

Abstract
Despite recent advances in crystallography of G protein-coupled receptors (GPCRs), little is known about the mechanism of their activation process, as only the β2 adrenergic receptor (β2AR) and rhodopsin have been crystallized in fully active conformations. Here, we report the structure of an agonist-bound, active state of the human M2 muscarinic acetylcholine receptor stabilized by a G-protein mimetic camelid antibody fragment isolated by conformational selection using yeast surface display. In addition to the expected changes in the intracellular surface, the structure reveals larger conformational changes in the extracellular region and orthosteric binding site than observed in the active states of the β2AR and rhodopsin. We also report the structure of the M2 receptor simultaneously binding the orthosteric agonist iperoxo and the positive allosteric modulator LY2119620. This structure reveals that LY2119620 recognizes a largely pre-formed binding site in the extracellular vestibule of the iperoxo-bound receptor, inducing a slight contraction of this outer binding pocket. These structures offer important insights into activation mechanism and allosteric modulation of muscarinic receptors.
Introduction
Muscarinic acetylcholine receptors (M1 – M5) are GPCRs that regulate the activity of a diverse array of central and peripheral functions in the human body, including the parasympathetic actions of acetylcholine1. The M2 muscarinic receptor subtype plays a key role in modulating cardiac function and many important central processes, such as cognition and pain perception1. As it was among the first GPCRs to be purified2 and cloned3, the M2 receptor has long served as a model system in GPCR biology and pharmacology. Muscarinic receptors have attracted particular interest due to their ability to bind small molecule allosteric modulators4. Since allosteric sites are often less conserved than the orthosteric binding site, some ligands binding to allosteric sites show substantial subtype selectivity5,6. Such agents hold great promise for the development of drugs for the treatment of conditions, including diseases of the central nervous system and metabolic disorders. Though crystal structures were recently obtained for inactive states of the M2 and M3 muscarinic receptors7,8, there are no structures of a GPCR bound to an allosteric modulator.
The binding of an agonist to the extracellular side of a GPCR results in conformational changes that enable the receptor to activate heterotrimeric G proteins. Despite the importance of this process, only the β2AR and rhodopsin have been crystallized in fully active conformations9-13. Crystallization of active-state GPCRs has been challenging due to their inherent conformational flexibility and biochemical instability14. To better understand the mechanistic details underlying GPCR activation and allosteric modulation, we solved X-ray crystal structures of the M2 receptor bound to the high affinity agonist iperoxo15 alone and in combination with LY2119620, a positive allosteric modulator.
Conformational selection of nanobodies
Initial crystallization attempts with M2 receptor bound to agonists were unsuccessful, likely due to the flexibility of the intracellular receptor surface in the absence of a stabilizing protein. We thus sought to obtain a ‘G protein mimetic’ nanobody for the M2 receptor to facilitate crystallization of the β2AR in an active conformation11. Llamas were immunized with M2 receptor bound to the agonist iperoxo, and a post-immune single variable domain (VHH) nanobody cDNA library was constructed and displayed on the surface of yeast (Fig. 1a).
Figure 1. Isolation of Nb9-8.
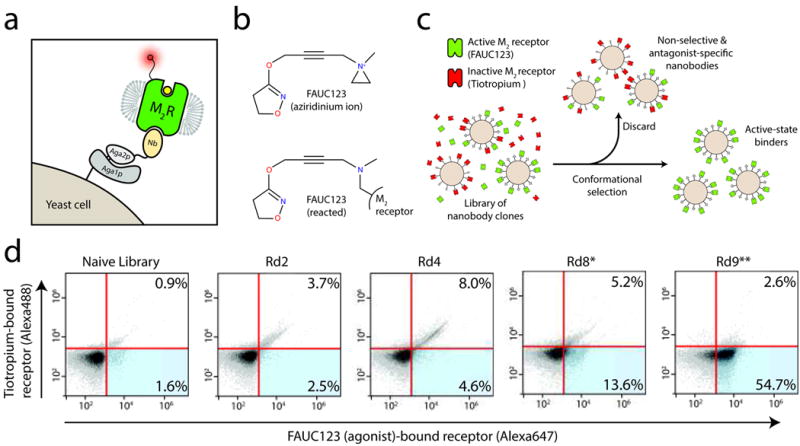
a, Nanobodies from a llama immunized with M2 receptor were displayed on yeast as an amino terminal fusion to Aga2p, and subjected to magnetic selection to enrich clones that bind preferentially to agonist-occupied receptor. b, For selections, an aziridinium ion derivative of iperoxo called FAUC123 was synthesized, allowing covalent modification of the receptor. c, Yeast were stained simultaneously with agonist-occupied M2 receptor and antagonist-occupied receptor labeled with distinct fluorophores. d, Yeast from each selection round (Rd. 1 – 9) were stained in this manner to assess selection progress, showing a clear enrichment first for non-selective binders (upper right quadrants) followed by specific enrichment for agonist-preferring clones (lower right quadrants). *Indicates a selection round employing conformational selection. **Indicates a selection round using FACS.
An essential component for the selection of active-state stabilizing nanobodies was simultaneous staining of yeast with both agonist and inverse-agonist occupied M2 receptor populations, which were distinguishably labeled with separate fluorophores. This allowed the use of fluorescence-activated cell sorting (FACS) to select those clones binding only agonist-occupied receptor (Fig. 1b; see Online Methods). To ensure that the different fluorophore-conjugated receptors represent distinct receptor populations requires that at least one receptor population must be bound to an exceptionally high-affinity or irreversible ligand. We therefore developed a covalent muscarinic receptor agonist for use in selection experiments. This has precedent in an acetylcholine mustard16, which is thought to react with the binding site residue Asp1033.32 to form a covalent adduct17. Accordingly, we synthesized an analogous “iperoxo mustard,” which we call FAUC123 (Supplementary Methods). We found that FAUC123 bound covalently and was able to induce activation of the M2 receptor (Extended Data Fig. 1), thereby allowing simultaneous staining of yeast with agonist- and antagonist-bound M2 receptor labeled with distinct fluorophores for each population.
Extended Data Figure 1. Characterization of FAUC123.

a, Activation of M2 receptor by the prototypical muscarinic agonist carbachol, the high affinity agonist iperoxo, and an irreversible iperoxo analog (FAUC 123) shows that iperoxo and FAUC 123 are exceptionally potent full agonists at the M2 muscarinic receptor. Points indicate mean ± SEM of three independent measurements, each performed in triplicate. b, Sf9 membranes expressing the human M2 receptor were incubated overnight at 4 °C with either no ligand, 100 μM iperoxo, or 100 μM FAUC 123. Membranes were then washed three times in buffer without ligand, and incubated with a saturating concentration (20 nM) of [3H]-NMS. Incubation with iperoxo had no effect on radioligand binding, while FAUC 123 blocked almost all [3H]-NMS binding sites. Bars indicate mean ± SEM of three independent measurements. c, FAUC 123 was tested for its ability to induce M2 receptor activation following covalent modification. While iperoxo-induced inositol phosphate production was blocked by 1 μM atropine, FAUC 123-induced activation was not susceptible to atropine blockade. Bars indicate mean ± SEM of three independent measurements.
After nine rounds of conformational selection, almost all remaining yeast cell clones preferentially bound FAUC123-occupied receptor (Fig. 1d). Three clones in particular, Nb9-1, Nb9-8, and Nb9-20 (Fig. 2a; see Online Methods) exhibited strong, conformationally-selective staining on yeast (Fig. 2b). All three nanobodies enhanced agonist affinity (Fig. 2c), indicating that they stabilize active states of the receptor. Nb9-8 was the most potent, with an EC50 of approximately 100 nM. At high concentrations, Nb9-8 enhanced the affinity of the M2 receptor for iperoxo to almost the same extent as that observed in the presence of the heterotrimeric G protein Gi (Fig. 2d).
Figure 2. M2 active-state specific nanobodies.
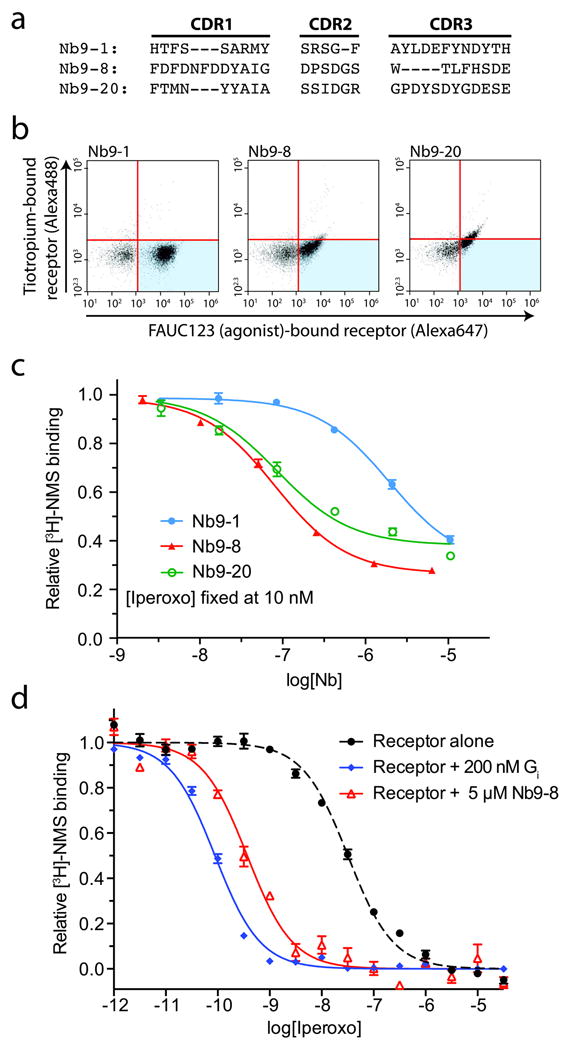
a, Three nanobodies were selected for detailed characterization, each with entirely unique complementarity determining region (CDR) sequences. These three nanobodies were expressed on the surface of yeast, and characterized by flow cytometry staining with FAUC123-bound (i.e., agonist-bound) M2 receptor and tiotropium-bound (i.e., antagonist-occupied) receptor. b, Each of the three clones displayed a preference for agonist-occupied receptor to varying degrees. c, Purified nanobodies were tested in a dose-response assay for their ability to suppress [3H]-NMS binding to the M2 receptor in the presence of 10 nM (IC20) iperoxo, with Nb9-8 being the most potent clone. d, Like the G protein Gi, Nb9-8 caused a substantial enhancement of iperoxo affinity in a competition binding assay.
M2 receptor was purified in the presence of 10 μM iperoxo, and we obtained crystals of iperoxo-bound M2 receptor in complex with Nb9-8 by lipidic mesophase crystallography. The structure was solved by microdiffraction at Advanced Photon Source beamline 23ID-D (Extended Data Table 1). Supplementing the optimized crystallization conditions with the positive allosteric modulator LY2119620 yielded crystals of M2 receptor simultaneously bound to both iperoxo and the modulator (see Online Methods). For all crystallization work, the agonist iperoxo was used rather than acetylcholine, as the latter is of lower affinity and is prone to hydrolysis.
Extended Data Table 1. Data collection and refinement statistics.
| M2 receptor:Nb9-8 complex | M2 receptor:Nb9-8 complex bound to LY2119620 | |
|---|---|---|
| Data collection* | ||
| Number of crystals | 17 | 18 |
| Space group | P212121 | P212121 |
| Unit cell dimensions | ||
| a, b, c (Å) | 62.9,78.1,163.5 | 59.0,77.4,163.8 |
| α,β,γ(°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 33 – 3.5(3.6 – 3.5) | 36 – 3.7(3.8 – 3.7) |
| Rmerge (%) | 18.8(74.4) | 19.5(60.5) |
| <l/σl> | 5.8(1.4) | 5.6(2.1) |
| CC1/2 (%) | 99.1 (35.0) | 99.0(54.1) |
| Completeness (%) | 95.9(83.1) | 93.0(80.1) |
| Redundancy | 4.8 | 4.8 |
| Refinement | ||
| Number of reflections | 10237 | 7867 |
| Rwork/Rfree (%) | 24.9 / 29.8 | 25.0 / 30.1 |
| No. atoms | ||
| Protein | 3020 | 3013 |
| Ligand(s) | 14 | 57 |
| Average B factors (Å2) | ||
| Receptor | 109.7 | 102.3 |
| Nb6B9 | 137.4 | 129.3 |
| Iperoxo | 105.5 | 107.6 |
| LY2119620 | - | 119.0 |
| RMS deviation from ideality | ||
| Bond length (Å) | 0.004 | 0.004 |
| Bond angles (°) | 0.86 | 0.81 |
| Ramachandran statistics† | ||
| Favored (%) | 97.0 | 95.9 |
| Allowed (%) | 3.0 | 4.1 |
| Outliers (%) | 0 | 0 |
Highest shell statistics in parentheses.
As calculated by Molprobity.
Cytoplasmic changes upon activation
A key feature of GPCR activation is an outward movement of the intracellular portion of transmembrane (TM) helices 5 and 6, creating a cavity large enough to accommodate the carboxy terminus of the G protein α subunit10,13. While several GPCRs have been crystallized in complex with agonists, only the β2AR and rhodopsin show a fully active state with adequate space to allow G protein binding (Extended Data Fig. 2). As anticipated based on functional studies (Fig. 2), Nb9-8 binds to the intracellular surface of the receptor (Fig. 3a). There is a significant outward displacement at the intracellular side of TM6, together with a smaller outward movement of TM5 and a rearrangement of TM7 around the NPxxY motif (Fig. 3b, d).
Extended Data Figure 2. Comparison to other active GPCR structures.

Structures of all activated GPCRs show similarities in conformational changes at the intracellular surface. In each case, the intracellular tip of transmembrane helix 6 (TM6) moves outward on activation, as seen in the view from the intracellular side (right panels). This creates a cavity to which a G protein can bind the receptor.
Figure 3. Intracellular changes on activation of the M2 receptor.
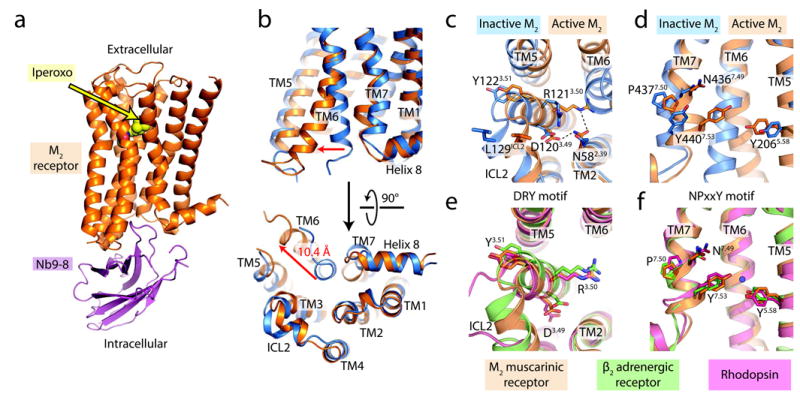
a, The overall structure of the active-state M2 receptor (orange) in complex with the orthosteric agonist iperoxo and the active-state stabilizing nanobody Nb9-8 is shown. b, Compared to the inactive structure of the M2 receptor (blue), transmembrane helix 6 (TM6) is substantially displaced outward, and TM7 has moved inward. Together, these motions lead to the formation of the G protein-binding site. c-d, Conserved motifs likewise show substantial changes on activation, and adopt conformations similar to those seen in the two other active GPCR structures (e-f). In particular, an interaction between two conserved tyrosines (Tyr5.58 and Tyr7.53) is likely mediated by a water molecule (blue circle), as seen in the high-resolution structure of the active β2AR [Ref. 18].
Like the active states of rhodopsin and the β2AR, the active M2 receptor shows rearrangements of the highly conserved DRY motif at the intracellular side of TM3 and the NPxxY motif in TM7 (Fig. 3c, d). In the active state of M2, Arg1213.50 of the DRY motif adopts an extended conformation virtually identical to that seen in metarhodopsin II and the β2AR-Gs complex (Fig. 3c, e, superscript numerals refer to the Ballesteros-Weinstein numbering system), and Asp1203.49 is stabilized by a hydrogen bond with Asn582.39 (Fig. 3c). To assess the importance of Asn582.39 for stabilization of the active conformation, we mutated it to alanine. The resulting mutant displayed normal ligand binding properties, but impaired ability to activate G protein (Extended Data Fig. 3a; Extended Data Table 2). Hence, it is likely that Asn582.39 either directly stabilizes the active conformation, or engages in direct interactions with G protein.
Extended Data Figure 3. Pharmacology.

