

Abstract
Aims/Introduction: Human islet polypeptide S20G mutation (hIAPPS20G) is associated with earlier onset type 2 diabetes and increased amyloidogenicity and cytotoxicity in vitro vs wild‐type hIAPP (hIAPPWT), suggesting that amyloidogenesis may be pathogenic for type 2 diabetes. We compared the contributions of hIAPPS20G and hIAPPWT toward intra islet amyloid formation and development of type 2 diabetes in a unique physiologic knock‐in mouse model.
Materials and Methods: We replaced the mouse IAPP gene (M allele) with hIAPPWT (W allele) and hIAPPS20G (G allele) via homologous recombination and backbred transgenic mice against C57Bl/6 strain 5 generations to minimize genetic variation. Mice (3 month old) were maintained on control (CD) or high fat diet (HFD) for 15 months and studied at 3 month intervals by oral glucose tolerance testing (OGTT) and pancreas histology to assess glucose homeostastis, amyloidogeneisis, islet mass, β cell replication, and apoptosis.
Results: IAPP blood levels were indistinguishable in all mice. WW and GW mice maintained on both diets lacked intraislet amyloid at all ages. On both diets relative to MM controls WW and GW mice exhibit glucose intolerance (P < 0.008) with no differences in insulin secretion. However, GW mice secreted significantly more insulin (P < 0.03 that WW mice on both diets throughout the study. By 12 months on the high fat diet all mice increased their β cell mass about 3‐fold and were indistinguishable.
Conclusions: Physiologic expression of hIAPPWT and hIAPPS20G in C57Bl/6 mice produces mild glucose intolerance with inappropriately normal insulin secretion that is independent of intraislet amyloid formation. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2011.00166.x, 2011)
Keywords: Islet amyloid polypeptide, Transgenic mice, Type 2 diabetes
Introduction
There has been renewed interest in the role of islet amyloidogenesis in the pathogenesis of type 2 diabetes.1,2 Pancreatic amyloid is found in approximately 90% of type 2 diabetes patients3 with loss of up to 50% of β cell mass.3 Studies in macaques4 and humans5,6 suggest that loss of β cell mass and islet amyloid accumulation are interdependent. The major constituent of islet amyloid in humans is derived from islet amyloid polypeptide (hIAPP).7 While hIAPP spontaneously forms fibrils in vitro, rodent IAPPs do not.8 We demonstrated that expression of hIAPP in COS‐1 cells results in the accumulation of large deposits of intracellular amyloid and cell death by apoptosis.9
Asians with premature onset type 2 diabetes harbor a mutation in the hIAPP gene (S20G), providing a causal link between this gene and disease.10 The mutation is rare affecting 1.9–2.6% of type 2 diabetics11,12 and 0.8% of non‐diabetic control subjects.11 We showed that hIAPPS20G is more cytotoxic than wild‐type hIAPP (hIAPPWT) when expressed in COS‐1 cells and this is correlated with the increased in vitro amyloidogenicity of this peptide.13 While the mechanism of amyloid‐associated cell death is unknown, recent evidence indicates that oligomeric intermediates forming nonselective, ion‐permeable channels in phospholipid membranes,14–16 lead to cell death.
Studies of hIAPP expression in transgenic mice support the hypothesis that islet amyloidogenesis plays a role in β cell loss. Mice homozygous for the hIAPP gene express high levels of hIAPP and develop diabetes mellitus.17 The islets of these mice exhibit amorphous hIAPP deposits but lack amyloid. Treatment of hIAPP+/− mice with growth hormone and dexamethasone to induce insulin resistance resulted in islet amyloidosis that preceded β cell dysfunction.18 Independent studies have demonstrated extensive islet amyloid deposits in male transgenic mice with approximately 50% of these animals becoming hyperglycemic.19,20 When hIAPP+/− mice were crossed with agouti viable yellow (Avy/a) mice that exhibit obesity and insulin‐resistance, hIAPP+/Avy males displayed fasting hyperglycemia at 90 days and progressed to severe hyperglycemia within 1 year.21 These animals exhibited 10‐ to 20‐fold lower plasma and pancreatic insulin levels, large islet amyloid deposits, and an 80% deficit in β cell mass.22 Also, hIAPP transgenic rats develop diabetes within 5–10 months of age and exhibit a 60% deficit in β cell mass due to increased β cell apoptosis.23 Recent studies have demonstrated an up‐regulation and nuclear localization of CHOP, suggesting that endoplasmic reticulum (ER) stress‐induced apoptosis accounts for loss of β cell mass in hIAPP transgenic animals.24
In order to compare the relative contributions of hIAPPWT and hIAPPS20G in a physiologic manner, we knocked‐in the corresponding expression constructs for each of these genes into the mouse (m) IAPP locus via homologous recombination. This replaces the non‐amyloidogenic mIAPP gene with the corresponding human IAPP and places each of these inserted genes under the control of the endogenous mIAPP promoter. This approach ensures that the human genes will be expressed at physiologic levels and avoids the confounding problems with traditional transgenic animal experiments, including multiple copy insertions that affect expression levels, integration of genes near other transcriptional control elements that can adversely influence expression, and/or random knock‐out of genes that affect phenotype.
