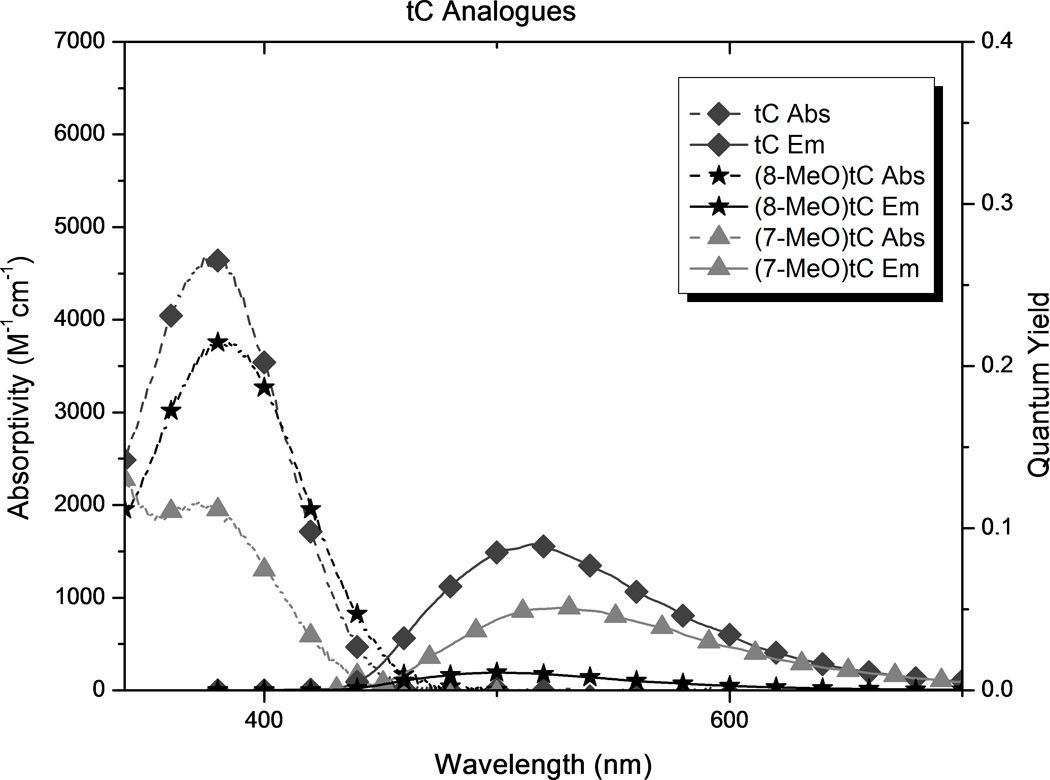
Spectroscopic properties of new analogues (see Figure 2) and comparison with tC and tCO as measured in 1X PBS buffer.
In all cases, substitution of the tC and tCO frameworks had little effect on λabs but a significant influence of λem. The addition of methoxy groups to either the 7- or 8-position of tCO red-shifted λem by 27 and 26 nm respectively ((7-MeO)tCO and (8-MeO)tCO) in buffer. The position of this substituent had a stronger effect on λem of the tC framework, with red-shifts of 35 nm for (8-MeO)tC and only 5 nm for (8-MeO)tC. In 1,4-dioxane, the pattern of the effects is the same with the overall magnitude of the influence diminished. While the influence of methoxy substitution is consistent for tC and tCO, the differences are largest at the 8-position, which is perhaps not surprising because this position is in direct resonance conjugation with the O or S atom.
Substitution of the tCO framework with chloro groups ((7-Cl)tCO and (8-Cl)tCO, 1e and 1d, respectively) resulted in a slight blue-shift of λem by 4–5 nm in aqueous buffer, but in 1,4-dioxane, λem for (7-Cl)tCO was red-shifted by 4 nm and λem for (8-Cl)tCO was blue-shifted by 25 nm. Fluoro substitution at the 7 position of tCO ((7-F)tCO)gives a 10 nm red shift of λem in dioxane but nearly matches the emission of the parent in buffer. Tetracyclic compound tetCO 1g has λem blue-shifted by 4 nm in dioxane and 26 nm in buffer with respect to the parent tCO.