a, Functional properties are shown for M2 receptors in which key residues were mutated. Agonist-induced increases in intracellular calcium levels were monitored via FLIPR using transfected COS-7 cells. Since some mutant receptors (N58A, D103E) were expressed at lower levels than the WT receptor, reference curves were obtained using cells transfected with either 3 μg DNA or 1 μg WT receptor DNA. The latter cells showed receptor expression levels comparable to those found with the N58A and D103E mutants (see Extended Data Table 2 for details). Data are given as means ± SEM of three independent experiments, each carried out in triplicate. AU, arbitrary units. b, The interaction between LY2119620 and iperoxo was measured by radioligand binding and functional assays. LY2119620 enhances the affinity of iperoxo (top graph) and its signaling potency (bottom graphs), and is also able to directly activate M2 receptor signaling as measured by [35S]GTPγS and ERK1/2 phosphorylation. Experiments were carried out with CHO cells stably expressing the human M2 receptor, and points are shown as mean ± SEM of three independent experiments, each carried out in duplicate.
Extended Data Table 2. Ligand binding properties of mutant M2 receptors.
Radioligand binding studies were carried out with membranes prepared from COS-7 cells transiently expressing the indicated mutant M2 receptor constructs. The WT M2 receptor was expressed at two different densities to allow for a more straightforward interpretation of the functional data shown in Supplementary Fig. 6 (see Online Methods for details). ACh and iperoxo binding affinities (Ki) were determined in radioligand competition binding assays as indicated. ACh and iperoxo binding affinities (Ki) were determined in [3H]-QNB competition binding assays for the N404Q6.52 mutant, which did not bind [3H]-NMS with sufficient affinity. Data are given as means ± SEM from two or three independent experiments, each performed in duplicate.
| Receptor | [3H]-NMS binding | ACh binding | Iperoxo binding | |
|---|---|---|---|---|
| KD | Bmax | Ki | Ki | |
| nM | pmol/mg of protein | μM | μM | |
| WT M2 | 1.48 ± 0.31 | 1.79 ± 0.17 | 2.74 ± 0.13 | 0.0073 ± 0.0006 |
| WT M2(1 μg DNA) | 1.40 ± 0.02 | 0.60 ±0.14 | ||
| N58A2.39 | 1.15 ± 0.05 | 0.62 ± 0.14 | 0.84 ± 0.21 | 0.0053 ± 0.0012 |
| D103E3.32 | 2.57 ± 0.58 | 0.51 ± 0.10 | 327 ± 91 | 2.76 ± 0.82 |
| Y206F5.58 | 1.67 ± 0.26 | 1.87 ± 0.26 | 31.8 ±0.71 | 0.52 ± 0.20 |
| N404Q6 52 | N.D.* | |||
| Receptor | [3H]-QNB binding | ACh binding | Iperoxo binding | |
|---|---|---|---|---|
| KD | Bmax | Ki | Ki | |
| nM | pmol/mg of protein | μM | μM | |
| WT M2 | 0.058 ± 0.015 | 1.63 ± 0.19 | 1.25 ± 0.07 | 0.0130 ± 0.0070 |
| N404Q6.52 | 9.47 ± 2.22 | 1.24 ± 0.12 | 34.3 ± 10.3 | 1.70 ± 0.29 |
no detectable [3H]-NMS binding activity
Similar to the DRY motif, the NPxxY region in TM7 shows significant rearrangements on activation (Fig. 3d). Most striking is a partial “unwinding” of TM7 around Tyr4407.53. This positions Tyr4407.53 of the NPxxY motif in close proximity to the highly conserved residue Tyr2065.58 (Fig. 3d). Although these two residues are not close enough to directly interact, their proximity may allow formation of a water-mediated hydrogen bond, as seen in the active-state structures of the β2AR18 and rhodopsin12. Indeed, the position of these two tyrosine residues is highly similar in the active structures of rhodopsin, β2AR, and the M2 receptor (Fig. 3f), suggesting that this feature represents a hallmark of GPCR activation. In addition, a molecular dynamics study recently predicted that Tyr2065.58 and Tyr4407.53 interact in the active conformation of the M2 receptor19, although this model was in other ways dissimilar from the structures presented here.
To assess the importance of this interaction for M2 receptor activation, we mutated Tyr2065.58 to phenylalanine, eliminating its ability to interact with Tyr4407.53 via a bridging water molecule. The Y206F mutant receptor could no longer be activated by acetylcholine (Extended Data Fig. 3a) and gave only a very weak functional response upon treatment with iperoxo. In addition, agonist affinity was reduced by greater than 10-fold (Extended Data Table 2), while antagonist binding was largely unaffected. These results suggest that the Tyr2065.58-Tyr4407.53 interaction stabilizes the active conformation of the receptor in a manner reminiscent of the “ionic lock” interaction20, which stabilizes the inactive conformation of family A GPCRs.
Activation mechanism
While activation of the β2AR and rhodopsin is associated with modest conformational changes in the orthosteric ligand-binding site, striking structural changes are observed in the M2 receptor. The activated M2 receptor shows a small orthosteric binding site, which completely occludes the agonist iperoxo from solvent (Fig. 4a, b). Indeed, the muscarinic inverse agonist quinuclidinyl benzilate (QNB) is too large to be accommodated in this binding cavity, perhaps accounting for its ability to suppress basal activity of the M2 receptor.
Figure 4. Orthosteric ligand binding site.
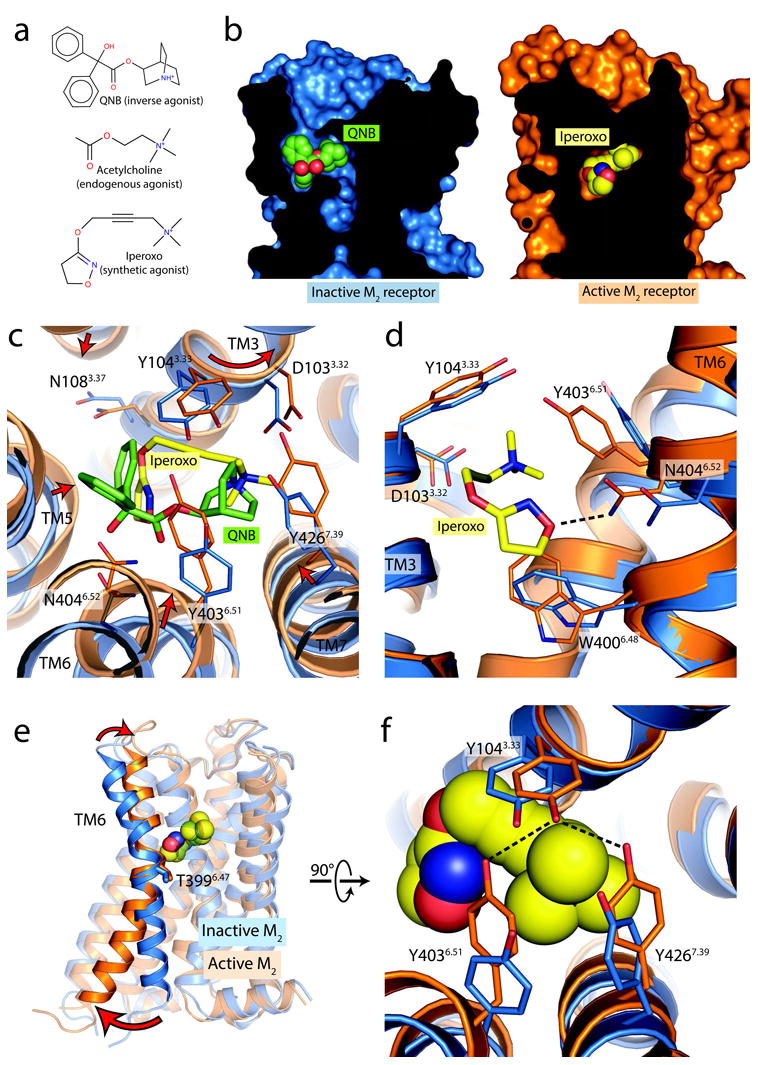
a, Orthosteric ligands used for crystallization of inactive and active M2 receptor are shown in. b, Cross-sections through the receptor are shown, with the interior in black. In the inactive conformation, the receptor (blue, at left) partially encloses the antagonist QNB, while the active conformation receptor encloses the agonist entirely, such that it is completely buried within the receptor (orange, at right). c, Conformational changes within the ligand binding pocket are shown from the extracellular side in, with changes highlighted as red arrows. d, A side view shows the inward motion of TM6, which is required for the formation of a hydrogen bond between Asn4046.52 and the agonist iperoxo. e, Activation thus involves a pivot of TM6, which moves inward in the orthosteric site and outward at the intracellular side. f, The closure of the binding pocket allows the formation of a hydrogen bonded tyrosine lid, located superficial to the agonist.
Within the active orthosteric binding pocket, the agonist iperoxo adopts a bent conformation (Fig. 4c; Extended Data Fig. 4). TM helices 5, 6, and 7 move inward, toward the agonist, in the active M2 conformation. TM3, in contrast, undergoes a slight rotation about its axis, but has almost no inward motion toward the ligand. The largest differences between inactive and active states of the M2 receptor involve TM6, where an inward movement of 2 Å at the α-carbon of Asn4046.52 allows for formation of a hydrogen bond between its side chain and iperoxo.
Extended Data Figure 4. Binding site diagram.

M2 receptor residues interacting with the orthosteric agonist iperoxo and the positive allosteric modulator LY2119620 are shown. Polar contacts are highlighted as red dotted lines, and hydrophobic contacts are in green solid lines.
Despite these activation-related structural changes, polar contacts between the agonist iperoxo and the receptor resemble those with QNB bound to the inactive M2 receptor. In particular, the conserved Asp1033.32 serves as a counter-ion to the ligand amine in both cases, and Asn4046.52 engages in hydrogen bonding with both ligands. The smaller size of iperoxo relative to QNB results in more limited hydrophobic contacts, however. This is particularly true along TM5, which engages the phenyl rings of QNB, but makes more limited hydrophobic contact with iperoxo in the active receptor conformation.
The hydrogen bond between Asn4046.52 and the iperoxo isoxazoline oxygen is analogous to the hydrogen bond between this residue and the QNB carbonyl in the inactive receptor state; however, the smaller size of iperoxo necessitates an inward motion of TM6 (Fig. 4d, e). To investigate the role of this hydrogen bond in receptor activation, we mutated Asn4046.52 to glutamine, which, due to the longer side chain, would allow TM6 to form a hydrogen bond with iperoxo in the inactive receptor. Consistent with a previous mutagenesis study21, the N404Q mutant receptor failed to bind detectable amounts of [3H]-NMS, but retained the ability to specifically bind [3H]-QNB, although with 163-fold reduced affinity (Extended Data Table 2). Similarly, the binding affinities for acetylcholine (ACh) and iperoxo were reduced, and while the N404Q mutant was able to activate G protein in response to both iperoxo and ACh, the concentration-response curves were shifted to the right by about 100-fold (Extended Data Fig. 3a), likely due to the reduced agonist binding affinities. Nevertheless, it remains possible that a structural reorientation of Asn4046.52 also contributes to M2 receptor activation.
Like Asn4046.52, Asp1033.32 plays a central role in receptor binding to iperoxo, engaging the trimethyl ammonium ion. Cation-π interactions with Tyr1043.33, Tyr4036.51 and Tyr4267.39 form an aromatic lid over the ligand amine (Fig. 4f). To assess the contribution of Asp1033.32 to receptor activation, we generated and analyzed the D103E mutant M2 receptor, which abolished agonist-induced M2 receptor activation (Extended Data Fig. 3a). The D103E mutant receptor bound [3H]-NMS with WT-like affinity but showed greatly reduced affinities for ACh (∼120-fold) and iperoxo (∼380-fold) (Extended Data Table 2), indicating that Asp1033.32 recognition of the ligand cation plays a critical role in both agonist binding and receptor activation.
In the active state of the M2 receptor, the inward motion of the upper portion of TM6 allows Tyr4036.51 to hydrogen bond with Tyr1043.33, which in turn hydrogen bonds to Tyr4267.39 (Fig. 4f), resulting in closure of the aforementioned tyrosine lid over the agonist. Hydrogen bonding of this lid appears to be an important feature of agonist binding and activation in muscarinic receptors: mutation of any of the three tyrosines to Phe leads to impaired agonist binding in the homologous M3 muscarinic receptor22, and mutation of Tyr1043.33 and Tyr4036.51 in the M2 receptor has a similar effect23,24. It should be noted that the structure of active M2 receptor bound to other agonists, including acetylcholine, might show differences as compared to the iperoxo-bound structure presented here.
Allosteric modulation
Muscarinic receptors have long served as important model systems for understanding allosteric modulation of GPCR signaling5,6,25. The structures of the inactive M2 and M3 receptors confirmed that these receptors possess a large extracellular vestibule, which has been shown to bind to allosteric modulators26,27. Situated directly above (i.e., extracellular to) the orthosteric site, this cavity also shows a substantial contraction upon activation of the M2 receptor due to the rotation of TM6 (Fig. 4b). The motion of TM6 thus provides a structural link among three regions of the receptor: the extracellular vestibule, the orthosteric binding pocket, and the intracellular surface. The structural coupling of these three regions likely accounts for the fact that allosteric modulators can affect the affinity and efficacy of orthosteric ligands and can also directly activate G proteins as allosteric agonists28.
To better understand how allosteric modulators act at GPCRs, we crystallized the iperoxo-occupied M2 receptor with LY2119620, a positive allosteric modulator (Fig. 5a). This agent has not been studied previously, so we characterized its affinity for the M2 receptor and its allosteric interaction with iperoxo (see Supplementary methods). Radioligand binding assays revealed that LY2119620 has similar pharmacological properties to its congener, LY203329829 (Extended Data Fig. 3b; Extended Data Table 3). It exhibits strong positive cooperativity with iperoxo, and mild negative cooperativity with the inverse agonist [3H]-NMS. While LY2119620 enhances the affinity of the M2 receptor for iperoxo, it does not significantly change the efficacy of this orthosteric agonist (Extended Data Table 3). We also observed that LY2119620 is capable of directly activating the M2 receptor, albeit with low potency and efficacy relative to iperoxo (Extended Data Table 3).
Figure 5. Structure of a GPCR allosteric modulator complex.
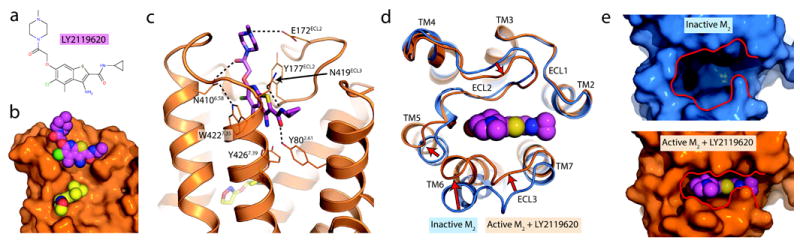
a, The M2 receptor occupied by the orthosteric agonist iperoxo was crystallized in complex with the positive allosteric modulator LY2119620. b, The allosteric ligand binds to the extracellular vestibule just above the orthosteric agonist. A cross-section through the membrane plane shows the relative positions of the two ligands. c, Several polar contacts are involved in LY2119620 binding, in addition to extensive aromatic stacking interactions with Trp4227.35 and Tyr177ECL2. d, Upon activation, the M2 receptor undergoes substantial conformational changes in the extracellular surface, leading to a contraction of the extracellular vestibule. e, This creates a binding site that fits tightly around the allosteric modulator, which would otherwise be unable to interact extensively with the extracellular vestibule in the inactive receptor conformation.
Extended Data Table 3. Pharmacological characterization of LY2119620.
a, Allosteric ternary complex model binding parameters for the interaction between LY2119620, iperoxo, and [3H]-NMS at the human M2 receptor. b, Operational model parameters for the functional allosteric interaction between iperoxo and LY2119620 at the human M2 receptor. Estimated parameter values represent the mean ± SEM of three experiments performed in duplicate.
| a
| ||
|---|---|---|
| Parameter | Value | |
| pKB* | 5.77 ± 0.10 | |
| pK|† | 8.51 ± 0.04 | |
| Logα‡ | 1.40 ± 0.09 (α = 25) | |
| Logα'§ | -0.26 ± 0.03 (α' = 0.6) | |
| b
| ||
|---|---|---|
| Parameter | [35S]GTPγS | ERK1/2 |
| pKB* | 5.73 ± 0.11 | 5.84 ± 0.18 |
| logαβ ‖ | 1.42 ± 0.09 | 1.30 ± 0.20 |
| (αβ = 26) | (αβ = 20) | |
| logτB¶ | -0.28 ± 0.05 | 0.33 ± 0.09 |
Negative logarithm of the equilibrium dissociation constant of LY2119620
Negative logarithm of the equilibrium dissociation constant of iperoxo
Logarithm of the binding cooperativity factor between LY2119620 and iperoxo (antilogarithm shown in parentheses)
Logarithm of the binding cooperativity factor between LY2119620 and [3H]-NMS (antilogarithm shown in parentheses)
Logarithm of the product of the binding cooperativity (α) and activation modulation (β) factors between iperoxo and LY2119620. Antilogarithm shown in parentheses.
Logarithm of the operational efficacy parameter of LY2119620 as an allosteric agonist.
Crystals of the M2 receptor bound to LY2119620 grew under identical conditions to those without the modulator, and the structure revealed unambiguous electron density for LY2119620 in the extracellular vestibule (Extended Data Fig. 5). The modulator is positioned directly above the orthosteric agonist (Fig. 5b), and it engages in extensive interactions with the extracellular vestibule. Specifically, the aromatic rings of the modulator are situated directly between Tyr177ECL2 and Trp4227.35, forming a three-layered aromatic stack. Importantly, a prior mutagenesis study implicated Tyr177ECL2 as a likely contact for the LY2119620 congener, LY2033298, at the M2 muscarinic receptor29. Several polar interactions are also seen (Fig. 5c). In particular, Tyr802.61, Asn4106.58, and Asn419ECL3 form hydrogen bonds to the modulator, and Glu172ECL2 engages in a charge-charge interaction with the ligand piperidine. LY2119620 binds at a site directly superficial to the orthosteric site, separated only by the tyrosine lid, with Tyr4267.39 interacting with both ligands.
Extended Data Figure 5. Electron density.

a-b, Fo-Fc omit maps are shown in gray, contoured at 2.5 σ within a 2.5 Å radius of the indicated ligand. c-f, 2Fo-Fc maps are shown in blue, contoured at 1.5 σ within a 2.0 Å radius of the indicated region.
The structure of the M2-iperoxo-LY2119620 complex is largely the same as that of receptor and agonist without LY2119620, suggesting that the allosteric binding site is largely pre-formed in the presence of agonist. The extracellular vestibule shows a slight additional contraction around the allosteric ligand (Extended Data Fig. 6). This subtle change stands in contrast to the substantial closure of the extracellular vestibule in the two active structures relative to the inactive conformation (Fig. 5d). A notable exception is Trp4227.35, which adopts a vertical conformation in the presence of LY2119620 and a horizontal conformation with iperoxo alone (Extended Data Fig. 6b). The vertical conformation of this residue in the M2-iperoxo-LY2119620 complex allows it to engage in an aromatic stacking interaction with the modulator, consistent with mutagenesis results implicating Trp4227.35 in the binding of other allosteric modulators30. The effect of mutagenesis of Trp4227.35 on LY2119620 affinity has not been tested, however. Closure of the LY2119620 binding site in the agonist-bound M2 receptor allows far more extensive interactions with the modulator than the inverse agonist-bound conformation (Fig. 5e), likely accounting for the ability of the modulator to enhance agonist binding affinity by preferentially slowing agonist dissociation.
Extended Data Figure 6. Comparison of M2 receptor structures with and without LY2119620 bound.

Comparison of the structure of active M2 receptor with and without the allosteric modulator LY2119620 reveals that there are few differences outside the extracellular vestibule. Within the extracellular vestibule, there is a slight contraction in the presence of the modulator, and Trp4227.35 undergoes a change of rotamer (panel b, red arrow).
The closed, active conformation of the extracellular vestibule is largely the consequence of the inward motion of TM6, which directly contacts the allosteric modulator, the orthosteric agonist, and likely the G protein as well. Stabilization of the closed extracellular vestibule by LY2119620 and other allosteric modulators may directly stabilize the open, active conformation of the intracellular side of TM6, accounting for the phenomenon of allosteric agonism in addition to positive cooperativity with orthosteric agonists. However, while the differences in TM6 between inactive and active structures can be described as a rigid-body motion, we cannot exclude the possibility that TM6 is flexible, allowing independent conformational changes in the G-protein binding site, the orthosteric site, and the extracellular vestibule.
Conclusions
The structures presented here offer insights into the structural basis for muscarinic receptor activation, and allosteric modulation by a drug-like molecule. In contrast to rhodopsin and the β2AR, extensive changes are seen in the orthosteric binding site and in the extracellular vestibule upon M2 receptor activation. The structure of active M2 receptor bound to the allosteric modulator LY2119620 definitively establishes the extracellular vestibule as an allosteric binding site, and shows that the allosteric modulator induces few additional structural changes as compared to those seen with orthosteric agonist alone. The structures presented here offer only a single view of an active muscarinic receptor; more work will be required to identify additional active states that may exist. Nonetheless, the information presented here provides a structural framework for future studies of GPCR activation and allostery, and may facilitate the development novel therapeutics.
Online Methods
Determination of M2 activation via inositol phosphate (IP) assays
Agonist-induced activation of the human M2 muscarinic receptor was studied in IP accumulation assays as described31. For M2 activation studies, HEK 293 cells were transiently cotransfected with cDNAs encoding the human M2 receptor (Missouri S&T cDNA Resource Center, Rolla, MO) and the hybrid G-protein Gαqi5 (Gαq protein with the last five amino acids at the C-terminus replaced by the corresponding sequence of Gαi; gift from The J. David Gladstone Institutes, San Francisco, CA)32. Twenty-four hours after transfection, cells were transferred into 24 well plates at a density of 100,000 cells per well in a volume of 270 μL. After addition of 30 μL of myo-[3H]inositol (specific activity = 22.5 Ci/mmol, PerkinElmer, Rodgau, Germany), cells were incubated for 15 hrs. Then, medium was aspirated, the cells were washed with serum-free medium supplemented with 10 mM LiCl, and test compounds (diluted in serum-free medium supplemented with 10 mM LiCl) were added at 37 °C for 60 min. Cells were then lysed by adding 150 μL of ice-cold 0.1 M NaOH for 5 min. After neutralisation with 50 μl of 0.2 M formic acid, the cell extract was diluted in buffer (5 mM sodium tetraborate, 0.5 mM Na-EDTA) and separated by anion-exchange chromatography using an AG1-X8 resin (Bio-Rad, Munich, Germany). After washing with water and elution-buffer A (5 mM sodium tetraborate, 60 mM sodium formate) and again with water, total IP was eluted with 2.5 mL elution-buffer B (1.0 M ammonium formate) and directly collected into scintillation counting vials. Radioactivity was measured by scintillation counting after adding 2.5 ml of Emulsifier-Safe (PerkinElmer, Rodgau, Germany). Data were analysed by normalizing dpm values with 0% for the non-stimulated receptor and 100% for the full effect of the reference iperoxo. Dose-response curves were calculated by non-linear regression using the Graphpad Prism 5 software.
Irreversible activation of the M2 receptor was tested at 1 nM FAUC123 in comparison to the reversible ligand iperoxo (1 nM). After incubation for 30 min, the antagonist atropine (1 μM) was added to one half of the sample (buffer was added to the other half) and incubations were continued for an additional 90 min. Total IP accumulation was determined as described above.
LY2119620 pharmacology
To characterize the allosteric interaction between LY2119620 and iperoxo, we performed radioligand binding and cellular functional assays at the wild-type human M2 muscarinic receptor stably expressed in a CHO FlpIn cell line. Increasing concentrations of LY2119620 caused a modest reduction in the specific binding of the orthosteric antagonist, [3H]-NMS, indicating weak negative cooperativity, but robustly enhanced the potency of iperoxo to compete for [3H]-NMS binding, indicating positive cooperativity with the agonist (Extended Data Fig. 3b). Application of an allosteric ternary complex model33,34 to these data yielded the values shown in Extended Data Table 3 for ligand affinity and cooperativities with agonist and antagonist. We then investigated the functional effect of LY2119620 on M2 muscarinic receptor signaling via monitoring receptor-mediated [35S]-GTPγS binding to activated G proteins, or phosphorylation of ERK1/2. [35S]-GTPγS binding was chosen as a proximal measure of receptor activation, while the pERK1/2 assay was chosen because it measures a downstream response that is also a point of convergence of multiple cellular pathways, some of them potentially G protein-independent. In both instances, LY2119620 caused receptor activation in its own right, indicating that the modulator can act as an allosteric agonist, while simultaneously enhancing the potency of iperoxo (Extended Data Fig. 3b). Application of an operational model of allosterism35 to these data yielded the parameter values shown in Extended Data Table 3. Comparison of the binding and functional data indicated that there was no significant difference between any of the pKB estimates of the affinity of LY2119620 for the allosteric site on the free receptor between assays. There was also no significant difference between the cooperativity factors with iperoxo across the assays, indicating that the molecular mechanism of action of LY2119620 is consistent with positive modulation of agonist affinity only, with minimal additional effects on agonist efficacy. This is in contrast to the more complex behavior previously noted with the congener, LY2033298, at the M2 receptor29. Full methods details are available in the Supplementary Methods.
