

Abstract
Histiocytic sarcoma is a rare, aggressive neoplasm that responds poorly to therapy. Histiocytic sarcoma is thought to arise from macrophage precursor cells via genetic changes that are largely undefined. To improve our understanding of the etiology of histiocytic sarcoma we conducted a forward genetic screen in mice using the Sleeping Beauty transposon as a mutagen to identify genetic drivers of histiocytic sarcoma. Sleeping Beauty mutagenesis was targeted to myeloid lineage cells using the Lysozyme2 promoter. Mice with activated Sleeping Beauty mutagenesis had significantly shortened lifespan and the majority of these mice developed tumors resembling human histiocytic sarcoma. Analysis of transposon insertions identified 27 common insertion sites containing 28 candidate cancer genes. Several of these genes are known drivers of hematological neoplasms, like Raf1, Fli1, and Mitf, while others are well-known cancer genes, including Nf1, Myc, Jak2, and Pten. Importantly, several new potential drivers of histiocytic sarcoma were identified and could serve as targets for therapy for histiocytic sarcoma patients.
Introduction
Histiocytic sarcoma (HS) is classified as a neoplastic proliferation with features of histiocytes/macrophages[1]. HS has also been called true histiocytic lymphoma or malignant histiocytosis, but these terms have been discontinued. Before 1990, the majority of patients diagnosed with HS were misdiagnosed due to a lack of antibodies specific for the histiocytic lineage. Retrospective analysis indicated the majority of these patients actually had B- or T-cell lymphomas[2]–[5]. Case studies have demonstrated that HS can occur in isolation or in the context of other hematological malignancies, such as B-cell lymphoma, to which the HS is sometimes clonally related[4]. HS may thus, in some cases, develop via trans-differentiation from a malignant, or premalignant, lymphoid neoplasm. HS is rare, with an incidence far less common than the non-Hodgkin lymphomas[1], [6]. Typically, patients present with advanced clinical disease and have a poor prognosis[1], [4], [5]. Since the genetic etiology of HS is largely unknown, HS is difficult to manage clinically and there is no standard therapy for patients with HS.
Currently, no precursor lesions or etiologic agents have been described for human HS[7]. Two cytogenetic case studies identified gains in chromosome 8 in human HS[8], [9], implicating MYC as a HS oncogene. Animal models have identified possible driver genetic lesions. ArrayCGH performed on over 100 canine HS samples revealed an average of 30 copy number alterations per tumor[10], while a genome wide association study in Bernese Mountain Dogs identified a strong association between HS and the MTAP-CDKN2A locus[11]. Pten and Ink4aARF are also implicated, as compound heterozygous mice develop HS and 60% of human HS examined for protein expression show a loss of PTEN, p16INK4A, or p14ARF [12]. Several other genetic mouse models have produced HS including Dok1/Dok2/Dok3 triple knockout animals[13], Cyp1b1 knockout mice[14], p21 knockout mice[15], and p19ARF/Bax mutant mice[16]. In addition, 50% of Cdkn2a deficient mice infected with Moloney murine leukemia virus developed HS, which was frequently accompanied by lymphoma[17].
To identify genetic drivers of HS we performed an unbiased forward genetic screen in mice using the Sleeping Beauty (SB) transposon as an insertional mutagen[18]–[20]. SB is capable of both activating proto-oncogenes and inactivating tumor suppressor genes and has been used to identify genetic drivers in a variety of cancers[21]–[32]. In this study we activated SB mutagenesis using the Lysozyme2 (Lyz2) promoter in a cohort of mice resulting in early mortality and a large percentage of mice developing HS. Analysis of transposon CISs identified 28 genes, including 2 miRNAs associated with HS. Several of these genes are known oncogenes and tumor suppressors including Nf1, Pten, Myc and Fli1, while many others have not been directly associated with cancer and could be potential targets for therapy.
Methods and Materials
Ethics Statement
All mice were bred, cared for and euthanized in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee (Protocol # 0901A56501).
Transgenic Mice
Lyz-Cre mice were obtained from Jackson Laboratories (Strain name: B6.129P2-Lyz2tm1(cre)Ifo/J, Cat # 004781)[33]. These mice were created using a knock-in allele that has a nuclear localized Cre recombinase cDNA inserted into the first coding ATG of the Lyz2 gene. This allele abolishes endogenous Lyz2 gene function and places NLS-Cre expression under the control of the endogenous Lyz2 promoter/enhancer elements.
Rosa26-LsL-SB11 mice backcrossed to C57BL/6J were a generous gift from Adam Dupuy (University of Iowa). These mice were described previously[22].
Three strains of T2/Onc transgenic mice were used. The first two strains, T2/Onc(chr1) and T2/Onc(chr15), contained roughly 25 transposons resident as a concatamer on mouse chromosomes (MMU) 1 and 15, respectively[19]. The third strain, T2/Onc2(chr4), contained roughly 214 transposons resident as a concatamer on MMU 4[20].
Genotyping and Excision PCR
We isolated tail biopsy DNA using a standard phenol-chloroform extraction method. PCR was performed using primer sequences for each transgene. Primer sequences are as follows: LyzM-Cre: primer oIMR3066 5′- CCCAGAAATGCCAGATTACG- 3′, primer oIMR3067 5′- CTTGGGCTGCCAGAATTTCTC-3′, primer oIMR3068 5′- TTACAGTCGGCCAGGCTGAC-3′; T2Onc or T2Onc2: Forward 5′- CGCTTCTCGCTTCTGTTCGC-3′, Reverse 5′- CCACCCCCAGCATTCTAGTT-3′; LsL-SB11: Wild-type Forward 5′- GGAGGGGAGTGTTGCAATACCTTT-3′; Wild-type Reverse 5′- AACTCGGGTGAGCATGTCTTTAATCTAC-3′; Transgenic Forward 5′-GGCATTGGGGGTGGTGATATAAACT-3′; and T2Onc Excision PCR was performed as previously described[19] using primer sequences: Forward 5′- GGGATGTGCTGCAAGGCGAT-3′; Reverse 5′- CAAGCTATGCATCCAACGCGTT-3′.
PCR analysis of VDJ rearrangement at Tcrb and Igh loci
DNA was isolated from eight representative tumors and control tissues from wild-type animals. For Tcrb analysis, two forward primers in the V locus and one forward primer in the D locus were used in conjunction with a reverse primer in the J locus. For IgH analysis, two forward primers in the D locus were used with a reverse primer in the J locus. Primer sequences are as follows: Vb8.2 5′-CTACCCCCTCTCAGACATCA-3′, Jb2 5′-TGAGAGCTGTCTCCTACTATCGATT-3′, Vb11.5 5′-TGCTGGTGTCATCCAAACACCTAG, Db2 5′-GTAGGCACCTGTGGGGAAGAAACT-3′, VHQ52 5′-CGGTACCAGACTGARCATCASCAAGGAC-3′, VH7183 5′-CGGTACCAAGAASAMCCTGTWCCTGCAAATGASC-3′, JH3 5′-GTCTAGATTCTCACAAGAGTCCGATAGACCCTGG-3′.
Kaplan-Meier Analysis
Survival was examined using a Kaplan-Meier curve (Prism Software, Graph Pad) and statistically analyzed using the logrank test controlling for multiple comparisons through the Sidak method[34].
Histopathology and Immunohistochemistry
Mice were necropsied when moribund or at 1.5 years of age, whichever came first. Lungs, heart, lymph nodes, spleen, pancreas, sternum and all abnormal tissues were removed and visually inspected for macroscopic tumors. Tissues were either fixed in 10% formalin or snap frozen in liquid nitrogen. Formalin-fixed samples underwent standard tissue processing, were paraffin-embedded, mounted and sectioned at 5 µm. Sections were adhered to glass slides by heat fixation. Slides were processed and stained with hematoxylin-eosin (HE). Immunohistochemistry was conducted with citrate-based antigen retrieval. Tissues were stained with antibodies for Mac2 and F4/80 (Cedarlane, Burlington, NC clones M3/38 and CI:A3-1), Lyz and CD3e (Dako, Carpinteria CA polyclonal), and Pax5 (Santa Cruz, Santa Cruz CA). Tissues were analyzed by a board-certified pathologist (ML, American Board of Pathology).
Transposon insertion analysis
Genomic DNA was isolated using standard phenol-chloroform extraction and ethanol precipitation. DNA was subjected to linker-mediated PCR as previously described[23], except that primer sequences were changed to include 12 bp barcodes and Illumina HiSeq 2000 platform-specific sequences (sequences available upon request). PCR amplicons were subjected to sequencing using the Illumina HiSeq 2000 platform following manufacturer's protocol.
Sequences were mapped to the mouse genome using BOWTIE[35] using the TAPDANCE[36] bioinformatics pipeline. TAPDANCE identifies CISs based on analysis of varying genomic window sizes, tested for significance using the Poisson distribution (p<0.05) utilizing a Bonferroni correction based on number of windows examined. Based on the 1,575 unique regions, 3 insertions in an 8.9 KB window or 4 or more insertions within a 263 KB window were considered a CIS.
qRT-PCR
RNA was extracted from 5 mg tissue with the RNeasy Minikit (Qiagen, Valencia, CA, USA). Tissues corresponded to match normal/tumor samples from liver and spleen. RNA concentrations were determined in an Epoch spectrophotometer system (BioTek, Winooski, VT, USA). 1 ug of RNA was converted to cDNA with the ABI High-Capacity cDNA Reverse Transcription Kit (#4368814) according to manufacturer conditions. Gene specific primers were designed from sequences retrieved from Genbank using Primer 3 v4.0 (http://frodo.wi.mit.edu/primer3/). All primer sequences are available upon request. Quantitative (q)PCR was carried out in an ABI 7500 system in triplicate using the FastStart Universal SYBR Green Master (Roche, Indianapolis, IN, USA) in 20 ul reactions containing 250 nm (Final concentration) for each forward and reverse primer, 5 ul of cDNA diluted mix (∼25 ng) and 10 ul of 2X SYBR Master Mix. Cycles parameters consisted in an initial denaturation step at 95° for 10 min followed by 40 cycles of amplification at 95° for 15 s and 60° for 1 min, and a dissociation step.
Association Analysis
Frequent itemset mining was performed to find groups of insertion regions, including larger sets of three or more regions, which frequently co-occur in the tumors. This represents a branch of data mining that originates from the analysis of market basket transaction data. More specifically, frequent itemset mining is a methodology that can efficiently determine items that are frequently purchased together from a binary transaction matrix, in which rows represent different transactions by customers at a store, columns represent the different items available, and entries in the matrix indicate whether or not that item was purchased in that transaction[37], [38]. For our purposes, tumors act as transactions (rows), genes are the items (columns), and frequent itemset mining is used to determine which sets of genes co-occur together across multiple tumors.
The set of unique insertion regions produced by TAPDANCE[36] were transformed into a set of genes by mapping each insertion region to its nearest gene. Each tumor is then represented by a set of genes containing at least one insertion in that tumor, which forms a binary transaction matrix in which rows are tumors and columns are affected genes. Closed frequent itemsets (a condensed form of frequent itemset results) were then extracted from the transaction matrix using an apriori-based algorithm to produce a list of candidate gene patterns[39]-[41]. This algorithm was run with a support threshold of three, meaning that only gene patterns that co-occur in three or more tumors are considered. Some support counts were then modified to reflect the number of unique mice that had the gene pattern, rather than the number of tumors. This is to correct for similar insertion sets in tumors originating from the same mouse.
A p-value is calculated for each candidate gene pattern by modeling the support of the pattern as the test statistic. The null distribution is modeled as a binomial with the number of trials equal to the number of tumors and the probability of success equal to the joint probability of the individual genes in the patterns occurring together (based on their individual frequencies in the dataset). In order to account for multiple hypotheses testing, the significance of each candidate pattern was determined by empirically estimating its q-value[42], which is the minimum False Discovery Rate (FDR) at which the test may be called significant[43]. Specifically, a set of 10,000 simulated results were generated by randomizing the tumor that each insertion appears in while preserving the overall set of insertion locations and the number of insertions in each tumor. The q-value for each candidate pattern was calculated as the percent of simulated results that had a p-value better or equal to the p-value of the candidate pattern divided by the percent of real patterns with a p-value equal to or better than the candidate pattern. The gene patterns with q-value ≤25% were deemed significant.
Statistics
The Cancer Gene Census and the COSMIC database were downloaded on 4/20/2013 from Sanger Institute website (ftp://ftp.sanger.ac.uk/pub/CGP/cosmic/data_export). A custom Perl script was used to extract haematopoietic_and_lymphoid_tissue mutations from the CosmicMutantExportIncFus_v64_270313.tsv file. The list of mutations in AML were derived from supplemental tables in the TCGA report on AML[44] combining the list of Tier 1 mutated genes with the list of fusion genes. Significance of association was computed using a 2-tailed Fisher's Exact Test[45]. MAPK pathway significance was determined by performing 10,000 iterations of randomly assigning mouse genes to libraries and calculating number of libraries with insertions in MAPK pathway genes using a custom perl script.
Results
SB Insertional Mutagenesis Promotes Histiocytic Sarcoma Formation
To perform a forward genetic screen for HS we generated mice harboring three elements required for activating SB mutagenesis in myeloid lineage cells. The first element was a nuclear localized Cre recombinase gene knocked into the myeloid-specific Lyz2 locus[46] (Fig S1-A). The Lyz2 promoter is expressed in granulocytes, macrophages, and splenic dendritic cells[33], [47]. The second element was a conditional SB11 transposase allele created by inserting a Lox-STOP-Lox-SB11-cDNA construct downstream of the ubiquitous Rosa26 promoter (Fig S1-B)[22], [23]. The third element was a concatamer of oncogenic SB transposons (T2/Onc). The SB transposon consists of terminal inverted and direct repeats required for SB transposition and an internal promoter, splice donor, splice acceptors and bidirectional polyA signal. The transposon was designed to be capable of overexpressing or disrupting genes, and these transposon-induced mutations provide cells with a selective advantage when they occur in oncogenes or tumor suppressors, respectively (Fig S1-C). The internal promoter within the transposon is highly active in hematopoietic stem cells[48]. We have shown that the SB transposon system is capable of generating insertional mutations leading to overexpression of oncogenes, overexpression of truncated genes, and disruption of genes [19], [20], [23], [49]. We designed a breeding scheme (Fig S2) and generated 73 experimental mice carrying all three elements and 117 littermate controls carrying only two of the three elements (Table S1).
Mice were sacrificed and necropsied when they became moribund or at 18 months, whichever came first. Triple transgenic mice became moribund at a faster rate than controls, beginning around one year of age (Fig 1). The majority of mice had malignancies occurring in multiple tissues throughout the mouse (Fig 2). Over 75% of mice examined had symptoms of disease with the majority being localized to spleen, pancreas, liver, thoracic cavity and peritoneum.
Figure 1. Kaplan Meier Survival Curve showing decreased survival in triple transgenic experimental animals compared to double transgenic controls.

Significance determined using Logrank test.
Figure 2. Representative images of tumor tissue.
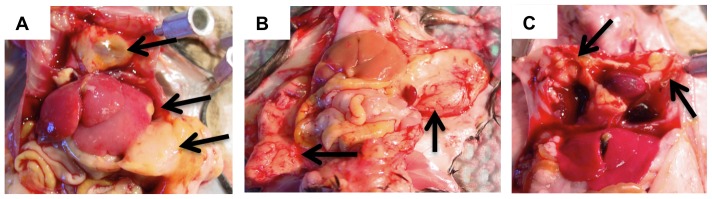
A) Disseminated HS in pancreas, liver, and thoracic cavity. B) HS adhering to peritoneum. C) HS within the thoracic cavity.
To classify the disease we prepared hematoxylin and eosin stained slides from multiple tissues from 51 animals (Fig 3A and Fig S3). Evidence of histiocytic neoplasm was visible in 33 of 51 mice (65%). Upon examination of the neoplasms by light microscopy, the tumors comprised a diffuse relatively non-cohesive proliferation of large cells. The neoplastic cells were large, round to oval in shape, with focal spindling, with large nuclei and abundant cytoplasm. The cytoplasm was eosinophilic, with fine granularity. The nuclei had vesicular chromatin, and many had prominent nucleoli. It is notable that some of the neoplasms have rather bland morphology, while others have marked pleomorphism and increased mitotic activity (Fig S3, panel G). The neoplasms invaded surrounding adjacent tissue, including muscle, spleen, liver, pancreas, lung, and bowel (Fig S3). Eight of these tissues were further analyzed by immunohistochemistry using a panel of antibodies to further confirm histiocytic differentiation (Mac2, F4/80 and Lyz) and exclude B-lineage (Pax5) or T-lineage (CD3) cells (Table S2 and Fig 3B-F). All eight tissues were strongly positive for Mac2, positive for Lyz, and negative for Pax5. Seven of the eight stained positive for F4/80, while three of eight were weakly positive for CD3; the level of CD3 staining was negligible in two of these and not diagnostic of T cell lineage. The immunophenotypic characteristics of these neoplasms in conjunction with the morphologic features are most consistent with the characteristic HS that occur in mice[7]. We also performed PCR on DNA from these same eight tumors using primers crossing VDJ boundaries in both the TCRb locus and the IgH locus. Multiple bands were amplified in control tissues (Thymus for TCRb and spleen for IgH locus) while no bands, or only germline bands were amplified in seven of eight HS tumors (representative images in Fig 4). The morphologic, immunophenotypic and molecular data support that the neoplasms are histiocytic in origin and do not have associated B- or T- lymphoid differentiation. Thus, they are best characterized as HS.
Figure 3. Typical morphologic and immunophenotypic characteristics of the murine histiocytic neoplasms generated by a forward genetic screen.

All images were captured using a 50X oil objective. The depicted neoplasm was present near the pancreas in one mouse (see supplementary figures for additional morphologic characterization). A) H&E – note abundant granular cytoplasm and large nuclei; B) MAC2 immunostain; C) F4/80 immunostain; D) Lysozyme immunostain; E) CD3 immunostain – single lymphocyte in lower left quadrant stains positively; F) PAX-5 – insert denotes on-slide positive control.
Figure 4. TCR and Ig genes are not rearranged in tumors.
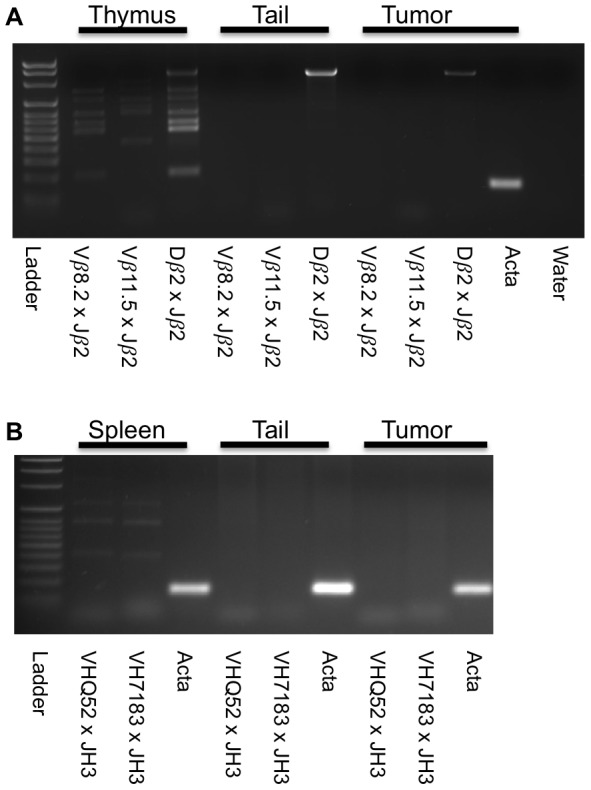
A) PCR amplification of TCR locus using genomic DNA from HS tumor (Lyz-728) indicates no rearrangement of TCR VDJ locus. B) PCR amplification of the IgH locus indicates no rearrangement of IgH DJ locus. Thymus, spleen, and tail DNA were from a wild-type control animal.
Identification of candidate driver genes and pathways in HS
To find genetic drivers of HS we analyzed transposon insertions in 92 tumors from 36 different mice. The tumors were distributed among eight different anatomical locations (Table S3). We were able to confirm that 35 of the 92 tumors were HS based on histology. The remaining tumors are assumed to be HS based on gross pathology, but we did not have enough tissue to confirm by histological examination. We performed linker-mediated PCR (LM-PCR) on purified DNA from these tumors to amplify transposon-genomic fragments and then sequenced the amplicons using the Illumina HiSeq 2000 platform. Sequences were analyzed using a bioinformatics pipeline we developed called TAPDANCE[36]. Approximately 13.8 million sequences were mapped to the genome. Redundant sequences and sequences mapping within 100 bases of each other were combined, resulting in 11,885 non-redundant mapped regions. The depth of sequence reads using the Illumina platform allowed us to filter regions based on the number of sequence reads that mapped to the region. We reasoned that regions with only one or a few reads could either be artifacts or only present in a minority of cells, while regions with a larger number of reads were more likely to be present in a majority of tumor cells. We set a read threshold of 0.01% of total reads mapping in a single tumor for each region. For example, one of our tumors had 227,882 reads in 365 regions. Using our threshold, a single region would require at least 23 mapped reads to be included in our analysis. Of the 365 regions mapping in this tumor, only 90 met the threshold. Out of the 11,885 non-redundant regions, 1,575 unique regions met the threshold (Table S4). A BED formatted version of the unique regions (Table S5) is also provided for use with the Integrated Genome Viewer (IGV) or for uploading to a genome browser to analyze insertion positions relative to exons. This works out to approximately 17 insertions per tumor, with a range of 1 to 90.
In previous screens we noted that transposon insertions mapping to the donor chromosome, where the original transposon transgene was located, constituted up to half of all the mapped transposon insertions[19], [23], [26], a phenomenon referred to as “local hopping”. In this experiment we generated experimental mice using three different founder strains with the donor concatamer on different chromosomes in each of the strains (chr1, chr4 and chr15). Surprisingly, in these tumors, we did not see a large bias of insertions in the donor concatamer. In general, the percentage of insertions on the donor chromosome for each of the three respective T2/Onc strains was 2 to 3 times higher than expected (Table S6) Because the insertion distribution was not heavily skewed towards the donor chromosome we performed four separate CIS analyses. The first three analyses eliminated the donor chromosomes (1, 4 & 15) in those respective tumors, while the fourth analysis included all chromosomes. Of the seven CISs identified on the donor chromosomes, five of the seven were still identified in the analyses even when the insertions in those chromosomes were excluded from the subset of tumor libraries with the corresponding donor concatamer. The other two CISs (Bach2 and Atp6v1c1) were not identified if the donor chromosome was excluded, indicating they may be biased by the donor concatamer. All of the CISs identified in the four analyses were merged into a single list resulting in a final list of 27 CISs (Table 1).
Table 1. Common transposon insertion sites in HS tumors.
| Candidate Gene | Entrez GeneID | Chr | Start | End | Predicted effect | # Mice | # Unique Regions in CIS |
| Bach2 * | 12014 | chr4 | 32237300 | 32487700 | Gain | 12 | 10 |
| Raf1 * | 110157 | chr6 | 115645700 | 115696200 | Gain | 12 | 8 |
| Fli1 * | 14247 | chr9 | 32467900 | 32521100 | Gain | 8 | 6 |
| Mitf * | 17342 | chr6 | 97865300 | 98065300 | Gain | 6 | 5 |
| No_gene_chr2 | NA | chr2 | 98662700 | 98675200 | Unknown | 3 | 5 |
| Pi4ka | 224020 | chr16 | 17107400 | 17307400 | Loss | 5 | 4 |
| Sin3a | 20466 | chr9 | 57093200 | 57113900 | Gain | 5 | 4 |
| Pten | 19211 | chr19 | 32781300 | 32831300 | Loss | 4 | 4 |
| Orai1 | 109305 | chr5 | 123014300 | 123026800 | Loss | 6 | 3 |
| Nf1 * | 18015 | chr11 | 79422000 | 79472000 | Loss | 4 | 3 |
| Jak2 | 16452 | chr19 | 29157700 | 29357700 | Gain | 4 | 3 |
| A330023F24Rik, Mir29b-2, and Mir29c | 320977 | chr1 | 194982400 | 195032400 | Gain | 3 | 3 |
| Rreb1 * | 68750 | chr13 | 37832600 | 37932600 | Gain | 3 | 3 |
| Rpgrip1 | 77945 | chr14 | 52114100 | 52314100 | Unknown | 3 | 3 |
| Atp6v1c1 † | 66335 | chr15 | 38680400 | 38705400 | Loss | 3 | 3 |
| Pvt1 | 19296 | chr15 | 62185400 | 62385400 | Unknown | 3 | 3 |
| Kctd5 | 69259 | chr17 | 24052300 | 24152300 | Unknown | 3 | 3 |
| Il20rb | 213208 | chr9 | 100256900 | 100456900 | Unknown | 3 | 3 |
| Kdm6a | 22289 | chrX | 18208700 | 18258700 | Loss | 3 | 3 |
| Ncoa2 | 17978 | chr1 | 13306200 | 13318700 | Gain | 7 | 2 |
| Serpinf1 | 20317 | chr11 | 75412500 | 75437500 | Gain | 5 | 2 |
| Myc | 17869 | chr15 | 61877000 | 62077000 | Unknown | 5 | 2 |
| Stim1 | 20866 | chr7 | 102313500 | 102338500 | Gain | 4 | 2 |
| Xpr1 | 19775 | chr1 | 155265800 | 155365800 | Loss | 3 | 2 |
| 2310035C23Rik | 227446 | chr1 | 105754300 | 105766800 | Loss | 3 | 2 |
| Arhgap18 | 73910 | chr10 | 26851600 | 26876600 | Loss | 3 | 2 |
| Erg | 13876 | chr16 | 95406500 | 95419000 | Gain | 3 | 1 |
* Identified as CIS in the subset of tumors with confirmed HS histopathology.
† Only identified when libraries with donor concatamer are included.
Because we could not positively diagnose all the tumors we sequenced via histology we performed a second analysis of CISs using only those tumors that had corresponding histological analysis confirming HS. Because there were fewer tumor libraries, only six CISs in this analysis were identified based on our criteria described above. All six of these CISs (Raf1, Mitf, Nf1, Fli1, Bach2, and Rreb1) were also present in the list of 27 CISs identified in the original analysis (Table 1).
To determine the clonality of tumors arising in a single animal we measured the overlap between tumors from the same animal. It was apparent that several tumors were clonal, based on the large percentage of shared insertions, although the majority of tumors did not share transposon insertions with tumors from the same animal (Table S7). To eliminate the bias these clonal tumors may have contributed to calculating CISs we required that all CISs consist of tumors from at least three separate mice. As a conservative test, we re-calculated CISs, this time considering all the tumors from each animal as a single tumor. This re-calculation still identified 24 of the 27 loci, indicating tumor clonality did not significantly affect CIS detection.
Manual analysis of the transposon insertion patterns in the 27 genomic loci allowed us to identify 28 candidate genes, including two micro-RNAs, and we could predict the effect (gain- or loss-of-function) for 21 of these genes based on the location and direction of the transposon insertions in the gene locus (Table 1). The three top hits, ranked by percentage of tumors contributing to the CIS, were Raf1 (alias C-Raf), Bach2 and Fli1. Over 25% of all tumors had a mutation in one of these three genes, and half of these tumors had mutations in at least two of the genes.
To measure the effect of the SB transposon insertions we selected a small subset of the tumors where we had sufficient frozen tumor tissue along with a matched normal tissue to extract RNA and perform qRT-PCR. We selected four tumors from three mice and measured the expression level of four genes (Fli1, Nf1, Mitf, and Raf1) in the tumors that had insertions in these four genes. Based on the transposon insertion pattern we predicted that Fli1, Mitf, and Raf1 would have gain of function mutations, while Nf1 would have a loss of function. Ten of the eleven comparisons possible in this set of tumor/normal tissue pairs indicated that the mRNA level changed in the predicted manner (Fig S4).
Network analysis and relevance to human HS
Because Raf1 was the site of transposon mutagenesis in over 20% of tumors analyzed, we checked for transposons inserted near other MAPK pathway genes in tumors without a Raf1 insertion. We found that 44 of the 92 tumors (48%) generated in our screen had a transposon insertion within 10 kb of an annotated MAPK pathway gene based on the KEGG[50] MAPK pathway gene list (Table S8). To measure the significance of this finding we analyzed randomly generated datasets. The average number of libraries with a MAPK insertion, from 10,000 randomly generated datasets, was 13 (st. dev. 3.0), which is significantly lower than the 44 libraries found in our set of tumors.
To identify cooperating mutations we tested for associations between CISs using Fisher's Exact Test. After correcting for multiple testing, we found significant associations between Fli1 and Bach2 and between Mitf and Raf1, suggesting these pairs of mutations may cooperate in HS tumorigenesis, although formal proof of cooperation would require further experiments. Interestingly, MITF is an oncogene in melanoma, and leads to cell survival via upregulation of BCL2 and other molecules[51]. This suggests the combination of a growth factor mutation and a cell survival mutation may be crucial for HS development.
We analyzed the overlap between our gene list, from which we could identify 25 human orthologs, and known human cancer genes. Ten of the 25 CIS human orthologs were in the list of 487 cancer genes annotated in the Sanger Institute's cancer gene census[52] (Table S9). This is a significant overlap (Fisher's Exact Test p<0.00001). Although all of the 25 CIS human orthologs had multiple documented somatic mutations in the COSMIC database[53], the significance of this comparison is difficult to ascertain, as over 96% of the ∼24,000 genes contained in COSMIC have documented mutations. If we limit our analysis to the 4,682 genes mutated in tumors classified as hematopoietic and lymphoid tissue in COSMIC, we find an overlap of 13 of our 25 CIS genes (Fisher's Exact Test, p<.001) (Table S9). There are no HS tumors documented in the COSMIC database. We also compared our gene list to the recent TCGA sponsored study of AML, because both HS and AML derive from the myeloid lineage. The AML study analyzed mutations and gene-fusions in 200 patient samples and identified 2,022 genes with mutations or gene fusions predicted to alter protein sequence[44]. Eleven of our 25 CIS genes overlapped with these AML genes (Table S9), which would not be expected by chance (Fisher's exact test, p<0.00001). These results support the hypothesis that our mouse model has discovered cancer genes relevant to human cancer, and myeloid malignancies specifically.
We analyzed networks associated with the 27 CIS human orthologs, including the two microRNAs using Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com). Six of the top ten canonical pathways associated with our gene set were cancer signaling (Table S10), while seven of the top ten functions involved death or proliferation of cancer cells (Table S11). The major proteins contributing to these associations were MYC, RAF1, JAK2, and PTEN. These findings suggest that agents that target these signaling pathways, such as ruxolitinib or sorafenib, could be effective in HS patients with a matching genetic profile.
Finally, we used a method of identifying cooperating mutations in our tumors that does not rely on defined CISs. Instead, we used an algorithm called frequent itemset mining[37], [38]. The algorithm identifies combinations of insertions that frequently co-occur in multiple tumors. These groups of genes can reach statistical significance, even though they do not reach significance as a CIS. Analysis of 1,575 transposon insertions (Table S4) using frequent itemset mining identified 38 sets of genes that were mutated in three or more mice, with an FDR≤0.25 (Table S12). A total of 28 genes comprise the 38 sets, with several genes appearing in multiple sets. The majority of the gene sets (24/38) contained three or more of the following genes: Pcf11, Dennd2c, Serpinf1, Ncoa2, Dctn4, Kif2c, Basp1 and Raf1. For example, three mice had tumors with transposon insertions in seven of these eight genes (See itemset #11 in Table S12). These results suggest that combinations of alterations in these genes may function coordinately to generate HS, although functional validation will require further experiments.
Discussion
HS is a rare human neoplasm that is difficult to diagnose and has a poor prognosis. To understand the genetics of HS, with the goal of expanding treatment options for these patients, we conducted a forward genetic screen in mice using the Sleeping Beauty DNA transposon as a mutagen. The majority of mice in the experimental cohort developed symptoms associated with HS.
CIS analysis identified 26 mouse protein-coding genes and two microRNAs that are putative drivers of HS in our model. We identified human orthologs for 25 of the genes, including both microRNAs. These candidate HS cancer genes were significantly enriched for human cancer genes based on the Sanger Institute's cancer gene census and COSMIC database. The list was also enriched in genes mutated in AML based on TCGA data. The significant overlap between genes identified in our screen and known human cancer genes suggests these genes are highly relevant as candidate cancer genes in HS.
The top three genes identified in our screen have been linked to human cancers. Raf1 is part of the MAP kinase pathway and is important for cell fate decisions. Altered RAF1 is associated with the development of Noonan and LEOPARD syndrome, AML, and pilocytic astrocytoma[54]-[56]. Fli1 is an ETS transcription factor and human FLI1 forms a fusion with EWS in 85% of Ewing sarcoma patients. Interestingly, the other major EWS fusion partner found in Ewing sarcoma patients is ERG, another gene identified in our screen[57]. FLI1 fusions have also been found in prostate cancer[58] and abnormal FLI1 expression in AML patients correlates with poor prognosis[59]. BACH2, paradoxically, is a suspected tumor suppressor in CML and Burkitts lymphoma[60], [61]. BACH2 is activated by oxidative stress and can inhibit proliferation and trigger apoptosis in cell lines[62]. Based on our screen we predict Bach2 is overexpressed in HS tumors, suggesting oncogenic activity in these tumors via aberrant activation of this transcriptional repressor in myeloid cells. In support of this hypothesis, BACH2 is significantly overexpressed in CLL and B-cell ALL[63]. Intriguingly, there are quite a few case reports of HS developing as a secondary cancer and/or morphologic variant in patients with B-cell lymphoma with evidence that the neoplasms are clonally related[64]-[66] suggesting similar genetic etiologies. BACH2 has recently been shown to be important for B-cell germinal center formation, where B cells undergo somatic hypermutation and extremely rapid proliferation[67]. It is possible that Bach2 overexpression in HS results in a transcriptional change that favors rapid proliferation in these cells.
Identifying effective targeted therapies for rare cancers is extremely difficult because it is impossible to conduct informative clinical trials due to the small number of patients. Our mouse model can be used to identify potential therapeutic targets in HS. Both Raf1 and Myc were identified as candidate genes in our study. Two case reports of cytogenetic analysis of human HS have identified extra copies of chromosome 8, where MYC resides, suggesting MYC is involved in human HS[8], [9]. We found that over 50% of tumors in our screen had transposon insertions near MAPK pathway genes, suggesting that MAPK pathway inhibitors or HDAC inhibitors, like FK228, that significantly decrease RAF1 levels[68] may be effective therapeutics for HS patients.
Another possible therapeutic target for HS patients, based on our findings, is FLI1 signaling. Abnormal expression of FLI1 is associated with AML and T-cell lymphoma[20], [59], while FLI1 fusion proteins are linked to Ewing sarcoma and prostate cancer[58], [69].
The “hallmarks of cancer” paradigm[70] posits that multiple pathways are disrupted in a single cancer. We used frequent itemset mining analysis of transposon insertions to identify multiple genes that were co-mutated in several tumors. This analysis identified 38 gene sets comprised of 28 genes. Analysis of these 38 gene sets indicates that different subsets of only eight genes heavily contribute to a majority of the itemsets (Pcf11, Dennd2c, Serpinf1, Ncoa2, Dctn4, Kif2c, Basp1 and Raf1). Based on the function of these eight co-occurring genes[71]–[78] we hypothesize that the combination of effects listed in Table 2 can cooperate to generate HS. The next step will be to directly test these combinations using in vitro and in vivo models where the set of genes are coordinately manipulated and the effect on cancer phenotypes is measured.
Table 2. Eight candidate cooperating genes in HS.
| Gene | Predicted functional effect of transposon insertion | Reference |
| Pcf11 | Gain: Increased transcription termination | 69 |
| Serpinf1 | Loss: Relief from angiogenesis inhibition | 71 |
| Dennd2c | Unknown: Disruption of Rab9a signaling | 70 |
| Kif2c | Loss: Altered chromosomal segregation | 72 |
| Raf1 | Gain: Activation of MAP kinase signaling | 73 |
| Dctn4 | Unknown: Altered trafficking along microtubules | 74 |
| Ncoa2 | Gain: Altered nuclear hormone signaling | 75 |
| Basp1 | Unknown: Altered WT1 transcription | 76 |
In conclusion, we have identified several candidate genetic drivers of HS using a transposon-based forward genetic screen in mice. The genes we identified are frequently associated with human cancer, including cancers highly related to HS. These findings lay the groundwork for testing new therapeutics to treat this rare neoplasm that currently has a very poor prognosis.
Supporting Information
Three elements for activating SB transposition in myeloid cells.
(TIF)
Breeding scheme for generating experimental animals and littermate controls.
(TIF)
Histiocytic neoplasms (HN) in multiple mice demonstrate involvement at multiple sites, local invasion/destruction, and aggressive morphology.
(TIF)
Change in mRNA levels of genes with transposon insertions comparing tumor to matched normal tissue.
(TIF)
Number and genotype of cohorts.
(XLSX)
IHC scoring for 5 markers in 8 tumors.
(XLSX)
List of tumors sequenced for transposon insertions.
(XLSX)
Non-redundant genomic regions containing transposon insertions (Excel version).
(XLSX)
Non-redundant genomic regions containing transposon insertions (BED formatted text version).
(BED)
Insertion distribution by donor chromosome.
(XLSX)
Clonality of multiple tumors from the same mouse based on insertion region overlap.
(XLSX)
Tumors with transposon insertions in/near MAPK pathway genes.
(XLSX)
CIS annotations.
(XLSX)
IPA Canonical Pathways.
(XLSX)
IPA Annotated Functions.
(XLSX)
Coordinately mutated genes based on frequent itemset mining.
(XLSX)
Acknowledgments
We would like to thank the Minnesota Supercomputing Institute and the Comparative Pathology Shared Resource Center for their assistance with the project. We would also like to thank Jamie Modiano, Joshua Schiffman, and Matthew Breen for their comments on the manuscript.
Funding Statement
RAB and DAL were supported by grants from the NCI (R01 CA113636) and the Leukemia and Lymphoma Society (LLS 7019-04). SL was supported by NIH Grant T32EB008389, Integrative Training in Neuroimaging, and by a University of Minnesota Interdisciplinary Doctoral Fellowship. JEA was supported by an SP3 grant from the Masonic Cancer Center. TKS was supported by a grant from the American Cancer Society (PF-06-282-01-MGO), the NIH (4R00CA151672-02), Masonic Cancer Center shared resources (NIH P30 CA077598) and generous assistance from the Masonic Cancer Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Swerdlow SH (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: International Agency for Research on Cancer. 439 p. [Google Scholar]
- 2. Arai E, Su WP, Roche PC, Li CY (1993) Cutaneous histiocytic malignancy. Immunohistochemical re-examination of cases previously diagnosed as cutaneous “histiocytic lymphoma” and “malignant histiocytosis”. J Cutan Pathol 20: 115–120. [DOI] [PubMed] [Google Scholar]
- 3. Copie-Bergman C, Wotherspoon AC, Norton AJ, Diss TC, Isaacson PG (1998) True histiocytic lymphoma: a morphologic, immunohistochemical, and molecular genetic study of 13 cases. Am J Surg Pathol 22: 1386–1392. [DOI] [PubMed] [Google Scholar]
- 4. Pileri SA, Grogan TM, Harris NL, Banks P, Campo E, et al. (2002) Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 41: 1–29. [DOI] [PubMed] [Google Scholar]
- 5. Hornick JL, Jaffe ES, Fletcher CD (2004) Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 28: 1133–1144. [DOI] [PubMed] [Google Scholar]
- 6. Vos JA, Abbondanzo SL, Barekman CL, Andriko JW, Miettinen M, et al. (2005) Histiocytic sarcoma: a study of five cases including the histiocyte marker CD163. Mod Pathol 18: 693–704. [DOI] [PubMed] [Google Scholar]
- 7. Hao X, Fredrickson TN, Chattopadhyay SK, Han W, Qi CF, et al. (2010) The histopathologic and molecular basis for the diagnosis of histiocytic sarcoma and histiocyte-associated lymphoma of mice. Veterinary pathology 47: 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alonso-Dominguez JM, Calbacho M, Talavera M, Villalon C, Abalo L, et al. (2012) Cytogenetics findings in a histiocytic sarcoma case. Case Report Hematol 2012: 428279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mori M, Matsushita A, Takiuchi Y, Arima H, Nagano S, et al. (2010) Histiocytic sarcoma and underlying chronic myelomonocytic leukemia: a proposal for the developmental classification of histiocytic sarcoma. Int J Hematol 92: 168–173. [DOI] [PubMed] [Google Scholar]
- 10. Hedan B, Thomas R, Motsinger-Reif A, Abadie J, Andre C, et al. (2011) Molecular cytogenetic characterization of canine histiocytic sarcoma: A spontaneous model for human histiocytic cancer identifies deletion of tumor suppressor genes and highlights influence of genetic background on tumor behavior. BMC Cancer 11: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shearin AL, Hedan B, Cadieu E, Erich SA, Schmidt EV, et al. (2012) The MTAP-CDKN2A locus confers susceptibility to a naturally occurring canine cancer. Cancer Epidemiol Biomarkers Prev 21: 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carrasco DR, Fenton T, Sukhdeo K, Protopopova M, Enos M, et al. (2006) The PTEN and INK4A/ARF tumor suppressors maintain myelolymphoid homeostasis and cooperate to constrain histiocytic sarcoma development in humans. Cancer Cell 9: 379–390. [DOI] [PubMed] [Google Scholar]
- 13.Mashima R, Honda K, Yang Y, Morita Y, Inoue A, et al.. (2010) Mice lacking Dok-1, Dok-2, and Dok-3 succumb to aggressive histiocytic sarcoma. Lab Invest. [DOI] [PubMed]
- 14. Ward JM, Nikolov NP, Tschetter JR, Kopp JB, Gonzalez FJ, et al. (2004) Progressive glomerulonephritis and histiocytic sarcoma associated with macrophage functional defects in CYP1B1-deficient mice. Toxicologic pathology 32: 710–718. [DOI] [PubMed] [Google Scholar]
- 15. Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M (2001) Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer research 61: 6234–6238. [PubMed] [Google Scholar]
- 16. Eischen CM, Rehg JE, Korsmeyer SJ, Cleveland JL (2002) Loss of Bax alters tumor spectrum and tumor numbers in ARF-deficient mice. Cancer research 62: 2184–2191. [PubMed] [Google Scholar]
- 17. Lund AH, Turner G, Trubetskoy A, Verhoeven E, Wientjens E, et al. (2002) Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nature genetics 32: 160–165. [DOI] [PubMed] [Google Scholar]
- 18. Ivics Z, Hackett PB, Plasterk RH, Izsvak Z (1997) Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91: 501–510. [DOI] [PubMed] [Google Scholar]
- 19. Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA (2005) Cancer gene discovery using Sleeping Beauty transposon-based somatic mutagenesis in the mouse. Nature 436: 272–276. [DOI] [PubMed] [Google Scholar]
- 20. Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA (2005) Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436: 221–226. [DOI] [PubMed] [Google Scholar]
- 21. Collier LS, Adams DJ, Hackett CS, Bendzick LE, Akagi K, et al. (2009) Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer research 69: 8429–8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, et al. (2009) A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res 69: 8150–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, et al. (2009) A Transposon-Based Genetic Screen in Mice Identifies Genes Altered in Colorectal Cancer. Science 323: 1747–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.March HN, Rust AG, Wright NA, Ten Hoeve J, de Ridder J, et al.. (2011) Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nature genetics. [DOI] [PMC free article] [PubMed]
- 25. McGrail M, Hatler JM, Kuang X, Liao HK, Nannapaneni K, et al. (2011) Somatic mutagenesis with a Sleeping Beauty transposon system leads to solid tumor formation in zebrafish. PLoS ONE 6: e18826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Starr TK, Scott PM, Marsh BM, Zhao L, Than BL, et al. (2011) A Sleeping Beauty transposon-mediated screen identifies murine susceptibility genes for adenomatous polyposis coli (Apc)-dependent intestinal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 108: 5765–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, et al. (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keng VW, Sia D, Sarver AL, Tschida BR, Fan D, et al.. (2012) Gender bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. [DOI] [PMC free article] [PubMed]
- 29. Koso H, Takeda H, Yew CC, Ward JM, Nariai N, et al. (2012) Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proceedings of the National Academy of Sciences of the United States of America 109: E2998–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, et al. (2012) Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 109: 5934–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Donnell KA, Keng VW, York B, Reineke EL, Seo D, et al.. (2012) A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed]
- 32.Quintana RM, Dupuy AJ, Bravo A, Casanova ML, Alameda JP, et al.. (2012) A Transposon-Based Analysis of Gene Mutations Related to Skin Cancer Development. The Journal of investigative dermatology. [DOI] [PubMed]
- 33. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277. [DOI] [PubMed] [Google Scholar]
- 34. Adams AT, Auersperg N (1981) Transformation of cultured rat ovarian surface epithelial cells by Kirsten murine sarcoma virus. Cancer research 41: 2063–2072. [PubMed] [Google Scholar]
- 35. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KA (2012) TAPDANCE: An Automated tool to identify and annotate Transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinformatics 13: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agrawal R, Imieliński T, Swami A (1993) Mining association rules between sets of items in large databases. ACM SIGMOD Record. ACM. pp. 207–216.
- 38. Agrawal R, Imielinski T, Swami A (1993) Database mining: A performance perspective. Knowledge and Data Engineering, IEEE Transactions on 5: 914–925. [Google Scholar]
- 39.Zaki MJ, Ogihara M (1998) Theoretical foundations of association rules. 3rd ACM SIGMOD Workshop on Research Issues in Data Mining and Knowledge Discovery. Citeseer. pp. 71–78.
- 40.Pasquier N, Bastide Y, Taouil R, Lakhal L (1999) Discovering Frequent Closed Itemsets for Association Rules. In: C Beeri and P Buneman, editors. Database Theory — ICDT'99. Springer Berlin Heidelberg. pp. 398–416. [Google Scholar]
- 41.Agrawal R, Srikant R (1994) Fast Algorithms for Mining Association Rules in Large Databases. Proceedings of the 20th International Conference on Very Large Data Bases. Morgan Kaufmann Publishers Inc. pp. 487–499.
- 42. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Storey JD (2002) A direct approach to false discovery rates. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 64: 479–498. [Google Scholar]
- 44.TCGARN (2013) Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. The New England journal of medicine. [DOI] [PMC free article] [PubMed]
- 45. Agresti A (1992) A Survey of Exact Inference for Contingency Tables. Staistical Science 7: 131–153. [Google Scholar]
- 46. el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, et al. (2004) Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39: 186–193. [DOI] [PubMed] [Google Scholar]
- 47. Faust N, Varas F, Kelly LM, Heck S, Graf T (2000) Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96: 719–726. [PubMed] [Google Scholar]
- 48. Cherry SR, Biniszkiewicz D, van Parijs L, Baltimore D, Jaenisch R (2000) Retroviral expression in embryonic stem cells and hematopoietic stem cells. Mol Cell Biol 20: 7419–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, et al. (2009) A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol 27: 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34: D354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haq R, Fisher DE (2011) Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 29: 3474–3482. [DOI] [PubMed] [Google Scholar]
- 52. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. (2004) A census of human cancer genes. Nat Rev Cancer 4: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, et al. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research 39: D945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, et al. (2007) Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nature genetics 39: 1007–1012. [DOI] [PubMed] [Google Scholar]
- 55. Zebisch A, Staber PB, Delavar A, Bodner C, Hiden K, et al. (2006) Two transforming C-RAF germ-line mutations identified in patients with therapy-related acute myeloid leukemia. Cancer research 66: 3401–3408. [DOI] [PubMed] [Google Scholar]
- 56. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, et al. (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28: 2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Riggi N, Stamenkovic I (2007) The Biology of Ewing sarcoma. Cancer letters 254: 1–10. [DOI] [PubMed] [Google Scholar]
- 58. Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, et al. (2012) FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes, chromosomes & cancer 51: 240–249. [DOI] [PubMed] [Google Scholar]
- 59. Kornblau SM, Qiu YH, Zhang N, Singh N, Faderl S, et al. (2011) Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood 118: 5604–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sakane-Ishikawa E, Nakatsuka S, