

Abstract
Aims/Introduction: Oral ingestion of carbohydrate triggers secretion of glucagon‐like peptide (GLP)‐1, which inhibits the postprandial rise in blood glucose levels. However, the mechanism of carbohydrate‐induced GLP‐1 secretion from enteroendocrine L cells remains unclear. In the present study, GLP‐1 secretion was examined by meal tolerance tests of healthy Japanese volunteers.
Materials and Methods: Twenty‐one healthy Japanese men participated in the study. The meal tolerance test was performed with modified nutrient compositions, with or without pretreatment with the α‐glucosidase inhibitor acarbose, or with substitution of sucrose with an equivalent dose of sweeteners in the meal. Blood concentrations of glucose, insulin, GLP‐1, and apolipoprotein (Apo) B‐48 were measured.
Results: GLP‐1 secretion started concomitant with the increase in blood glucose levels 10 min after meal ingestion. Insulin secretion started at 5 min, before the increase in blood glucose levels, reflecting the contribution of direct nutrient stimulation on the former parameter and neural regulation in the latter. Carbohydrate retention in the gut lumen induced by acarbose pretreatment extended postprandial GLP‐1 secretion and negated the increase in serum ApoB‐48 levels. GLP‐1 secretion was markedly decreased by a reduction in the amount of sucrose in the meal and was not restored by an equivalent dose of sweeteners used to compensate for the sweet taste.
Conclusions: The results indicate that direct stimulation of L cells with sugar, but not sweetener, is required for carbohydrate‐induced GLP‐1 secretion. In addition, inhibition of digestion of dietary carbohydrate by α‐glucosidase inhibitors may prevent postprandial hyperglycemia by increasing GLP‐1 secretion and by inhibiting glucose absorption. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2011.00163.x, 2011)
Keywords: Acarbose, Apolipoprotein B‐48, Glucagon‐like peptide‐1
Introduction
Enteroendocrine cells in the gastrointestinal tract are able to sense ingested nutrients so as to trigger secretion of digestive fluid (e.g. trypsin and hydrochloride secretion by gastrin and pancreatic juice secretion by cholecystokinin) to modulate gut motility (e.g. activation and inhibition by vasoactive intestinal peptide [VIP] and cholecystokinin [CCK], respectively) and to maintain glucose homeostasis1.
Maintenance of glucose homeostasis by enteroendocrine cells is accomplished principally by two incretin hormones, namely glucagon‐like peptide (GLP)‐1 and glucose‐dependent insulinotropic polypeptide (GIP)2–6. GLP‐1 and GIP are secreted from enteroendocrine L cells and K cells, respectively. Their biological effects partly overlap and both contribute to inhibition of blood glucose increases after meal ingestion through a variety of mechanisms. Of these, the insulinotropic effect has been studied extensively, and both GLP‐1 and GIP potentiate insulin secretion from pancreatic β‐cells in a glucose‐dependent manner. In addition, GLP‐1, but not GIP, has unique biological effects, such as suppression of appetite, glucagon secretion, and gastric empting, all of which contribute to inhibition of the postprandial rise in plasma glucose concentrations. For these reasons, GLP‐1 mimetics and dipeptidyl peptidase (DPP) 4 inhibitors that block GLP‐1 inactivation are widely used in treatment of type 2 diabetes mellitus2,7,8. Secretion of GLP‐1 is triggered by oral ingestion of food; fat and protein induce GLP‐1 secretion and orally ingested carbohydrates are potent triggers of secretion. Both disaccharide and monosaccharide glucose trigger GLP‐1 secretion when given orally9. However, the mechanism by which L cells sense luminal carbohydrates remains largely unknown. A lack of suitable cell lines to study the mechanism of GLP‐1 secretion and the difficulty in measuring plasma GLP‐1 levels in vivo have hindered clarification of the mechanism involved and many inconclusive studies have been performed10.
To investigate the mechanism underlying GLP‐1 secretion in the present study, we used a novel method to measure active GLP‐1 in healthy Japanese volunteers and found that direct stimulation from the gut lumen by sugar, but not sweetener, is critical for triggering GLP‐1 secretion in response to carbohydrate ingestion.
Materials and Methods
Subjects
Twenty‐one healthy Japanese men participated in the study. None of the men had chronic diseases or was receiving any form of chronic treatment and no medications were taken in the week prior to the experiment. All subjects provided written informed consent and the study protocol was approved by the Ethics Committee of Chiba University.
Study Design
All studies started at 0900 h after 12‐h fast. None of the men had HbA1c ≥6.0% or fasting plasma glucose levels ≥110 mg/dL or more was included. The HbA1c (%) values in the present study were estimated as NGSP equivalent values (%)11.
Meal Tolerance Test with Three Different Meal Types
The men (n = 3) were subjected to three consecutive meal tolerance tests, with one of three different meal types each time, for comparison of metabolic parameters. The three meal types were as follows: (i) a low glycemic index (GI) meal12 of chicken cream soup (Kewpie, Tokyo, Japan) and a biscuit (Yamazaki‐Nabisco, Tokyo, Japan), comprised of 41.7 g carbohydrate, 12.1 g protein, and 16.7 g fat in a total of 365 kcal; (ii) a high‐GI meal of chicken cream soup and biscuit (as above) supplemented with 50 g sucrose (in 200 mL aqueous solution), comprised of 91.7 g carbohydrate, 12.1 g protein, and 16.7 g fat in a total of 565 kcal; and (iii) 50 g sucrose solution (200 mL of 25% w/v sucrose solution), comprised of 50 g carbohydrate in 200 kcal. After 12 h fasting, the subjects ate one of the three meal types and blood samples were obtained for plasma glucose and serum insulin measurement at the time points indicated in the figures. Tolerance tests with these different meals were repeated consecutively in the same subjects for 3 days or more.
Meal Tolerance Test With or Without Acarbose Pretreatment
To evaluate the effect of acarbose, after a 12‐h fast, subjects (n = 12) were given a sucrose‐supplemented meal (as above) with or without 100 mg acarbose (Bayer Yakuhin, Osaka, Japan) pretreatment just before meal ingestion. The clinical features of the subjects are listed in (Table 1). Blood samples were obtained for plasma glucose, serum insulin, plasma intact GLP‐1, serum triglycerides, and serum apolipoprotein (Apo) B‐48 at the time points indicated in the figures.
Table 1. Subject characteristics (note, all subjects in the present study were male).
| n | 12 |
| Age (years) | 34.7 ± 1.3 |
| BMI (kg/m2) | 22.10 ± 0.66 |
| Abdominal circumference (cm) | 79.4 ± 1.9 |
| HbA1c (%) | 4.88 ± 0.07 |
| FPG (mg/dL) | 95.8 ± 3.62 |
| HOMA‐β | 52.9 ± 9.4 |
| HOMA‐IR | 0.78 ± 0.15 |
HOMA‐β was calculated as IRI0 × (FPG − 63), where IRI0 is the fasting serum insulin concentration.
HOMA‐IR was calculated as FPG × IRI0/405.
BMI, body mass index; FPG, fasting plasma glucose.
RT–PCR
Expression of the GLP‐1 receptor (GLP‐1R) was determined in mouse ileal mucosa by RT–PCR. The mucosal layer of the ileum was mechanically stripped from a male C57Bl/6 mouse and RT‐PCR was performed using standard methods, as described in detail previously13. The pancreatic β‐cell line MIN6 was used as a positive control for GLP‐1R. The cDNAs were subjected to PCR of GLP‐1R, sodium–glucose cotransporter 1 (SGLT1; to demonstrate successful sampling of the mucosal layer), and hypoxanthine guanine phosphoribosyl transferase (HPRT; to demonstrate successful cDNA synthesis). The primers used for the PCR are available from the authors on request.
Oral Sweetener Tolerance Test
For the oral sweetener tolerance test, subjects (n = 6) were challenged with test meal (chicken cream soup and biscuit, as above) supplemented with either 5 g sucrose (in 200 mL aqueous solution) or 5 g sucrose plus mixed sweeteners (sucralose, acesulfame, aspartame, and erythritol). The sweeteners were prepared by mixing two types of commercially available sweetener so as to have the sweetness equivalent of 45 g sucrose and were served in 200 mL aqueous solution. Blood samples were obtained to measure plasma glucose, serum insulin, and plasma intact GLP‐1 at the time points indicated in the figures.
Assays
Blood samples were withdrawn through an indwelling catheter directly into BD P700 Blood Collection Tubes (BD, Franklin Lakes, NJ, USA) containing DPP‐4 inhibitor to prevent breakdown of intact GLP‐1 (for GLP‐1) or were withdrawn and aliquotted immediately (for the measurement of parameters other than GLP‐1). Plasma glucose concentrations were determined by the glucose oxidase method; serum insulin concentrations were determined using the Insulin Ultrasensitive ELISA Kit (Mercodia, Uppsala, Sweden). Plasma intact GLP‐1 concentrations were measured by the Mitsubishi Chemical Medience Corporation (Tokyo, Japan) according to the methods of Yabe et al.14. Briefly, plasma aliquots (300 μL) were extracted using an Oasis column (Waters, Milford, MA, USA) and the dried extract was reconstituted in 250 μL assay buffer prior to measurements. Intact GLP‐1 was measured using an Active GLP‐1 Assay Kit (Millipore, Billerica, MA, USA) as described by Yabe et al.14. Antibody‐linked enzyme activity was detected as chemiluminescent intensity. Serum ApoB‐48 concentrations were measured with a chemiluminescent enzyme immunoassay method using ApoB‐48 CLEIA (Fujirebio, Tokyo, Japan).
Statistical Analysis
Data are expressed as the mean ± SE. Statistical significance was calculated by paired or unpaired t‐test. P < 0.05 was considered significant. Correlations between GLP‐1 secretion and serum triglyceride (TG) or ApoB‐48 levels were analyzed by Stat View‐J version 5.0 (SAS Institute, Cary, NC, USA).