Because the substituents affected λem much more than λabs, the Stokes shift is strongly correlated to λem (r = 0.98). As a result of its significantly red-shifted λem, (8-MeO)tC 2b has the greatest Stokes shift of the tC family in water, 159 nm. Tetracyclic analogue 1g has the smallest Stokes shift of the series of compounds in buffer, 73 nm.
Using mixtures of 1X PBS buffer and 1,4-dioxane, we measured the effect of solvent polarity on Stokes shift, calibrated to the ET(30) scale of Reichardt’s dye (Table 1 and supporting information).[18] The results show that substituents impart significant differences to polarity sensitivity. Substitution generally decreases the polarity sensitivity with the tCO derivatives, whereas it increases polarity sensitivity of the tC compounds. (8-Cl)tCO is now the least solvent-sensitive known member of the tC family, both in terms of this Stokes shift measurement and Φem. In contrast, (8-MeO)tC has the most solvent sensitive photophysical properties.
The introduction of substituents, including extending the conjugated framework, in general causes a decrease in the observed absorptivity of the compounds. An exception to this trend is seen with the chloro substituents in 1,4-dioxane. The decrease is greatest for the methoxy groups when the functionality is in the 7 position, but in buffer the smallest absorptivity is seen when a chloro group is in the 8 position. In contrast, the absorptivity of the methoxy analogues is relatively insensitive to solvent polarity.
Although tC and tCO are known to maintain fluorescence in duplex oligonucleotides (tC becomes slightly brighter and tCO slightly less bright), these analogues show variance in quantum yield depending on the solvent. tC and tCO were found to have a 3-fold and 1.5-fold increase in quantum yield, respectively, when changing from buffer to 1,4-dioxane. Similar increases in Φem on going to non-hydrogen bonding solvents have been reported previously for tC.[52] The presence of the chloro substituent, in either position on tCO, attenuated solvent effects on quantum yield. The opposite is true when the substituents are methoxy groups. Although methoxy substituents attenuated the solvent sensitivity of ε, Φem for the analogues with methoxy groups is much more sensitive to solvent polarity than that for the corresponding parent compounds, with the greatest difference observed for 8-MeO- tC or -tCO (1b and 2b). The analogues with methoxy groups have a relatively high Φem in 1,4-dioxane, but much less so in buffer. Notable, (8-MeO)tC increases in fluorescence intensity by almost 30-fold on going from buffer to dioxane. The presence of a fourth aromatic ring (tetCO) shows solvent sensitivity for Φem comparable to that of parent tCO. In dioxane, (8-Cl)tCO shows a greater quantum yield of emission than tCO. In buffer, tetCO, (7-Cl)tCO, and (8-Cl)tCO, all have greater Φem than the tCO parent.
As a result of the combined effects of substitution on absorptivity and quantum yield, both chloro analogues (7-Cl)tCO and (8-Cl)tCO are brighter than the parent tCO. (7-Cl)tCO is now the brightest known member of the tC family in buffer, more than 40% brighter than tCO. In 1,4-dioxane, the chloro analogues (8-Cl)tCO and (7-Cl)tCO are also the brightest known fluorophores of this family. The least bright analogues in each family are those which have the methoxy group in the 8 position ((8-MeO)tC and (8-MeO)tCO), but these analogues are exciting prospective molecular probes because of the large increase in brightness on going to a less-polar environment. This property should be useful for probing molecular recognition of nucleic acids and may also be useful for reporting on the environmental changes during hybridization of oligonucleotides to form a duplex.
Conclusion
While the chemical literature reports many examples of nucleobase-derived fluorophores, there is a dearth of data for substituent effects on the photophysical properties of the most important of these probes. In this work, we have designed and synthesized a new set of analogues of the tC family using synthetic procedures re-optimized for improved yields and access to greater structural diversity. Photophysical characterization of the analogues in buffer, 1,4-dioxane, and mixtures thereof reveals that substituents have similar effects whether attached to tC or tCO. Generally, chloro compounds gave brighter fluorescence and methoxy compounds gave dramatically increased solvent sensitivity. Because the patterns of effects hold across tC and tCO, future derivatives can be screened using whichever framework offers easier synthetic access, and then the most desired substituents can be added to the other framework, if desired, with predictable effect.
The results of this work encourage a full study of the compatibility of the new probes with DNA- and RNA-processing and polymerizing enzymes and a full photophysical characterization in single-stranded and duplex oligonucleotides. We anticipate that they will make highly valuable additions to the growing toolkit of fluorescent probes for the study of nucleic acid structure, recognition, and metabolism.
Experimental Section
Full details of the synthesis and characterization of new compounds, in addition to modified procedures for the synthesis of tC 2a and tCO 1a, are provided in the supporting information. Photophysical measurements were performed at least in triplicate using samples dissolved in 1X PBS buffer, 1,4-dioxane (spectrophotometric grade), or mixtures thereof. Absorption spectra were recorded using a Varian Cary 100 Bio UV-visible spectrophotometer and fluorescence spectra were recorded using a Varian Cary Eclipse Fluorescence Spectrophotometer. Fluorescence spectra were corrected using published standard curves for the commercially available fluorophores 1,1,4,4-tetraphenyl-1,3-butadiene, Coumarin 153, and 4-(dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran.[53] Absorption and emission spectra for all compounds 1 and 2 are included in the supporting information. Quantum yields were determined using the comparative method of Williams et. al. and a solution of quinine sulfate dissolved in 0.1 M H2SO4 as a reference with an accepted Φem = 0.54.[54] All quantum yield measurements were performed with tC 2a as a second standard to ensure the accuracy of comparisons. Example plots for quantum yield measurements are shown in the supporting information. Polarity sensitivity was measured as the slope of a line defined by plotting Stokes shift against solvent polarity from the Dimroth-Reichardt ET(30) scale. The accuracy of this determination was in some cases limited by changes to the vibrational fine structure that occurred when changing solvents, sometimes manifest in abrupt changes to λmax, em (all relevant spectra are shown in the supporting information).
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the support of the NIH for this research (grant GM093943 to B.W.P. and AI59764 to R.D.K.).
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Wilson JN, Kool ET. Org. Biomol. Chem. 2006;4:4265–4274. doi: 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Z, Chao J, Yu H, Waggoner AS. Nucl. Acids Res. 1994;22:3418–3422. doi: 10.1093/nar/22.16.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barhate N, Cekan P, Massey AP, Sigurdsson ST. Angew. Chem. Int. Ed. 2007;46:2655–2658. doi: 10.1002/anie.200603993. [DOI] [PubMed] [Google Scholar]
- 4.Metzker ML. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 5.Sinkeldam RW, Greco NJ, Tor Y. Chem. Rev. 2010;110:2579–2619. doi: 10.1021/cr900301e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilhelmsson LM. Quart. Rev. Biophys. 2010;43:159–183. doi: 10.1017/S0033583510000090. [DOI] [PubMed] [Google Scholar]
- 7.Riedl J, Ménová P, Pohl R, Orság P, Fojta M, Hocek M. J. Org. Chem. 2012;77:8287–8293. doi: 10.1021/jo301684b. [DOI] [PubMed] [Google Scholar]
- 8.Srivatsan SG, Weizman H, Tor Y. Org. Biomol. Chem. 2008;6:1334. doi: 10.1039/b801054d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivatsan SG, Tor Y. J. Am. Chem. Soc. 2007;129:2044–2053. doi: 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krueger AT, Kool ET. J. Am. Chem. Soc. 2008;130:3989–3999. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]
- 11.Saito Y, Matsumoto K, Takeuchi Y, Bag SS, Kodate S, Morii T, Saito I. Tet. Lett. 2009;50:1403–1406. [Google Scholar]
- 12.Kimoto M, Mitsui T, Harada Y, Sato A, Yokoyama S, Hirao I. Nucl. Acids Res. 2007;35:5360–5369. doi: 10.1093/nar/gkm508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seela F, Jiang D, Xu K. Org. Biomol. Chem. 2009;7:3463. doi: 10.1039/b908017a. [DOI] [PubMed] [Google Scholar]
- 14.Inoue H, Imura A, Ohtsuka E. Nucl. Acids Res. 1985;13:7119–7128. doi: 10.1093/nar/13.19.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingale SA, Pujari SS, Sirivolu VR, Ding P, Xiong H, Mei H, Seela F. J. Org. Chem. 2012;77:188–199. doi: 10.1021/jo202103q. [DOI] [PubMed] [Google Scholar]
- 16.Dierckx A, Miannay F-A, Ben Gaied N, Preus S, Björck M, Brown T, Wilhelmsson LM. Chem. Eur. J. 2012;18:5987–5997. doi: 10.1002/chem.201103419. [DOI] [PubMed] [Google Scholar]
- 17.Shin D, Sinkeldam RW, Tor Y. J. Am. Chem. Soc. 2011;133:14912–14915. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noé MS, Ríos AC, Tor Y. Org. Lett. 2012;14:3150–3153. doi: 10.1021/ol3012327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dziuba D, Postupalenko VY, Spadafora M, Klymchenko AS, Guérineau V, Mély Y, Benhida R, Burger A. J. Am. Chem. Soc. 2012;134:10209–10213. doi: 10.1021/ja3030388. [DOI] [PubMed] [Google Scholar]
- 20.Kim K, Kim H, Moon D, Rhee Y, Kim B. Org. Biomol. Chem. 2013;11:5605–5614. doi: 10.1039/c3ob41222a. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki A, Takahashi N, Okada Y, Saito I, Nemoto N, Saito Y. Bioorg. Med. Chem. Lett. 2013;23:886–892. doi: 10.1016/j.bmcl.2012.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Segal M, Yavin E, Kafri P, Shav-Tal Y, Fischer B. J. Med. Chem. 2013;56:4860–4869. doi: 10.1021/jm301838y. [DOI] [PubMed] [Google Scholar]
- 23.Liu W, Shin D, Tor Y, Cooperman BS. ACS Chem. Biol. 2013 doi: 10.1021/cb400256h. 130718131602008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singer M, Jäschke A. J. Am. Chem. Soc. 2010;132:8372–8377. doi: 10.1021/ja1024782. [DOI] [PubMed] [Google Scholar]
- 25.Pawar MG, Nuthanakanti A, Srivatsan SG. Bioconjugate Chem. 2013;24:1367–1377. doi: 10.1021/bc400194g. [DOI] [PubMed] [Google Scholar]
- 26.Astakhova IV, Ustinov AV, Korshun VA, Wengel J. Bioconjugate Chem. 2011;22:533–539. doi: 10.1021/bc1005027. [DOI] [PubMed] [Google Scholar]
- 27.Singleton SF, Shan F, Kanan MW, McIntosh CM, Stearman CJ, Helm JS, Webb KJ. Org. Lett. 2001;3:3919–3922. doi: 10.1021/ol0167863. [DOI] [PubMed] [Google Scholar]
- 28.St Amant AH, Bean LA, Guthrie JP, Hudson RHE. Org. Biomol. Chem. 2012;10:6521. doi: 10.1039/c2ob25678a. [DOI] [PubMed] [Google Scholar]
- 29.Dallmann A, Dehmel L, Peters T, Mügge C, Griesinger C, Tuma J, Ernsting NP. Angew. Chem. Int. Ed. 2010;49:5989–5992. doi: 10.1002/anie.201001312. [DOI] [PubMed] [Google Scholar]
- 30.Dash C. Nucl. Acids Res. 2004;32:1539–1547. doi: 10.1093/nar/gkh307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson JP, Angerer B, Loeb LA. BioTechniques. 2005;38:257–264. doi: 10.2144/05382RR02. [DOI] [PubMed] [Google Scholar]