Materials and Methods
Transgenic Mice
All experiments with mice were approved by the Mayo Institutional Animal Care and Use Committee. The complete description of vector construction, transformation of ES cells and mice generation are provided in the supplemental data (Appendix S1). The homozygous hIAPP wild‐type knock‐in mice are designated WW (where W = the wild‐type hIAPP allele) and the homozygous S20G mutant hIAPP knock‐in mice are designated GG (where G = the S20G mutant hIAPP allele). GW knock‐in mice represent the heterozygous mice containing a wild‐type and S20G mutant hIAPP allele. The control mice designated MM (for mouse IAPP allele) were derived from the matings of MW × MW and MG × MG heterozygotes. All mice (MM, WW and GG) were backbred five generations against C57Bl/6 mice to 96.9% congenicity to diminish potential confounding effects that can arise due to 129Sv/E chimaerism.25,26
Oral Glucose Tolerance Testing
Animals were fasted for 12 h and given 1 g/kg glucose via oral gavage. Blood (30 μL) was obtained from the tail vein at 0, 5, 10, 15 and 30 min for glucose and insulin determinations. Blood (5 μL) was obtained at 60 and 120 min for glucose determinations. Insulin was determined using a mouse insulin ELISA assay (Crystal Chem, Inc., Downers Grove, IL, USA). Glucose was determined by glucometer (OneTouch Ultra, Lifescan, Milpitas, CA, USA).
Dietary Regimes and Experimental Design
Male MM, WW and GW mice were divided into groups (48 animals/group) and fed either a control diet consisting of standard mouse chow (PicoLab Rodent Diet 20, Brentwood, MO, USA), containing 4.5% fat or a high fat diet, consisting of 42% fat (TD.88137; Harlan Laboratories, Madison, WI, USA) beginning at 3 months of age. At each time point (3, 9, 6, 12, and 15 months) a minimum of four mice were killed and analyzed. GW mice were employed to reflect the genotype of humans carrying a single copy of the mutated S20G hIAPP allele. Animals were continued on this diet for a period of 15 months and were subjected to OGTT examination at 3 month intervals.
Pancreas Isolation and Tissue Fixation
Tissue samples were collected from four mice of each group at 3, 6, 9, 12, 15 and 18 months of age. Mice were anesthetized with 2.0% isoflurane during harvesting to minimize degradation of critical cellular components. The left lobe of the pancreas was teased free of surrounding tissue and fat. The pancreas was rapidly weighed, immediately fixed in 10% phosphate buffered formalin, and embedded in paraffin. In some cases, the pancreas was divided by transverse sectioning with half of the pancreas being placed in Trumps solution for electron microscopy processing and the other half fixed in 10% phosphate buffered formalin and embedded in paraffin.
Immunohistochemistry
To evaluate β cell mass, β cell apoptosis, and β cell replication, adjacent serial 5 μm sections were immunostained for insulin, caspase‐3, and Ki67, respectively. Insulin immunostaining was achieved using polyclonal guinea pig anti‐porcine insulin (DAKO, Carpenteria, CA, USA). Anti‐activated caspase‐3 antibody (CP 229) detecting the large fragment (17/19 kDa) of activated caspase‐3 was obtained from Biocare Medical (Concord, CA, USA).27 The anti‐Ki67 antibody (M7240) was obtained from DAKO. Immunostaining was performed at the Mayo TACMA core laboratories. Stained slides were then scanned with a NanoZoomer Digital Pathology System (C9600) instrument. In each case pancreata from four animals were obtained and 3–4 sections separated by 200 μm were analyzed for each animal, resulting in the assessment of a total of 100–300 islets for each condition. The areas of all islets were measured in each section and the results were reported as events/μm2 × 106.
Determination of β Cell Mass
Assessment of β cell mass at 6 and 12 months was determined by measuring the relative cross sectional area of insulin‐positive tissue compared to the area of exocrine tissue. The percent islet area was then multiplied by the weight of the pancreas.28 Five 5 μm sections of each pancreas obtained at 200 μm intervals were stained for insulin and digital images captured as described above. Islet and total pancreas areas were analyzed using WebSlide Browser 4.00 software (Bacus Laboratories, Lombard, IL, USA). Total β cell mass was then determined by the equation: Massβ cell = ∑ [(islet area/total pancreas area)]sections× pancreas weight (mg).
Sequence Analysis
Blood from randomly selected 6 month old male and female mice was obtained by heart puncture for hIAPP analysis and the pancreas resected for total RNA isolation (TRIzol; Invitrogen, Carlsbad, CA, USA). Circulating levels of IAPP were determined in blood samples using a human amylin ELISA (Millipore, Billerica, MA, USA) assay kit. Total RNA was reverse transcribed using AMV reverse transcriptase and the cDNAs were sequenced.
Congo Red and Thioflavin S staining
Thioflavin S staining was performed on 5 μm sections according to the method of Hull et al.29 Congo Red staining was performed according to the method of Puchtler et al.30
Statistical Analyses
All timed series data was subjected to repeated measures analysis of variance (anova). All other data was analyzed by anova and differences among groups were determined by post hoc Bonferroni‐Dunn t tests. Differences were deemed significant with P < 0.05. Statistical analyses were performed using SAS software (SunOS 9.1.3) (SAS Institute Inc., Cary, NC, USA).
Results
Characterization of WW and GG‐KI Mice
RNA from various tissues obtained from the WW mice were analyzed by RT‐PCR for the presence of hIAPP and mouse insulin mRNA (Figure 1a). Human IAPPWT and mouse insulin were detected only in the pancreas of WW mice. Identical results were obtained with RT‐PCR analyses of RNA from GG‐KI mice (not shown). We verified the sequences of the human IAPPWT and hIAPPS20G mRNAs via RT‐PCR and cDNA sequencing in the WW and GG mice (Figure 1b). These data confirm the appropriate genotypic and tissue‐specific expression patterns of the WW and GG mice. Using an IAPP‐specific ELISA, which detects rat IAPP (identical to mouse IAPP) and both hIAPPWT and hIAPPS20G equally well13, circulating hIAPP levels were determined in 6 month old male and female animals in the fed state (Figure 1c). No significant differences in circulating IAPP levels were observed in the MM, WW and GG mice.
Figure 1.
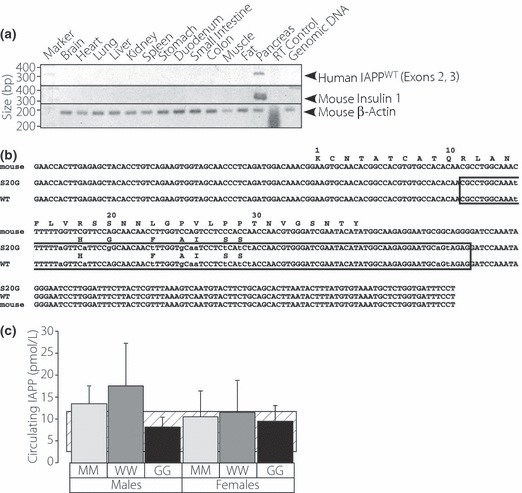
Characterization of hIAPP expression in WW mice. (a) Distribution of hIAPPWT mRNA in various tissues from the W/W knock in mouse via RT‐PCR using IAPP (upper panel), insulin (middle panel) and β‐actin (lower panel) primers. (b) Sequences of mouse IAPP, hIAPPWT, and hIAPPS20G mRNAs derived from cDNA sequence of RT‐PCR products of WW and GG mouse pancreas RNAs. Sequences within the boxed region represent the sequences engineered into the mouse genomic IAPP locus. (c) Circulating levels of randomly selected wild‐type and S20G mutant hIAPP and mIAPP in MM, WW and GG mice that were maintained on control diet. The striped area represents the typical range of values from the literature.33–35 Error bars represent S.D.
Effect of Diet on Glucose Homeostasis in WW and GW Mice
Male MM, WW and GW mice were maintained on either standard mouse chow or a high fat diet and followed for a period of 15 months. GW animals gained about 8% (P < 0.003) more weight than either MM or WW mice throughout the period of observation when maintained on control diet (Figure 2a and b). When maintained on high fat diet WW mice were slightly, but significantly smaller than either MM or GW mice (P < 0.0086) (Figure 2a and b). The reasons for these differences in weight gain among the WW and GW animals are unknown.
Figure 2.
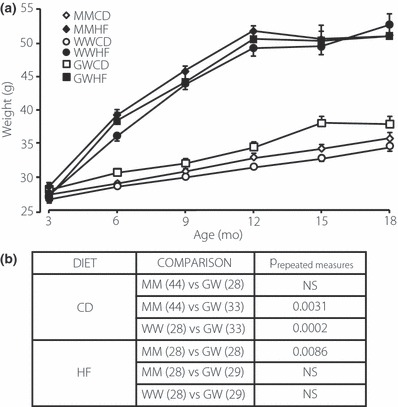
(a) Weights of MM, WW and GW mice on control (open symbols) vs high fat diet (solid symbols) over a period of 15 months. (b) Outcomes of repeated measures anova of the data in A. Numbers within parentheses represent the number of animals in each group.
Figure 3a and b show glucose and insulin values, respectively, from representative OGTT analyses for all animals on control and high fat diet at 9 months. Figure 3c and d show the areas under the glucose curves for mice on control and high fat diet, respectively, and were compiled from the OGTT analyses over the entire study period. WW and GW mice exhibit mild glucose intolerance on both control (P <0.007) (Figure 3c) and high fat diet (P < 0.004) (Figure 3d). This mild glucose intolerance manifests early by 3–6 months of age and persists throughout the study period. Figure 3e and f show the areas under the insulin curves for mice on control and high fat diet, respectively and were compiled from the OGTT analyses over the entire study period. On both control and high fat diet there were no significant differences in insulin secretion between MM and WW or MM and GW mice. However, under both dietary regimes, the WW mice did secrete significantly less insulin (15.5%, P < 0.0297 on control diet and 31.2%, P < 0.0132 on high fat diet) than the GW mice.
Figure 3.
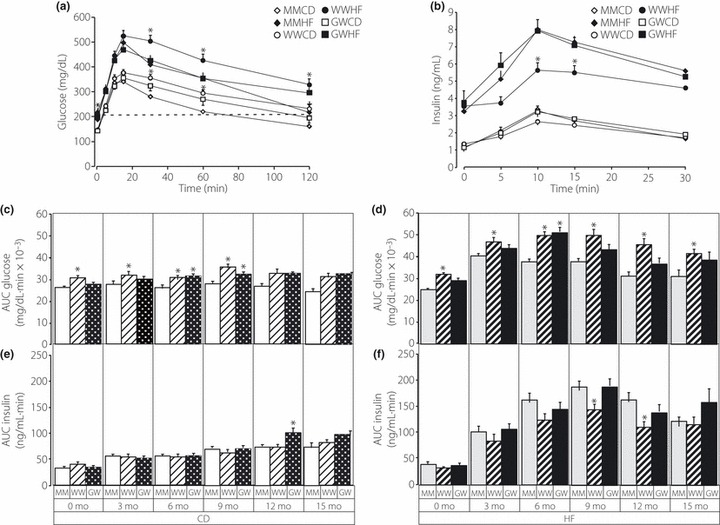
(a) Blood OGTT glucose values of MM, WW and GW mice on control (CD) or high fat (HF) diet after 9 months of treatment. (b) Blood insulin values of MM, WW and GW mice on control or high fat diet after 9 months of treatment. Asterisks indicate significance at P < 0.05 determined from anova and post hoc Bonferroni‐Dunn t tests. Comparisons are within group exclusively for control and high fat diet. (c) Total circulating glucose derived from the areas under the curve (AUC) of individual OGTT curves for MM (open diamond), WW (open circle) and GW (open square) mice maintained on normal diet. The numbers of MM, WW and GW mice were 24, 12 and 22, respectively. (d) Total circulating glucose derived from the areas under the curve (AUC) of individual OGTT curves for MM (solid diamond), WW (solid circle) and GW (solid square) mice maintained on high fat diet. The numbers of MM, WW and GW mice were 15, 11 and 10, respectively. (e) Total circulating insulin derived from the areas under the curve (AUC) of individual OGTT curves for MM (open diamond), WW (open circle) and GW (open square) mice maintained on normal diet. The numbers of MM, WW and GW mice were 24, 16 and 22, respectively. (f) Total circulating insulin derived from the areas under the curve (AUC) of individual OGTT curves for MM (solid diamond), WW (solid circle) and GW (solid square) mice maintained on high fat diet. The numbers of MM, WW and GW mice were 15, 11 and 14, respectively. Individual repeated measures anova comparisons are shown in the insets where prm (p repeated measures) is the probability of difference.
Figure 4a and b depict fasting blood glucose levels of mice prior to administration of glucose at 3‐month intervals throughout the study period. No differences were observed in fasting blood glucose levels for MM, WW and GW mice on the control diet (Figure 4b). On the high fat diet both WW and GW mice exhibited significantly elevated fasting blood glucose levels (15–20%) compared to MM mice. For the WW mice on a high fat diet the fasting blood glucose remained at about 200 mg/dL between 3–15 months. However, fasting blood glucose levels for both the MM and GW mice began to decline at about 9 months and decreased to approximately 150 mg/dL at 15 months of age, equivalent to the levels of 3‐month‐old animals. While the MM and GW mice adjust to the high fat diet by lowering their fasting blood glucose level, the WW appear to be less capable of responding.
Figure 4.
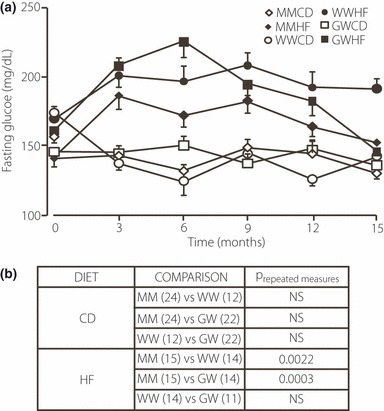
(a) Fasting blood glucose levels of MM, WW and GW mice fed control (open symbols) or high fat (solid symbols) diet obtained at 3, 6, 9, 12 and 15 months. (b) Outcomes of repeated measures anova of the data in A. Numbers within parentheses represent the number of animals in each group.
Islet Histomorphometry
Figure 5 shows representative slides of insulin, activated caspase‐3 and Ki67 immunohistochemical staining that were used for histomorphometric analysis of β cell mass,28 islet cell apoptosis,31 and islet cell replication,32 respectively (Figure 6). At 6 months on the high fat diet only the GW mice had significantly elevated their β cell mass by 1.8‐fold, whereas the MM and WW mice displayed 1.5‐ and 1.1‐fold increases in β cell mass, respectively, that were not significant (Figure 6a). While the data suggest there might have been a delay in acquiring increased β cell mass in the WW mice at 6 months, and thus inadequate compensation, by 12 months it had increased to the same level as the MM control mice. Thus the MM, WW and GW mice increased their β cell mass by 3.2‐, 3.4‐, and 1.8‐fold, respectively, after 12 months on a high fat diet (Figure 6a) and did not differ, although the GW mice showed a trend for somewhat decreased β cell mass relative to MM and WW mice.
Figure 5.
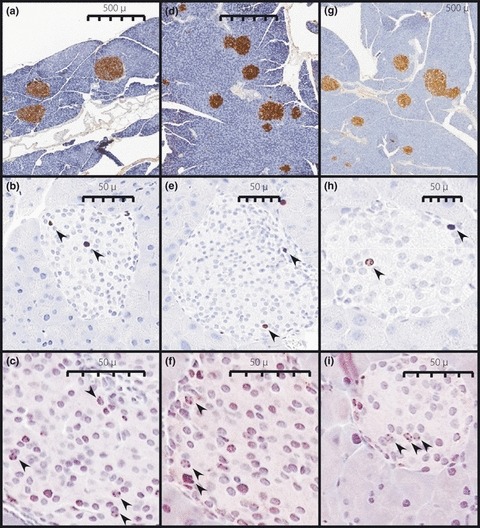
Representative insulin (a, d, g), Ki67 (b, e, h) and activated caspase‐3 (c, f, g) immunostaining. Arrows indicate positively stained nuclei. Images are from MM (a–c), WW (d–f), and GW (g–i) mice maintained on high fat diet for 12 months.
Figure 6.
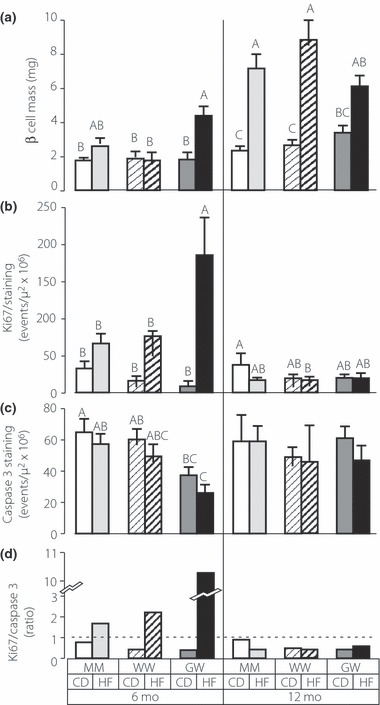
Pancreas histomorphometry to assess β cell mass determination, replication index (Ki67) and apoptosis index (caspase‐3) of MM, WW and GW mice maintained on control (CD) or high fat (HF) diet for 6 months or 12 months. β cell mass (a) Ki67 immunostaining (b) activated caspase‐3 immunostaining (c) and ratio of Ki67 to caspase‐3 (d). Ki67 and caspase 3 staining was only assessed within islets. β cell Mass = Insulin‐Stained Islet Area/Total Tissue Area x Pancreas Weight. β cell and tissue areas were assessed using WebSlide Browser Software (Bacus Laboratories, Inc., Lombard, IL, USA). Data is from examination of five tissue sections at 200 μm intervals for each of four mice in each genotype and diet. Upper case letters represent the statistical classes derived from anova and post hoc Bonferonni‐Dunn t tests. Groups with unique letter designations are significantly different at the P < 0.05 level. For example, A is significantly different from B, but neither A or B is different from AB.
β cell replication and apoptosis in the pancreata of the MM, WW and GW mice were assessed by immunostaining for the Ki67 antigen and activated caspase‐3 (Figure 6b and c). At 6 months there was increased replication in islets of MM, WW, and GW mice maintained on high fat diet, but the increase was only significant for the GW mice. The increased islet replication in the GW mice was reflected in significantly increased β cell mass at 6 months (Figure 6a). By 12 months no differences were observed in islet replication of MM, WW or GW mice, although at this time the MM and WW animals had clearly increased their β cell mass significantly (Figure 6a). No differences could be detected in the apoptotic rates of islet cells in the MM or WW mice. However, islet apoptosis rates of the GW mice on high fat diet at 6 months were significantly lower than the MM or WW mice. Thus increased proliferation and decreased apoptosis have likely contributed to the increased β cell mass at 6 months for the GW animals on high fat diet. The relative ratio of islet cell proliferation to apoptosis is shown in Figure 6d. Only MM mice on control diets have islet proliferation‐apoptosis ratios that approach unity. At 6 months on a high fat diet all groups of mice exhibit markedly increased proliferation, whereas at 12 months on a high fat diet all groups exhibit markedly decreased proliferation and increased apoptosis.
Islet tissue sections stained with Congo Red and thioflavin S were negative for islet amyloid at all ages (3, 6, 9, 12 and 15 months). Separate tissue samples were prepared for electron microscopy (EM) and stained by immunogold for either insulin, hIAPP, or both, to identify islets. Examination of over 300 independent EM images failed to reveal evidence for amyloid fibers within islets.
Discussion
We have previously demonstrated with in vitro experiments that the mutant hIAPPS20G is more amyloidogenic and cytotoxic than hIAPPWT when expressed in COS‐1 cells.13 We have hypothesized that the increased amyloidogenicity and cytotoxicity of hIAPPS20G are interdependent pathogenic factors that contribute to the early onset type 2 diabetes seen in some individuals that possess the hIAPP S20G mutation.13 In order to directly test this hyposthesis in a physiologic model, we knocked‐in the hIAPPS20G and hIAPPWT genes into the mIAPP locus, thereby replacing the mIAPP with the two human counterparts. This model provides control of hIAPP gene copy number and insertion site, ensuring equivalent expression of WT and S20G mutant hIAPPs via the endogenous mouse IAPP promoter, which was directly demonstrated (Figure 1) and shown to be comparable to previous reports.33–35 All mice have been backbred five generations against C57Bl/6 mice to minimize the confounding contributions of genetic chimaerism associated with the generation of targeted gene replacement in mice.25,26
Contrary to our expectations, intraislet amyloid was undetectable in either the WW or GW animals using traditional Congo Red and Thioflavin S staining, and extensive analysis of electron microscopic images of insulin and/or hIAPP immunogold‐labeled sections (not shown). This result contrasts with that of Westermark et al.,36 who demonstrated islet amyloid formation in hIAPP transgenic mice that were crossed with a mouse IAPP (mIAPP) knock‐out mouse to yield mice expressing hIAPP without mIAPP. In this case the male hIAPP(+/+)/mIAPP(−/−) mice developed amyloid sooner than hIAPP transgenic mice retaining the mIAPP gene. We do not know what explains the difference between these two models, although it may be related to efficiency of hIAPP expression, since hIAPP is under the direction of the more efficient rat insulin II promoter in the transgenic hIAPP(+/+)/mIAPP(−/−) animals. Alternatively, differences in genetic background may contribute to amyloidogenesis in ways that are not yet understood. In this regard it is noteworthy that mouse strain introduces marked differences in the appearance of islet amyloid deposits among several hIAPP transgenic models.1 Also in support of the concept that genetic factors contribute to amyloidogenesis, we did observe amyloid fibrils in our 15 month old 129Sv/E chimaeric founder animals. Amyloid fibrils were observed within WW and GW founders on both control and high fat diet as shown in the supplemental data (Appendix S2). It is important to note that the penetrance of the S20G mutation with premature onset type 2 diabetes is low and appears to be restricted to a genetic background predisposing to normal onset type 2 diabetes,10 although it is unknown whether these genetic differences affect amyloidogenesis in these patients.
Despite the absence of amyloid within the knock‐in mice, both WW and GW mice exhibit mild glucose intolerance without evidence of insulin insufficiency on both control and high fat diets. Based on the fasting blood glucose data (Figure 4), the WW mice appear to be somewhat more glucose intolerant up to 15 months, while GW mice appear to be able to ameliorate their glucose intolerance after 9 months. The WW mice secreted significantly less insulin than GW mice, but not MM control mice, and no differences in β cell mass was observed except that at 6 months GW mice had significantly elevated β cell mass (Figure 6a). The latter effect may help to explain the decreased insulin secretion observed in WW mice compared to GW mice. In addition, the decreased insulin secretion in WW vs GW might be explained if the S20G mutation exhibited suppressed insulinostatic actions relative to wild‐type hIAPP.37,38 All mice compensated equally to the high fat diet by increasing their β cell mass approximately 3‐fold. This result contrasts to several transgenic rodent models whereby over‐expression of hIAPP results in loss of β cell mass.22,23,39,40 These differences may be the result of over‐expression of hIAPP in the transgenic models and/or due to differences in genetic background that may contribute to amyloidogenesis.
After 6 months on high fat diet replication rates (Figure 6b) and β cell mass (Figure 6a) were significantly elevated only in GW animals. The rates of islet apoptosis did not differ significantly among the MM and WW mice at 6 months and did not differ by diet. However, the GW mice exhibited significantly lower rates of islet apoptosis at 6 months compared to MM mice on control and high fat diet, respectively (Figure 6c). This, coupled with higher replication in the GW animals at 6 months, may account for the significant gain in β cell mass in this group. In addition, islet replication exceeded islet apoptosis at 6 months (Figure 6d), indicating that at this time β cell mass appears to be controlled largely at the level of replication. At 12 months replication rates in all groups on the high fat diet had decreased and were not different from animals on the control diet. Moreover, at 12 months islet apoptosis exceeded replication, suggesting that the maximal β cell mass may have been achieved before 12 months. While the shift in favor of apoptosis would predictably lead to decreasing β cell mass as the mice continue to age, this did not lead to abnormal control of fasting blood glucose the MM and GW animals at 15 months (Figure 4).
In summary, both wild‐type and mutant S20G hIAPP induce glucose intolerance in mice when expressed at physiologic levels under conditions that do not result in intraislet amyloid deposition. These data suggest that that hIAPP is a factor that contributes to the pre‐diabetic condition, particularly glucose intolerance. The hIAPP‐induced glucose intolerance in the absence of compensatory insulin secretion resembles diabetes in humans. The reasons for lack of compensatory insulin secretion remain unknown, but could be related to differences in physiologic actions of hIAPP or a β cell dysfunction. A major question that is raised by the current studies is whether certain genetic and/or environmental conditions exist that may enhance the toxic potential of the amyloidogenic pathway.
Supplementary Material
Appendix S1 Materials and Methods, providing detailed description of vector construction for the homologous replacement of the mouse IAPP gene for the wild‐type and S20G mutant hIAPP in mouse embryonic stem cells and subsequent generation of the wild‐type and S20G mutant hIAPP knock‐in mice.
Appendix S2 Intraislet amyloidogenesis in WW and GW chimaeric 129Sv/E founder mice at 15 months.
Supporting info item
Acknowledgements
This work was supported by National Institutes of Health grant DK56890 (N.L.E.) and funds from the Mayo Foundation. Drs H.J. Hiddinga and S. Sakagashira are co‐equal first authors on this manuscript. None of the authors of this manuscript has any financial interests or conflicts of interests with respect to this manuscript to declare. The authors extend their appreciation to Dr Michael C. Blanco and the Department of Comparative Medicine staff for help with animal care. We are indebted to Dr Wilma L. Lingle and the staff of the Mayo Tissue and Cell Molecular analysis core, who performed all of the immunohistochemical procedures. We thank Jon Charlesworth and the staff of the Mayo Electron Microscopy Core Facility for helping us with obtaining electron micrographs and immunogold staining of pancreata. We are grateful to Mike Deeds for providing expert instruction for retrieving pancreata with minimal damage.
References
- 1.Matveyenko AV, Butler PC. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J 2006; 47: 225–233 [DOI] [PubMed] [Google Scholar]
- 2.Haataja L, Gurlo T, Huang CJ, et al. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 2008; 29: 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Brien TD, Butler PC, Westermark P, et al. Islet amyloid polypeptide: a review of its biology and potential roles in the pathogenesis of diabetes mellitus. Vet Pathol 1993; 30: 317–332 [DOI] [PubMed] [Google Scholar]
- 4.Howard CF Jr. Longitudinal studies on the development of diabetes in individual Macaca nigra. Diabetologia 1986; 29: 301–306 [DOI] [PubMed] [Google Scholar]
- 5.Rocken C, Linke RP, Saeger W. Immunohistology of islet amyloid polypeptide in diabetes mellitus: semi‐quantitative studies in a post‐mortem series. Virchows Arch A Pathol Anat Histopathol 1992; 421: 339–344 [DOI] [PubMed] [Google Scholar]
- 6.O’Brien TD, Rizza RA, Carney JA, et al. Islet amyloidosis in a patient with chronic massive insulin resistance due to antiinsulin receptor antibodies. J Clin Endocrinol Metab 1994; 79: 290–292 [DOI] [PubMed] [Google Scholar]
- 7.Westermark P, Wernstedt C, Wilander E, et al. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide‐like protein also present in normal islet cells. Proc Natl Acad Sci USA 1987; 84: 3881–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westermark P, Engstrom U, Johnson KH, et al. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA 1990; 87: 5036–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien TD, Butler PC, Kreutter DK, et al. Human islet amyloid polypeptide expression in COS‐1 cells. A model of intracellular amyloidogenesis. Am J Pathol 1995; 147: 609–616 [PMC free article] [PubMed] [Google Scholar]
- 10.Sakagashira S, Sanke T, Hanabusa T, et al. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996; 45: 1279–1281 [DOI] [PubMed] [Google Scholar]
- 11.Seino S. S20G mutation of the amylin gene is associated with Type II diabetes in Japanese. Study Group of Comprehensive Analysis of Genetic Factors in Diabetes Mellitus. Diabetologia 2001; 44: 906–909 [DOI] [PubMed] [Google Scholar]
- 12.Cho YM, Kim M, Park KS, et al. S20G mutation of the amylin gene is associated with a lower body mass index in Korean type 2 diabetic patients. Diabetes Res Clin Pract 2003; 60: 125–129 [DOI] [PubMed] [Google Scholar]
- 13.Sakagashira S, Hiddinga HJ, Tateishi K, et al. S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild‐type amylin. Am J Pathol 2001; 157: 2101–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anguiano M, Nowak RJ, Lansbury PT Jr, et al. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore‐like mechanism that may be relevant to type II diabetes. Biochemistry (Mosc) 2002; 41: 11338–11343 [DOI] [PubMed] [Google Scholar]
- 15.Kayed R, Sokolov Y, Edmonds B, et al. Permeabilization of lipid bilayers is a common conformation‐dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 2004; 279: 46363–46366 [DOI] [PubMed] [Google Scholar]
- 16.Green JD, Kreplak L, Goldsbury C, et al. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J Mol Biol 2004; 342: 877–887 [DOI] [PubMed] [Google Scholar]
- 17.Janson J, Soeller WC, Roche PC, et al. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 1996; 93: 7283–7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couce M, Kane LA, O’Brien TD, et al. Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and beta‐cell dysfunction. Diabetes 1996; 45: 1094–1101 [DOI] [PubMed] [Google Scholar]
- 19.D’Alessio DA, Verchere CB, Kahn SE, et al. Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes 1994; 43: 1457–1461 [DOI] [PubMed] [Google Scholar]
- 20.Verchere CB, D’Alessio DA, Palmiter RD, et al. Transgenic mice overproducing islet amyloid polypeptide have increased insulin storage and secretion in vitro. Diabetologia 1994; 37: 725–728 [DOI] [PubMed] [Google Scholar]
- 21.Soeller WC, Janson J, Hart SE, et al. Islet amyloid‐associated diabetes in obese A(VY)/A mice expressing human islet amyloid polypeptide. Diabetes 1998; 47: 743–750 [DOI] [PubMed] [Google Scholar]
- 22.Butler AE, Janson J, Soeller WC, et al. Increased beta‐cell apoptosis prevents adaptive increase in beta‐cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 2003; 52: 2304–2314 [DOI] [PubMed] [Google Scholar]
- 23.Butler AE, Jang J, Gurlo T, et al. Diabetes due to a progressive defect in beta‐cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes 2004; 53: 1509–1516 [DOI] [PubMed] [Google Scholar]
- 24.Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta‐cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007; 56: 2016–2027 [DOI] [PubMed] [Google Scholar]
- 25.Suzuki R, Tobe K, Terauchi Y, et al. Pdx1 expression in Irs2‐deficient mouse beta‐cells is regulated in a strain‐dependent manner. J Biol Chem 2003; 278: 43691–43698 [DOI] [PubMed] [Google Scholar]
- 26.Terauchi Y, Matsui J, Suzuki R, et al. Impact of genetic background and ablation of insulin receptor substrate (IRS)‐3 on IRS‐2 knock‐out mice. J Biol Chem 2003; 278: 14284–14290 [DOI] [PubMed] [Google Scholar]
- 27.Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem 2002; 50: 449–454 [DOI] [PubMed] [Google Scholar]
- 28.Bonner‐Weir S. Beta‐cell turnover: its assessment and implications. Diabetes 2001; 50 (Suppl. 1): S20–S24 [DOI] [PubMed] [Google Scholar]
- 29.Hull RL, Shen ZP, Watts MR, et al. Long‐term treatment with rosiglitazone and metformin reduces the extent of, but does not prevent, islet amyloid deposition in mice expressing the gene for human islet amyloid polypeptide. Diabetes 2005; 54: 2235–2244 [DOI] [PubMed] [Google Scholar]
- 30.Puchtler H, Sweat F, Kuhns JG. On the binding of direct cotton dyes by amyloid. J Histochem Cytochem 1964; 12: 900–907 [DOI] [PubMed] [Google Scholar]
- 31.Kamada S, Kikkawa U, Tsujimoto Y, et al. Nuclear translocation of caspase‐3 is dependent on its proteolytic activation and recognition of a substrate‐like protein(s). J Biol Chem 2005; 280: 857–860 [DOI] [PubMed] [Google Scholar]
- 32.Gerdes J, Lemke H, Baisch H, et al. Cell cycle analysis of a cell proliferation‐associated human nuclear antigen defined by the monoclonal antibody Ki‐67. J Immunol 1984; 133: 1710–1715 [PubMed] [Google Scholar]
- 33.Vine W, Blase E, Koda J, et al. Plasma amylin concentrations in fasted and fed rats quantified by a monoclonal immunoenzymometric assay. Horm Metab Res 1998; 30: 581–585 [DOI] [PubMed] [Google Scholar]
- 34.Arnelo U, Reidelberger R, Adrian TE, et al. Sufficiency of postprandial plasma levels of islet amyloid polypeptide for suppression of feeding in rats. Am J Physiol 1998; 275: R1537–R1542 [DOI] [PubMed] [Google Scholar]
- 35.van Hulst KL, Hackeng WH, Hoppener JW, et al. An improved method for the determination of islet amyloid polypeptide levels in plasma. Ann Clin Biochem 1994; 31: 165–170 [DOI] [PubMed] [Google Scholar]
- 36.Westermark GT, Gebre‐Medhin S, Steiner DF, et al. Islet amyloid development in a mouse strain lacking endogenous islet amyloid polypeptide (IAPP) but expressing human IAPP. Mol Med 2000; 6: 998–1007 [PMC free article] [PubMed] [Google Scholar]
- 37.Ohsawa H, Kanatsuka A, Yamaguchi T, et al. Islet amyloid polypeptide inhibits glucose‐stimulated insulin from isolated rat pancreatic islets. Biochem Biophys Res Comm 1998; 28: 961–967 [DOI] [PubMed] [Google Scholar]
- 38.Gebre‐Medhin S, Mulder H, Pekny M, et al. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem Biophys Res Comm 1998; 250: 271–277 [DOI] [PubMed] [Google Scholar]
- 39.Matveyenko AV, Butler PC. Beta‐cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 2006; 55: 2106–2114 [DOI] [PubMed] [Google Scholar]
- 40.Hull RL, Andrikopoulos S, Verchere CB, et al. Increased dietary fat promotes islet amyloid formation and beta‐cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes 2003; 52: 372–379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Materials and Methods, providing detailed description of vector construction for the homologous replacement of the mouse IAPP gene for the wild‐type and S20G mutant hIAPP in mouse embryonic stem cells and subsequent generation of the wild‐type and S20G mutant hIAPP knock‐in mice.
Appendix S2 Intraislet amyloidogenesis in WW and GW chimaeric 129Sv/E founder mice at 15 months.
Supporting info item