Results
Glucose and Insulin Responses to Three Types of Meal Ingestion
To evaluate the effects of nutrient components on glucose and insulin responses, the subjects (n = 3) were fed a low‐GI meal composed of chicken cream soup and a biscuit (41.7 g carbohydrate in a total of 365 kcal) and changes in plasma glucose and serum insulin were monitored (Figure 1). As expected, plasma glucose levels showed only a slight increase up to 15 min after meal intake. To increase the plasma glucose upsurge and the consequent rise in insulin secretion, we modified the meal by adding 50 g sucrose (high‐GI meal; Figure 1). Ingestion of the high‐GI meal induced a significant increase in plasma glucose as soon as 10 min after the meal and serum insulin was also markedly increased. To determine whether the added sucrose was responsible for the early rise in plasma glucose and serum insulin, we measured these parameters again after loading with 50 g sucrose alone (Figure 1). The shift in plasma glucose was almost identical to that seen after the high‐GI meal tolerance test, even though insulin secretion up to 15 min after the high‐GI meal was higher than that after sucrose alone. Similarly, plasma intact GLP‐1 levels after the high‐GI meal and sucrose alone did not differ up to 15 min after the meal (data not shown), indicating that the sucrose in the high‐GI meal plays a central role in eliciting early phase GLP‐1 secretion. Because there was only a small increase in plasma glucose and serum insulin levels in the early phase (up to 15 min) after ingestion of the low‐GI meal (Figure 1), we used the high‐GI meal to assess GLP‐1 secretion.
Figure 1.
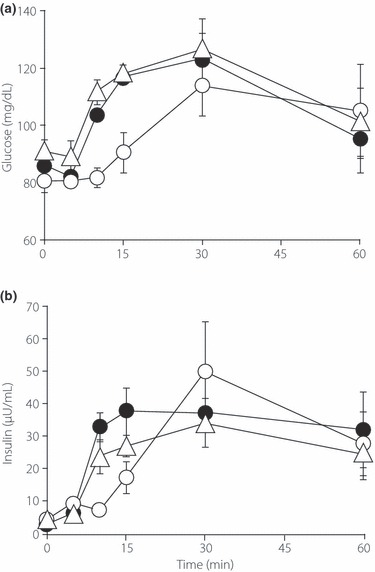
Changes in (a) plasma glucose and (b) serum insulin after of a meal tolerance test with either a low glycemic index (GI) meal (○), a high‐GI meal (•), or sucrose (△). Data are the mean ± SE.
GLP‐1 Secretion in Response to Meal Ingestion
Ingestion of the high‐GI meal induced an early rise in plasma glucose (Figure 2a) and plasma insulin (Figure 2b) concentrations. As expected, ingestion of the high‐GI meal elicited a significant rise in plasma GLP‐1 concentrations as soon as 10 min (1.15 ± 0.20 and 2.76 ± 0.41 pmol/L at 0 and 10 min, respectively; n = 12; P < 0.0005). Plasma glucose concentrations were not increased at 5 min, but did increase after 10 min, whereas serum insulin concentrations started to increase as early as 5 min after the meal, before the increase in plasma glucose concentrations (Figure 2a,b, insets), suggesting that the secretion is triggered by cephalic regulation, as proposed originally by Berthoud et al.15. In contrast, plasma GLP‐1 started to increase at 10 min, coinciding with the rise in plasma glucose concentrations.
Figure 2.
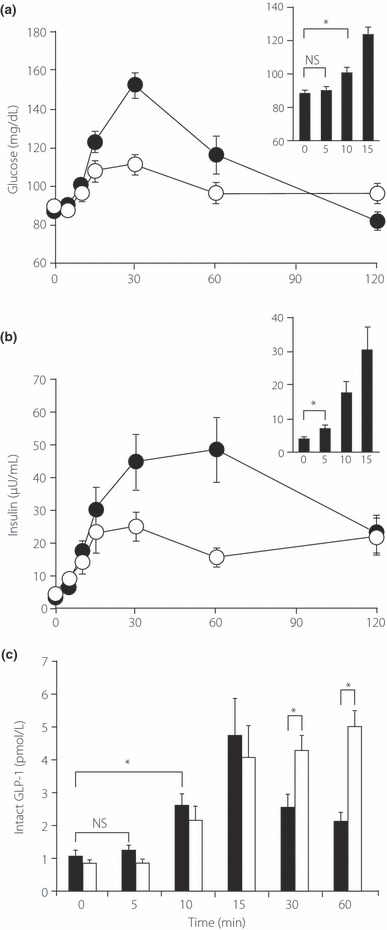
Glucagon‐like peptide (GLP)‐1 secretion during the meal tolerance test. Changes in (a) plasma glucose and (b) serum insulin and (c) plasma intact GLP‐1 levels during the meal tolerance test with (open symbols) or without (filled symbols) acarbose pretreatment. Insets show only early phase (0–15 min) responses of (a) plasma glucose and (b) serum insulin from meal tolerance tests without acarbose pretreatment. Data are the mean ± SE. *P < 0.05, **P < 0.0005.
Effect of Acarbose on Plasma Glucose, Serum Insulin and Plasma GLP‐1 Concentrations after Meal Ingestion
We then examined the effect of the α‐glucosidase inhibitor acarbose on changes in plasma glucose and serum insulin concentrations after ingestion of the high‐GI meal in the same subjects. Pretreatment with 100 mg acarbose prevented the increase in plasma glucose at or later than 15 min (Figure 2a), indicating that hydrolysis of carbohydrate was suppressed by acarbose after 15 min, the undigested carbohydrate remaining in the gut lumen. The peak serum insulin concentrations were also decreased significantly at or later than 30 min (Figure 2b). Nevertheless, there was a very good correlation between increases in plasma glucose and plasma GLP‐1 (Figure 2a, inset,c). Without acarbose pretreatment, the plasma concentration of intact GLP‐1 (which reflects GLP‐1 secretion) returned to basal levels 60 min after the meal (Figure 2c); acarbose pretreatment prevented the cessation of GLP‐1 secretion at 60 min (Figure 2c). This is compatible with our hypothesis that only sucrose and glucose in the gut lumen stimulate GLP‐1 secretion from L cells.
Because chronic treatment with acarbose has been reported to lower serum TG levels16, in the present study we measured acute changes in the serum lipid profile (TG and ApoB‐48) after single administration of acarbose (Figure 3a–d). Meal ingestion failed to increase serum TG as a whole (n = 12; Figure 3a). However, when we assessed the relationship between GLP‐1 secretion (as assessed by AUC60 min) and the increase in serum TG levels at 120 min, we found a negative correlation (correlation coefficient, R, 0.627; n = 12; P = 0.029; Figure 3b). Acarbose pretreatment both increased GLP‐1 secretion and suppressed the increase in serum TG levels in all but one subject (Figure 3b). With all data combined (with and without acarbose pretreatment), the overall correlation coefficient was 0.355 (n = 24; P = 0.089). The same trend was observed between GLP‐1 secretion and the postprandial increase (at 120 min) in serum ApoB‐48 levels, although it failed to reach statistical significance (R = 0.305 in total; n = 24; P = 0.14; Figure 3d). Interestingly, meal ingestion increased ApoB‐48 as a whole (n = 12; P = 0.020), whereas acarbose negated the postprandial increase in ApoB‐48 (Figure 3c), suggesting that acarbose suppresses the postprandial release of chylomicron.
Figure 3.
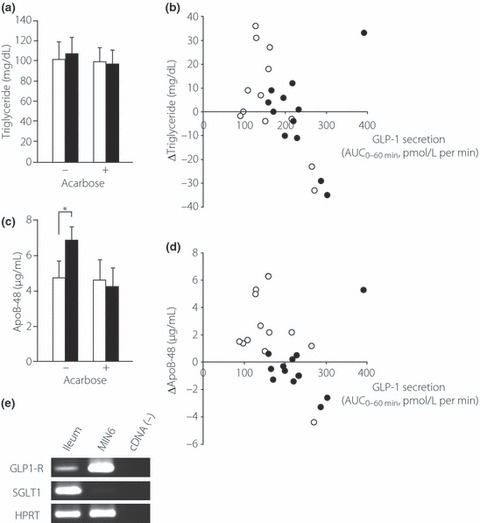
Postprandial (a,b) triglyceride and (c,d) apolipoprotein (Apo) B‐48 levels in the blood and (e) mRNA expression of the glucagon‐like peptide (GLP)‐1 receptor (GLP‐1R) in mouse ileal mucosa. (a,c) Serum triglyceride (a) and ApoB‐48 (c) concentrations, with or without acarbose pretreatment, as indicated. (□), values at 0 min; ( ), values at 120 min after the meal. (b,d) Relationship between GLP‐1 secretion and increases in serum triglyceride (b) or ApoB‐48 (d). GLP‐1 secretion is expressed as AUC0–60min of plasma intact GLP‐1 levels, whereas increases in serum triglyceride or ApoB‐48 are expressed as change at 120 min after meal. Symbols represent data from individuals without (○) or with (•) acarbose pretreatment. Data are the mean ± SE. *P < 0.05. (e) mRNA expression of GLP‐1R in mouse ileal mucosa as determined by RT‐PCR. Data are shown for GLP‐1R, sodium–glucose cotransporter 1 (SGLT1), and hypoxanthine guanine phosphoribosyl transferase (HPRT) in mouse ileal mucosa and MIN6 cells.
), values at 120 min after the meal. (b,d) Relationship between GLP‐1 secretion and increases in serum triglyceride (b) or ApoB‐48 (d). GLP‐1 secretion is expressed as AUC0–60min of plasma intact GLP‐1 levels, whereas increases in serum triglyceride or ApoB‐48 are expressed as change at 120 min after meal. Symbols represent data from individuals without (○) or with (•) acarbose pretreatment. Data are the mean ± SE. *P < 0.05. (e) mRNA expression of GLP‐1R in mouse ileal mucosa as determined by RT‐PCR. Data are shown for GLP‐1R, sodium–glucose cotransporter 1 (SGLT1), and hypoxanthine guanine phosphoribosyl transferase (HPRT) in mouse ileal mucosa and MIN6 cells.
Such a relationship between GLP‐1 secretion (from L cells) and postprandial chylomicron release (from enterocytes) suggests a local function of GLP‐1 in the gut mucosa. We therefore assessed the role of GLP‐1 as a cytokine by examining the expression of GLP‐1R in mouse ileal mucosa. Using RT‐PCR, we demonstrated that the GLP‐1R is expressed in the ileal mucosa, which is comprised to a large extent by enterocytes (Figure 3e). Therefore, GLP‐1 may act as a paracrine as well as endocrine factor to inhibit postprandial chylomicron release from enterocytes.
Effects of Sweeteners on GLP‐1 Secretion
We then compared the effects of an equivalent sweet taste using non‐nutritive sweeteners on L cells and GLP‐1 secretion. We first reduced the amount of sucrose (to one‐tenth) in the test meal to determine the importance of sucrose in the high GI‐test meal on GLP‐1 secretion. The large reduction of sucrose in the test meal significantly lowered the increase in plasma glucose (Figure 4a), indicating that sucrose plays a major role in determining the postprandial glucose levels in our experiments. The reduction in sucrose in the test meal also decreased GLP‐1 secretion at 15 min after meal ingestion (1.91 ± 0.31 pmol/L; n = 7; P < 0.05 vs the high‐GI test meal), demonstrating that sucrose in the test meal plays a central role in triggering GLP‐1 secretion under the present experimental conditions.
Figure 4.
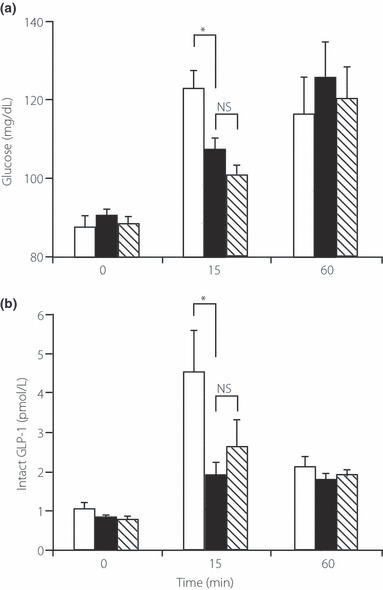
Effects of sucrose and sweeteners on glucagon‐like peptide (GLP)‐1 secretion during the meal tolerance test. Plasma (a) glucose and (b) active GLP‐1 levels at 0, 15, and 60 min after challenge with meals supplemented with high (50 g) sucrose (□), as shown in Figure 2, low (5 g) sucrose (), and mixed sweeteners with low (5 g) sucrose ( ). Data are the mean ± SE. *P < 0.05.
). Data are the mean ± SE. *P < 0.05.
We then added mixed sweeteners to the low (5 g)‐sucrose meal to complement the sweet taste. The attenuated GLP‐1 secretion on the low (5 g)‐sucrose meal was not restored following the addition of mixed sweeteners (2.62 ± 0.65 pmol/L at 15 min; n = 7; P < 0.05 vs high‐GI test meal; NS vs low [5 g]‐sucrose meal). These results indicate that the sweeteners in the test meal did not have a significant role in eliciting GLP‐1 secretion.
Discussion
The early insulin secretory response to a meal is critical for maintaining glucose homeostasis17–19 and a defect in this response is a characteristic feature of type 2 diabetes mellitus20. Although the rise in blood glucose and serum insulin levels in the case of the low‐GI meal were too small, the high‐GI meal elicited an early plasma glucose upsurge and insulin secretion, with the added sucrose clearly playing a substantial role. The high‐GI meal was therefore used as a test meal to assess early phase insulin and GLP‐1 secretion responses.
The temporal patterns of blood glucose were almost identical between the high‐GI meal and sucrose tolerance tests, despite the large difference in energy content between the high‐GI meal (565 kcal) and sucrose solution (200 kcal; Figure 1a). This also suggests that the sucrose contained in our high‐GI meal is responsible for the early (up to 15 min) rise in plasma glucose concentrations. However, the total amount of insulin secreted following the high‐GI meal was higher than that following the ingestion of sucrose alone. These results indicate that insulin secretion is regulated not only by changes in plasma glucose, but also by the composition of the meal via nutrient sensing in the gut. Because the incretin system is likely to participate in such regulation, we examined GLP‐1 secretion in the early phase after meal ingestion.
There are many reports regarding the GLP‐1 secretory profile after meal ingestion. However, it is difficult to accurately measure plasma GLP‐1: its values differ considerably as absolute plasma intact GLP‐1 levels and there are also large variations among samples. This hampers the evaluation of GLP‐1 secretion in both health and disease. In the present study, the plasma GLP‐1 concentration was first purified by solid phase extraction and then determined using an ELISA kit from Millipore. Solid phase extraction markedly reduced the non‐specific cross‐reaction with adulterated substance, which causes the large variation among samples. As a result, we obtained a relatively low level of fasting plasma GLP‐1 with small variation among samples (1.15 ± 0.20 pmol/L; n = 12).
The temporal pattern of GLP‐1 secretion was then compared with that of blood glucose and serum insulin. Interestingly, insulin secretion started prior to the increase in plasma glucose (Figure 2b), suggesting that the secretion is triggered through cephalic regulation, as proposed originally by Berthoud et al.21. In contrast, plasma GLP‐1 started to rise at 10 min, coinciding with the increase in plasma glucose concentrations. Because both glucose‐absorbing enterocytes and GLP‐1‐secreting enteroendocrine L cells exist side‐by‐side in the intestine, the L cells are thought to secrete GLP‐1 in response to direct stimulation by nutrients in the gut lumen. This is compatible with the concept proposed by Holst et al.4 that neural regulation is limited in humans. However, our novel method for measuring intact GLP‐1 has enabled us to show a significant increase in GLP‐1 secretion 10 min after meal ingestion.
In addition, GLP‐1 is thought to participate in the prevention of the glycemic upsurge in the early phase (∼15 min) after meal ingestion, because GLP‐1 secretion begins as early as 10 min after a meal. The difficulty in measuring GLP‐1 has also hampered determination of impaired GLP‐1 secretion in type 2 diabetes mellitus22,23 and investigations into whether there are ethnic differences in GLP‐1 secretion24.
We found that acarbose pretreatment prevented the increase in both plasma glucose and insulin at or later than 15 min (Figure 2a,b), which seems to reflect a substantial blockade of glucose absorption by acarbose pretreatment. Without acarbose pretreatment, plasma concentrations of intact GLP‐1, which closely reflect GLP‐1 secretion, returned to basal levels 60 min after the meal, but acarbose pretreatment prevented this cessation of GLP‐1 secretion (Figure 2c). These results are compatible with our hypothesis that sugar in the gut lumen stimulates GLP‐1 secretion from L cells. Although suppression of the increase in postprandial glucose by acarbose is considered to result from delayed and inadequate digestion of glucose due to poor processing of carbohydrates, our data suggest that potentiation of GLP‐1 secretion may also contribute to the anti‐diabetic effect of acarbose.
Both glucose and sucrose evoke GLP‐1 secretion when administrated orally and both are substrates for the sweet receptors on the taste buds on the tongue25. The sweetness is sensed by the heterodimeric T1R2 + T1R3 sweet taste receptor25,26. Stimulation of the sweet receptors with sugars and sweeteners leads to activation of the trimeric G‐protein gastducin27. The T1R2 and T1R3 receptors are expressed in rodent gut and the enteroendocrine tumor cell line STC‐1 and α‐gastducin is also present in these L cells28. Several papers have reported a role for sweet sensation in GLP‐1 secretion28,29. In addition, the involvement of sweet sensation in GLP‐1 secretion has been demonstrated in mice lacking α‐gastducin28. Recently, the sweet receptor has been shown to be present in pancreatic β‐cells and to participate in glucose‐stimulated insulin secretion30. Thus, the sweet receptor in L cells is expected to contribute to carbohydrate‐induced GLP‐1 secretion. However, other studies have reported that the oral ingestion of sugar and not sweeteners evokes GIP or GLP‐1 secretion31–33. Therefore, the contribution of the sweet receptors to GLP‐1 secretion remains unclear. The results of the present study indicate a negligible contribution of sweeteners in GLP‐1 secretion. Because the increases in plasma glucose and GLP‐1 occur within the same time frame after meal ingestion, sucrose and/or its hydrolyzed derivative glucose at the apical process of the L cells is the most likely trigger of GLP‐1 secretion. In addition, our results with a reduced amount of sucrose and sweeteners indicate that GLP‐1 is secreted in proportion to the rise in plasma glucose and that sweeteners do not mimic sucrose as a GLP‐1secretagogue. Interestingly, GLP‐1 secretion at 15 min was not suppressed by acarbose pretreatment, suggesting that luminal sucrose and/or glucose, but not sweetener, in the gut lumen is sensed by SGLT134 or an unidentified sucrose receptor in the L cells other than T1R2+ T1R3 to dose‐dependently trigger GLP‐1 secretion (Figure 5).
Figure 5.
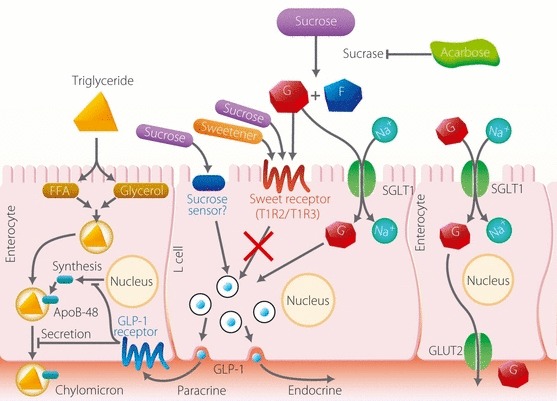
A model for the secretion and action of glucagon‐like peptide (GLP)‐1 in the gut epithelium. See text for details. G, glucose; F, fructose; GLUT2, glucose transporter 2; FFA, free fatty acid; ApoB‐48, apolipoprotein B‐48; SGLT1, sodium–glucose cotransporter 1.
ApoB‐48 is an apolipoprotein generated in the absorptive epithelium and is required to generate chylomicron, which is essential for exporting meal‐derived TG. Because serum levels of ApoB‐48 reflect the level of chylomicron release after meal ingestion, acarbose is thought to inhibit the increase in chylomicron that occurs after fatty meal ingestion. Interestingly, GLP‐1 has recently been reported to suppress ApoB‐48 excretion from enterocytes35. Considering the potential relationship between GLP‐1 and the lipid profile, and the effect of acarbose on these parameters, the effect of acarbose on ApoB‐48 may be mediated through an increase in GLP‐1 secretion. We propose a model in which GLP‐1 secreted from the L cells acts on neighboring enterocytes as a paracrine factor to suppress postprandial chylomicron release (Figure 5). This is compatible with the finding that intact GLP‐1 is inactivated promptly in the blood by DPP4, a peptidase involved in the inactivation of several chemokines, such as CCL and CXCL5,36.
In conclusion, we found that GLP‐1 secretion coincides with the increase in plasma glucose concentrations, whereas insulin secretion precedes the increase in glucose. Thus, although insulin secretion can be initiated by neuronal regulation, GLP‐1 secretion is triggered by direct stimulation of the L cells by nutrients. We also found that acarbose pretreatment extended meal‐induced GLP‐1 secretion and that sweeteners played a negligible role in GLP‐1 secretion. These results all suggest the importance of the direct stimulation of L cells by luminal sugar, but not sweetener, in GLP‐1 secretion.
Acknowledgements
The authors thank Dr Hideki Tanzawa, Chiba University, and Dr Yutaka Seino, Kansai Electric Power Hospital, for helpful suggestions for the study. This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture, and Technology (T.M.). The authors declare no conflict of interest.
References
- 1.Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature 2006; 444: 854–859 [DOI] [PubMed] [Google Scholar]
- 2.Seino Y, Fukushima M, Yabe D. GIP and GLP‐1, the two incretin hormones: similarities and differences. J Diabetes Invest 2010; 1: 8–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157 [DOI] [PubMed] [Google Scholar]
- 4.Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev 2007; 87: 1409–1439 [DOI] [PubMed] [Google Scholar]
- 5.Wideman RD, Kieffer TJ. Mining incretin hormone pathways for novel therapies. Trends Endocrinol Metab 2009; 20: 280–286 [DOI] [PubMed] [Google Scholar]
- 6.Ahren B. Islet G protein‐coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 2009; 8: 369–385 [DOI] [PubMed] [Google Scholar]
- 7.Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta‐analysis. JAMA 2007; 298: 194–206 [DOI] [PubMed] [Google Scholar]
- 8.Peters A. Incretin‐based therapies: review of current clinical trial data. Am J Med 2011; 123(Suppl): S28–S37 [DOI] [PubMed] [Google Scholar]
- 9.Shima K, Suda T, Nishimoto K, et al. Relationship between molecular structures of sugars and their ability to stimulate the release of glucagon‐like peptide‐1 from canine ileal loops. Acta Endocrinol 1990; 123: 464–470 [DOI] [PubMed] [Google Scholar]
- 10.Nauck MA, Vardarli I, Deacon CF, et al. Secretion of glucagon‐like peptide‐1 (GLP‐1) in type 2 diabetes: what is up, what is down? Diabetologia 2011; 54: 10–18 [DOI] [PubMed] [Google Scholar]
- 11.Seino Y, Nanjo K, Tajima N, et al. Report of the Committee on the Classification and Diagnostic Criteria of Diabetes Mellitus. Diabetol Int 2010; 1: 2–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins DJ, Kendall CW, McKeown‐Eyssen G, et al. Effect of a low‐glycemic index or a high‐cereal fiber diet on type 2 diabetes: a randomized trial. JAMA 2008; 300: 2742–2753 [DOI] [PubMed] [Google Scholar]
- 13.Miki T, Taira M, Hockman S, et al. Characterization of the cDNA and gene encoding human PDE3B, the cGIP1 isoform of the human cyclic GMP‐inhibited cyclic nucleotide phosphodiesterase family. Genomics 1996; 36: 476–485 [DOI] [PubMed] [Google Scholar]
- 14.Yabe D, Watanabe K, Kuwata H, et al. Comparison of incretin immunoassays with or without plasma extraction: incretin secretion in Japanese patients with type 2 diabetes. J Diabetes Invest 2011; (in press). doi: 10.1111/j.2040‐1124.2011.00141.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berthoud HR, Bereiter DA, Trimble ER, et al. Cephalic phase, reflex insulin secretion. Neuroanatomical and physiological characterization. Diabetologia 1981; 20(Suppl): 393–401 [DOI] [PubMed] [Google Scholar]
- 16.Ogawa S, Takeuchi K, Ito S. Acarbose lowers serum triglyceride and postprandial chylomicron levels in type 2 diabetes. Diabetes Obes Metab 2004; 6: 384–390 [DOI] [PubMed] [Google Scholar]
- 17.Luzi L, DeFronzo RA. Effect of loss of first‐phase insulin secretion on hepatic glucose production and tissue glucose disposal in humans. Am J Physiol 1989; 257: E241–E246 [DOI] [PubMed] [Google Scholar]
- 18.Mitrakou A, Kelley D, Mokan M, et al. Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med 1992; 326: 22–29 [DOI] [PubMed] [Google Scholar]
- 19.Basu A, Alzaid A, Dinneen S, et al. Effects of a change in the pattern of insulin delivery on carbohydrate tolerance in diabetic and nondiabetic humans in the presence of differing degrees of insulin resistance. J Clin Invest 1996; 97: 2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruce DG, Chisholm DJ, Storlien LH, et al. Physiological importance of deficiency in early prandial insulin secretion in non‐insulin‐dependent diabetes. Diabetes 1988; 37: 736–744 [DOI] [PubMed] [Google Scholar]
- 21.Berthoud HR, Trimble ER, Siegel EG, et al. Cephalic‐phase insulin secretion in normal and pancreatic islet‐transplanted rats. Am J Physiol 1980; 238: E336–E340 [DOI] [PubMed] [Google Scholar]
- 22.Vilsboll T, Krarup T, Deacon CF, et al. Reduced postprandial concentrations of intact biologically active glucagon‐like peptide 1 in type 2 diabetic patients. Diabetes 2001; 50: 609–613 [DOI] [PubMed] [Google Scholar]
- 23.Vilsboll T, Krarup T, Sonne J, et al. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003; 88: 2706–2713 [DOI] [PubMed] [Google Scholar]
- 24.Yabe D, Kurose A, Lee S, et al. Little enhancement of meal‐induced glucagon‐like peptide 1 secretion in Japanese: ‐comparisonof type 2 diabetes patients and healthy controls. J Diabetes Invest 2010; 1: 56–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson G, Hoon MA, Chandrashekar J, et al. Mammalian sweet taste receptors. Cell 2001; 106: 381–390 [DOI] [PubMed] [Google Scholar]
- 26.Zhang F, Klebansky B, Fine RM, et al. Molecular mechanism of the sweet taste enhancers. Proc Natl Acad Sci USA 2010; 107: 4752–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandrashekar J, Hoon MA, Ryba NJ, et al. The receptors and cells for mammalian taste. Nature 2006; 444: 288–294 [DOI] [PubMed] [Google Scholar]
- 28.Jang HJ, Kokrashvili Z, Theodorakis MJ, et al. Gut‐expressed gustducin and taste receptors regulate secretion of glucagon‐like peptide‐1. Proc Natl Acad Sci USA 2007; 104: 15069–15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown RJ, Walter M, Rother KI. Ingestion of diet soda before a glucose load augments glucagon‐like peptide‐1 secretion. Diabetes Care 2009; 32: 2184–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa Y, Nagasawa M, Yamada S, et al. Sweet taste receptor expressed in pancreatic beta‐cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS ONE 2009; 4: e5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujita Y, Wideman RD, Speck M, et al. Incretin release from gut is acutely enhanced by sugar but not by sweeteners in vivo. Am J Physiol Endocrinol Metab 2009; 296: E473–E479 [DOI] [PubMed] [Google Scholar]
- 32.Ma J, Bellon M, Wishart JM, et al. Effect of the artificial sweetener, sucralose, on gastric emptying and incretin hormone release in healthy subjects. Am J Physiol Gastrointest Liver Physiol 2009; 296: G735–G739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reimann F, Habib AM, Tolhurst G, et al. Glucose sensing in L cells: a primary cell study. Cell Metab 2008; 8: 532–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriya R, Shirakura T, Ito J, et al. Activation of sodium‐glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab 2009; 297: E1358–E1365 [DOI] [PubMed] [Google Scholar]
- 35.Hsieh J, Longuet C, Baker CL, et al. The glucagon‐like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia 2010; 53: 552–561 [DOI] [PubMed] [Google Scholar]
- 36.Drucker DJ. Dipeptidyl peptidase‐4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care 2007; 30: 1335–1343 [DOI] [PubMed] [Google Scholar]