- 32.Kato T, Kashida H, Kishida H, Yada H, Okamoto H, Asanuma H. J. Am. Chem. Soc. 2013;135:741–750. doi: 10.1021/ja309279w. [DOI] [PubMed] [Google Scholar]
- 33.Iqbal A, Arslan S, Okumus B, Wilson TJ, Giraud G, Norman DG, Ha T, Lilley DM. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin K-Y, Jones RJ, Matteucci M. J. Am. Chem. Soc. 1995;117:3873–3874. [Google Scholar]
- 35.Wilhelmsson LM, Holmén A, Lincoln P, Nielsen PE, Nordén B. J. Am. Chem. Soc. 2001;123:2434–2435. doi: 10.1021/ja0025797. [DOI] [PubMed] [Google Scholar]
- 36.Engman KC, Sandin P, Osborne S, Brown T, Billeter M, Lincoln P, Nordén B, Albinsson B, Wilhelmsson LM. Nucl. Acids Res. 2004;32:5087–5095. doi: 10.1093/nar/gkh844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandin P, Wilhelmsson LM, Lincoln P, Powers VEC, Brown T, Albinsson B. Nucl. Acids Res. 2005;33:5019–5025. doi: 10.1093/nar/gki790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stengel G, Gill JP, Sandin P, Wilhelmsson LM, Albinsson B, Nordén B, Millar D. Biochemistry. 2007;46:12289–12297. doi: 10.1021/bi700755m. [DOI] [PubMed] [Google Scholar]
- 39.Gardarsson H, Kale AS, Sigurdsson ST. ChemBioChem. 2011 doi: 10.1002/cbic.201000478. [DOI] [PubMed] [Google Scholar]
- 40.Sandin P, Borjesson K, Li H, Martensson J, Brown T, Wilhelmsson LM, Albinsson B. Nucl. Acids Res. 2008;36:157–167. doi: 10.1093/nar/gkm1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Börjesson K, Preus S, El-Sagheer AH, Brown T, Albinsson B, Wilhelmsson LM. J. Am. Chem. Soc. 2009;131:4288–4293. doi: 10.1021/ja806944w. [DOI] [PubMed] [Google Scholar]
- 42.Preus S, Kilså K, Wilhelmsson LM, Albinsson B. Phys. Chem. Chem. Phys. 2010;12:8881. doi: 10.1039/c000625d. [DOI] [PubMed] [Google Scholar]
- 43.Stengel G, Purse BW, Wilhelmsson LM, Urban M, Kuchta RD. Biochemistry. 2009;48:7547–7555. doi: 10.1021/bi9006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stengel G, Urban M, Purse BW, Kuchta RD. Anal. Chem. 2010;82:1082–1089. doi: 10.1021/ac902456n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin K-Y, Matteucci MD. J. Am. Chem. Soc. 1998;120:8531–8532. [Google Scholar]
- 46.Koga Y, Fuchi Y, Nakagawa O, Sasaki S. Tetrahedron. 2011;67:6746–6752. [Google Scholar]
- 47.Sandin P, Lincoln P, Brown T, Wilhelmsson LM. Nat Protoc. 2007;2:615–623. doi: 10.1038/nprot.2007.80. [DOI] [PubMed] [Google Scholar]
- 48.De Napoli L, Messere A, Montesarchio D, Piccialli G, Santacroce C. Nucleosides Nucleotides. 1991;10:1719–1728. [Google Scholar]
- 49.Hoffer M. Chem. Ber. 1960;93:2777–2781. [Google Scholar]
- 50.Niedballa U, Vorbrüggen H. J. Org. Chem. 1974;39:3654–3660. doi: 10.1021/jo00939a008. [DOI] [PubMed] [Google Scholar]
- 51.Hwang GT, Romesberg FE. J. Am. Chem. Soc. 2008;130:14872–14882. doi: 10.1021/ja803833h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilhelmsson LM, Sandin P, Holmén A, Albinsson B, Lincoln P, Nordén B. J. Phys. Chem. B. 2003;107:9094–9101. [Google Scholar]
- 53.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; 2006. [Google Scholar]
- 54.Williams ATR, Winfield SA, Miller JN. Analyst. 1983;108:1067–1071. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.