M2 muscarinic receptor expression and purification
The human M2 muscarinic receptor gene was modified to remove glycosylation sites, and to add an amino-terminal FLAG epitope tag and a carboxy-terminal 8×His tag. In addition, residues 233 – 374 of intracellular loop 3 were deleted. This region has previously been shown to be unstructured36 and is not essential for G protein coupling in the homologous M1 muscarinic receptor37. This construct was expressed in Sf9 insect cells using the BestBac baculovirus system (Expression Systems; Davis, CA). Cells were infected at a density of 4 × 106 cells/mL and then incubated for two days at 27 °C. Receptor was extracted and purified in the manner described previously for the M3 muscarinic receptor8. Briefly, receptor was purified by Ni-NTA chromatography, FLAG affinity chromatography and size exclusion chromatography.
Llama immunization samples
M2 receptor was prepared as described above, and bound to iperoxo by including it at 10 μM starting at FLAG wash steps and in all subsequent buffers. Receptor was reconstituted into phospholipid vesicles composed of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) and Lipid A in a 10:1 (w:w) ratio, then aliquoted at 1 mg/mL receptor concentration and frozen in 100 μL aliquots prior to injection.
Yeast display samples
M2 receptor was purified as described above with 1 μM atropine included in all buffers. Receptor was then labeled with a 5-fold molar excess of biotin-NHS ester (Sigma-Aldrich; St. Louis, MO) in buffer containing 25 mM HEPES pH 7.2. Following a 30 min incubation at room temperature and a 30 min incubation on ice, unreacted label was quenched with 50 mM tris pH 8. Directly labeled samples with fluorophore-NHS esters were prepared in a similar manner. Receptor was then desalted into buffer containing either 10 μM tiotropium, 10 μM iperoxo, or buffer containing no ligand. Receptor eluted in buffer containing no ligand was treated with 50 μM iperoxo mustard (FAUC123; see Supplementary Information for details) for 20 min at room temperature. Samples were then concentrated, aliquoted, and flash frozen with 20% (v/v) glycerol.
Crystallization samples
M2 receptor for crystallization was prepared as described above. When bound to FLAG resin, the sample was washed with a mix of dodecyl maltoside buffer (DDM) and buffer containing 0.2% lauryl maltose neopentyl glycol detergent (MNG; Anatrace). These buffers were mixed first in a 1:1 ratio (DDM:MNG buffer), then 1:4, and 1:10 ratios. At each step the 5 mL column was washed with 10 mL of buffer at a 1 mL/min flow rate, and all buffers contained 1 μM atropine. Finally, the column was washed with 10 mL MNG buffer, and then 10 mL of low detergent buffer with agonist (0.01% MNG, 0.001% cholesterol hemisuccinate, 20 mM HEPES pH 7.5, 100 mM NaCl, 10 μM iperoxo). The sample was eluted, mixed with a 1.5-fold stoichiometric excess of Nb9-8 and a second nanobody, NbB4. This nanobody binds to an epitope different from Nb9-8, but was not resolved in the crystal structure. Following mixing, the sample was incubated 30 min on ice, then concentrated and purified by size exclusion in low detergent buffer. Eluted protein was concentrated to A280 = 96, and frozen in liquid nitrogen in 7 μL aliquots.
Llama immunization
One Llama (Lama glama) was immunized for six weeks with 1 mg receptor in total. Peripheral blood lymphocytes were isolated from the immunized animal to extract total RNA. cDNA was prepared using 50 μg of total RNA and 2.5 μg of oligo-dN6primer. Nanobody open reading frames were amplified as described38.
Post-immune M2 receptor llama nanobody library construction
Nanobody VHH fragments were amplified by PCR using the primers pYalNB80AMPF (CATTTTCAAT TAAGATGCAG TTACTTCGCT GTTTTTCAAT ATTTTCTGTT ATTGCTAGCG TTTTAGCAAT GGCCCAGGTG CAGCTGCAGG AG) and pYalNB80AMPR (CCACCAGATC CACCACCACC CAAGTCTTCT TCGGAGATAA GCTTTTGTTC GGATCCTGAG GAGACGGTGA CCTGGGTCCC). The PCR products were then co-transformed with linearized pYal into yeast strain EBY100 as for the Nb80 affinity-maturation library, yielding a library size of 0.6×108 transformants.
Selection of M2 Gi-mimetic nanobodies from post-immune M2 llama nanobody library
For the first round of selection, counter-selection was performed against the β2 receptor to remove yeast clones that bind non-specifically to membrane proteins or to secondary staining reagents. 1.0×109 of induced yeast were washed with PBEM buffer and then stained in 5 mL of PBEM buffer containing 1 μM biotinylated β2 receptor liganded with carazolol for 1 hr at 4 °C. Yeast were then stained with streptavidin-647 as a secondary reagent and magnetically-labeled with anti-647 microbeads (Miltenyi) as described previously18. Positively-labeled yeast were then removed by the use of an LD column (Miltenyi); the cleared flow-through was then used for subsequent selection. Positive selection for clones recognizing the active state of the M2 receptor was performed by staining with 2 μM biotinylated M2 receptor bound to the agonist iperoxo in 5 mL PBEM buffer supplemented with 2 μM iperoxo for 1 hr at 4°C. Yeast were then washed, stained with streptavidin-647, and magnetically-labeled with anti-647 microbeads, including 1 μM iperoxo in the PBEM buffer at all steps. Magnetic separation of M2 receptor-binding yeast clones was performed using an LS column (Miltenyi) following the manufacturer's instructions. Magnetically sorted yeast were resuspended in SDCAA medium and cultured at 30°C. Rounds 2-4 were selected in a similar manner, counter-selecting against 1 μM biotinylated β2 receptor bound to carazolol and positively selecting using 1 μM biotinylated M2 receptor bound to iperoxo. For these rounds, the scale was reduced ten-fold to 1 × 108 induced yeast and staining volumes of 0.5 mL.
Conformational selection was performed for rounds 5-9. For rounds 5-8, yeast were stained with 1 μM biotinylated M2 receptor pre-incubated with the high-affinity antagonist tiotropium for 1 hr at 4°C. Yeast were then fluorescently labeled with either streptavidin-647 or streptavidin-PE, and magnetically labeled with the corresponding anti-647 or anti-PE microbeads (Miltenyi). Depletion of inactive-state binders was carried out using an LS column. The cleared yeast were then positively selected by staining with 0.5 μM (rounds 5-7) or 0.1 μM (round 8) M2 receptor pre-bound to iperoxo for 1 hr at 4° C. Yeast were then fluorescently-labeled with either streptavidin-PE or streptavidin-647, using a fluorophore distinct from that used in the previous counter-selection step. Magnetic separation of agonist-occupied M2 receptor was performed using an LS column, as for steps 1-4. For round 9, two-color FACS was performed. Induced yeast were simultaneously stained with 1 μM Alexa647-labeled M2 receptor reacted with iperoxo mustard and 1 μM Alexa488-labeled M2 receptor pre-bound with tiotropium for 1 hr at 4°C. Alexa647 positive/Alexa488 negative yeast were purified using a FACS Jazz cell (BD Biosciences) sorter. Post-sorted yeast were plated onto SDCAA-agar plates and the nanobody-encoding sequences of several colonies were sequenced. Full sequences of clones confirmed to enhance agonist affinity are below:
| Clone | Full amino acid sequence |
| Nb9-1 | QVQLQESGGG LVQAGGSLRL SCAASGHTFS SARMYWVRQA PGKEREFVAA ISRSGFTYSA DSVKGRFTIS RDIANNTVYL QMNSLQPEDT AIYTCYAAYL DEFYNDYTHY WGLGTQVTVS S |
| Nb9-8 | QVQLQESGGG LVQAGDSLRL SCAASGFDFD NFDDYAIGWF RQAPGQEREG VSCIDPSDGS TIYADSAKGR FTISSDNAEN TVYLQMNSLK PEDTAVYVCS AWTLFHSDEY WGQGTQVTVS S |
| Nb9-20 | QVQLQESGGG LVQPEGSLTL ACDTSGFTMN YYAIAWFRQA PEKEREGLAT ISSIDGRTYY ADSVKGRFTI SRDSAKNMVY LQMNNLRPED TAVYYCSAGP DYSDYGDESE YWGQGTQVTV SS |
Expression of MBP-nanobody fusions in E. coli
Nanobody sequences were subcloned into a modified pMalp2x vector (New England Biolabs), containing an amino-terminal, 3C protease-cleavable maltose binding protein (MBP) tag and a carboxy-terminal 8×His tag. Plasmids were transformed into BL21(DE3) cells and protein expression induced in Terrific Broth by addition of IPTG to 1 mM at an OD600 of 0.8. After 24 hr of incubation at 22°C, cells were harvested and periplasmic protein was obtained by osmotic shock. MBP-nanobody fusions were purified by Ni-NTA chromatography and MBP was removed using 3C protease. Cleaved MBP was separated from the 8×His tagged nanobodies by an additional Ni-NTA purification step. The 8×His tag was subsequently removed using carboxypeptidase A.
Expression and purification of G protein
Heterotrimeric Gi was prepared by expression using a single baculovirus for the human Gαi1 subunit and a second, bicistronic virus for human Gβ1 and Gγ2 subunits. G protein was expressed in HighFive insect cells, and then purified as described previously for Gs10. In brief, G protein was extracted with cholate, purified by Ni-NTA chromatography, detergent exchanged into dodecyl maltoside buffer, and then purified by ion exchange and dialyzed prior to use.
M2 receptor radioligand binding assays with G protein and nanobody
M2 receptor was expressed and purified as described above. Receptor was then reconstituted into HDL particles consisting of apolipoprotein A1 and a 3:2 (mol:mol) mixture of the lipids POPC:POPG (1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine: 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) respectively, Avanti Polar Lipids). Binding reactions contained 50 fmol functional receptor, 0.6 nM [3H] N-methyl scopolamine (NMS), 100 mM NaCl, 20 mM HEPES pH 7.5, 0.1% BSA, and ligands and nanobodies as indicated. Concentration-dependent effects of nanobodies were measured in the presence of 10 nM iperoxo. All reactions were carried out in a 500 μL volume. For samples containing G protein, purified Gi heterotrimer from insect cells was added to the reactions at a 1000-fold dilution from a 200 μM stock, resulting in a large stoichiometric excess over receptor and diluting G protein below the detergent CMC to allow incorporation into HDL particles, essentially as described previously39. Reactions were mixed and then incubated for 2 hr. Samples were then filtered on a 48-well harvester (Brandel) onto a filter which had been pre-treated with 0.1% polyethylenimine. All measurements were taken by liquid scintillation counting, and experiments were performed at least in triplicate.
Site-directed mutagenesis
A mammalian expression plasmid coding for the human M2 muscarinic receptor (M2R-pcDNA3.1+) was obtained from the Missouri S&T cDNA Resource Center. Mutant M2 receptors were generated by using the QuikChange site-directed mutagenesis kit (Stratagene, Foster City, CA, USA) according to the manufacturer's instructions. The identity of all mutant M2 receptor constructs was confirmed by DNA sequencing.
Transient expression of receptor constructs in COS-7 cells
WT and mutant M2 receptors were transiently expressed in COS-7 cells grown in 100 mm dishes, as described previously40. For functional studies, the various receptor constructs (3 μg each) were co-expressed with a chimeric G protein α subunit (Gqi5; 3 μg plasmid DNA) in which the last five amino acids of Gαq were replaced with the corresponding Gαi sequence41.
Radioligand binding studies of mutant and WT M2 receptors
Acetylcholine (ACh) bromide was purchased from Sigma (St. Louis, MO, USA). Iperoxo was a generous gift of Bristol Myers Squibb (New York, NY). [3H]-NMS (85.5 Ci/mmol) and 3-quinuclidinyl benzilate ([3H]-QNB; 47.4 Ci/mmol) were from PerkinElmer Life Sciences (Downers Grove, IL, USA). Radioligand binding studies were carried out with membranes prepared from transfected COS-7 cells as described41. Forty-eight hours after transfection, cells were harvested and resuspended in 25 mM sodium phosphate buffer (pH 7.4) containing 5 mM MgCl2. Membrane homogenates were prepared and resuspended in the same buffer. [3H]-NMS or [3H]-QNB binding reactions were carried out in the presence of 9 μg of membrane protein for 3 hr at room temperature (total volume of the incubation mixture: 0.5 ml). In saturation binding studies, six different concentrations of the radioligand were used ([3H]-NMS, 0.3 nM to 10 nM; [3H]-QNB, 0.05 nM to 20 nM). In competition binding assays, membrane homogenates were incubated with ten different concentrations of ACh (13 nM to 1 mM) or iperoxo (0.13 nM to 10 μM) in the presence of a fixed concentration of radioligand (2 nM [3H]-NMS for all receptors except N404Q; 15 nM [3H]-QNB for N404Q and 0.5 nM [3H]-QNB for WT M2 receptor). Non-specific binding was determined in the presence of 10 μM atropine. Reactions were stopped by rapid filtration through GF/C filters. Data were analyzed using Prism 4.0 software (GraphPad Software, Inc., San Diego, CA).
Calcium mobilization assay
COS-7 cells co-expressing WT or mutant M2 receptor and the hybrid G protein, Gqi541, were incubated with increasing concentrations of agonists (ACh, 5 nM to 50 μM; iperoxo, 50 pM to 0.5 μM), and increases in intracellular calcium levels were determined in 96-well plates using FLIPR technology (Molecular Devices, Sunnyvale, CA), as described in detail previously42,43. Agonist concentration-response curves were analyzed using Prism 4.0 software.
Crystallization
Purified M2 receptor was reconstituted into lipidic cubic phase by mixing with a 1.5-fold excess by mass of 10:1 (w:w) monoolein cholesterol lipid mix. Protein and lipid were loaded into glass syringes (Art Robbins Instruments, Sunnyvale, CA), and then mixed 100 times by the coupled syringe method44. Samples of 30 – 100 nL in volume were spotted onto 96 well glass plates and overlaid en bloc with 600 nL precipitant solution for each well. Precipitant solution consisted of 10 – 20% PEG300, 100 mM HEPES pH 7.2 – 7.9, 1.2% 1,2,3-heptanetriol, and 20 – 80 mM EDTA pH 8.0. Identical conditions were used to crystallize LY2119620-receptor complexes, except that the overlay precipitant solution was supplemented with 500 μM LY2119620. Crystals grew in 24 hr, and reached full size within two days. Crystals were then harvested in mesh grid loops (MiTeGen, Ithaca, NY) with 10 – 50 crystals per loop and stored in liquid nitrogen prior to use.
Data collection
Grids of crystals were rastered at Advanced Photon Source beamlines 23ID-B and 23ID-D. Initial rastering was performed with an 80 μm by 30 μm beam with 5-fold attenuation and 1 sec exposure, and regions with strong diffraction were sub-rastered with a 10 μm collimated beam with equivalent X-ray dose. Data collection was similarly performed with a 10 μm beam, but with no attenuation and exposures of typically 1 – 5 s. An oscillation width of 1 – 2 degrees was used in each case, and wedges of 5 – 10 degrees were compiled to create the final data sets.
Data reduction and refinement
Diffraction data were processed in HKL200045, and statistics are summarized in Extended Data Table 1. The structure was solved using molecular replacement with the structure of the inactive M2 receptor (PDB ID: 3UON) and Nb80 (PDB ID: 3P0G) as search models in Phaser46. The resulting structure was iteratively refined in Phenix47 and manually rebuilt in Coot48. Final refinement statistics are summarized in Extended Data Table 1. Figures were prepared in PyMol (Schrödinger).
Supplementary Material
Acknowledgments
We acknowledge support from the National Science Foundation (graduate fellowship to A.C.K., and Award 1223785 to B.K.K) , the Stanford Medical Scientist Training Program (A.M. and A.M.R.), the American Heart Association (A.M.), the Ruth L. Kirschstein National Research Service Award (A.M.R.), National Institutes of Health grants NS02847123 and GM08311806 (B.K.K.), the Mathers Foundation (B.K.K., W.I.W., and K.C.G), the Deutsche Forschungsgemeinschaft for the grant GM 13/10-1 (K.E., H.H., P.G.), the National Health and Medical Research Council (NHMRC) of Australia program grant 519461 (P.M.S. and A.C.), NHMRC Principal Research Fellowships (P.M.S. and A.C.), and the Howard Hughes Medical Institute (K.C.G.). This work was supported in part by the Intramural Research Program, NIDDK, NIH, US Department of Health and Human Services (J.H., K.H., and J.W.). We thank Katie Leach for performing ERK assays, Briana Davie and Peter Scammells for synthesis of iperoxo. We thank Hongling Xiao, Carrie H. Croy, Douglas A. Schober for functional characterization of LY2119620. We thank Tong Sun Kobilka for preparation of affinity chromatography reagents and Foon Sun Thian for help with cell culture.
Footnotes
Full Methods and associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions: A.C.K. expressed and purified M2 receptor for yeast display and crystallographic experiments, performed crystallization, data collection, and structure refinement, and performed radioligand binding assays to validate nanobody activity. A.C.K. A.M.R. and A.M. designed experiments to identify nanobodies by yeast display. A.M.R. performed all yeast selections, and expressed and purified Nb9-8 and other nanobodies. J.H. and K.H. performed site-directed mutagenesis and characterization of resulting mutants. K.E. synthesized FAUC123. H.H. performed cell assays and radioligand binding to characterize FAUC123. C.V. performed pharmacological characterization of LY2119620. P.M.S. and A.C. supervised pharmacological characterization of LY2119620. C.C.F. designed key solubility, physical chemistry and ligand analysis to select LY2119620 as an appropriate co-crystallization candidate for the M2 receptor. P.G. supervised synthesis and characterization of FAUC123. E.P. and J.S. performed llama immunization, cDNA production, and performed selections by phage display. W.I.W. supervised structure refinement. K.C.G. supervised yeast selection experiments. J.W. supervised mutagenesis experiments and analyzed results. B.K.K. provided overall project supervision, and with A.C.K., A.M.R., and A.M. wrote the manuscript with assistance from A.C. and J.W.
Author Information: Coordinates and structure factors for the active M2 receptor in complex with Nb9-8 and iperoxo are deposited in the Protein Data Bank under accession code 4MQS, and the coordinates and structure factors of the same complex bound additionally to the allosteric modulator LY2119620 are deposited under accession code 4MQT.
Reprints and permission information is available at www.nature.com/reprints. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 2.Peterson GL, Herron GS, Yamaki M, Fullerton DS, Schimerlik MI. Purification of the muscarinic acetylcholine receptor from porcine atria. Proc Natl Acad Sci USA. 1984;81:4993–4997. doi: 10.1073/pnas.81.15.4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kubo T, et al. Primary structure of porcine cardiac muscarinic acetylcholine receptor deduced from the cDNA sequence. FEBS Lett. 1986;209:367–372. doi: 10.1016/0014-5793(86)81144-9. [DOI] [PubMed] [Google Scholar]
- 4.Mohr K, Trankle C, Holzgrabe U. Structure/activity relationships of M2 muscarinic allosteric modulators. Receptors Channels. 2003;9:229–240. [PubMed] [Google Scholar]
- 5.Digby GJ, Shirey JK, Conn PJ. Allosteric activators of muscarinic receptors as novel approaches for treatment of CNS disorders. Mol Biosyst. 2010;6:1345–1354. doi: 10.1039/c002938f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Haga K, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruse AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe HW, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deupi X, et al. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc Natl Acad Sci USA. 2012;109:119–124. doi: 10.1073/pnas.1114089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 14.Nygaard R, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kloeckner J, Schmitz J, Holzgrabe U. Convergent, short synthesis of the muscarinic superagonist iperoxo. Tetrahedron Lett. 2010;51:3470–3472. [Google Scholar]
- 16.Hudgins PM, Stubbins JF. A comparison of the action of acetylcholine and acetylcholine mustard (chloroethylmethylaminoethyl acetate) on muscarinic and nicotinic receptors. J Pharmacol Exp Ther. 1972;182:303–311. [PubMed] [Google Scholar]
- 17.Spalding TA, Birdsall NJ, Curtis CA, Hulme EC. Acetylcholine mustard labels the binding site aspartate in muscarinic acetylcholine receptors. J Biol Chem. 1994;269:4092–4097. [PubMed] [Google Scholar]
- 18.Ring AM, et al. Adrenaline-activated structure of the β2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013 doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao Y, Nichols SE, Gasper PM, Metzger VT, McCammon JA. Activation and dynamic network of the M2 muscarinic receptor. Proc Natl Acad Sci USA. 2013;110:10982–10987. doi: 10.1073/pnas.1309755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballesteros JA, et al. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 21.Heitz F, et al. Site-directed mutagenesis of the putative human muscarinic M2 receptor binding site. Eur J Pharmacol. 1999;380:183–195. doi: 10.1016/s0014-2999(99)00439-2. [DOI] [PubMed] [Google Scholar]
- 22.Wess J, Maggio R, Palmer JR, Vogel Z. Role of conserved threonine and tyrosine residues in acetylcholine binding and muscarinic receptor activation. A study with m3 muscarinic receptor point mutants. J Biol Chem. 1992;267:19313–19319. [PubMed] [Google Scholar]
- 23.Vogel WK, Sheehan DM, Schimerlik MI. Site-directed mutagenesis on the m2 muscarinic acetylcholine receptor: the significance of Tyr403 in the binding of agonists and functional coupling. Mol Pharmacol. 1997;52:1087–1094. doi: 10.1124/mol.52.6.1087. [DOI] [PubMed] [Google Scholar]
- 24.Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–7474. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Amici M, Dallanoce C, Holzgrabe U, Trankle C, Mohr K. Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med Res Rev. 2010;30:463–549. doi: 10.1002/med.20166. [DOI] [PubMed] [Google Scholar]
- 26.Gregory KJ, Sexton PM, Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr Neuropharmacol. 2007;5:157–167. doi: 10.2174/157015907781695946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bock A, et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat Commun. 2012;3:1044. doi: 10.1038/ncomms2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.May LT, et al. Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007;72:463–476. doi: 10.1124/mol.107.037630. [DOI] [PubMed] [Google Scholar]
- 29.Valant C, Felder CC, Sexton PM, Christopoulos A. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol. 2012;81:41–52. doi: 10.1124/mol.111.074872. [DOI] [PubMed] [Google Scholar]
- 30.Prilla S, Schrobang J, Ellis J, Holtje HD, Mohr K. Allosteric interactions with muscarinic acetylcholine receptors: complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol Pharmacol. 2006;70:181–193. doi: 10.1124/mol.106.023481. [DOI] [PubMed] [Google Scholar]
- 31.Chee MJ, et al. The third intracellular loop stabilizes the inactive state of the neuropeptide Y1 receptor. J Biol Chem. 2008;283:33337–33346. doi: 10.1074/jbc.M804671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broach JR, Thorner J. High-throughput screening for drug discovery. Nature. 1996;384:14–16. [PubMed] [Google Scholar]
- 33.Ehlert FJ. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol. 1988;33:187–194. [PubMed] [Google Scholar]
- 34.Canals M, et al. A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem. 2012;287:650–659. doi: 10.1074/jbc.M111.314278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Ichiyama S, et al. The structure of the third intracellular loop of the muscarinic acetylcholine receptor M2 subtype. FEBS Lett. 2006;580:23–26. doi: 10.1016/j.febslet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 37.Shapiro RA, Nathanson NM. Deletion analysis of the mouse m1 muscarinic acetylcholine receptor: effects on phosphoinositide metabolism and down-regulation. Biochemistry. 1989;28:8946–8950. doi: 10.1021/bi00448a039. [DOI] [PubMed] [Google Scholar]
- 38.Conrath KE, et al. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother. 2001;45:2807–2812. doi: 10.1128/AAC.45.10.2807-2812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu J, et al. Structural basis of G protein-coupled receptor-G protein interactions. Nat Chem Biol. 2010;6:541–548. doi: 10.1038/nchembio.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Conklin BR, Blin N, Yun J, Wess J. Identification of a receptor/G-protein contact site critical for signaling specificity and G-protein activation. Proc Natl Acad Sci USA. 1995;92:11642–11646. doi: 10.1073/pnas.92.25.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, et al. Rapid identification of functionally critical amino acids in a G protein-coupled receptor. Nat Methods. 2007;4:169–174. doi: 10.1038/nmeth990. [DOI] [PubMed] [Google Scholar]
- 43.McMillin SM, Heusel M, Liu T, Costanzi S, Wess J. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J Biol Chem. 2011;286:28584–28598. doi: 10.1074/jbc.M111.259788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. In: Methods in Enzymology. Carter Charles W., Jr, editor. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr Biol Crystallogr. 2012;D68:352–367. doi: 10.1107/s0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Biol Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.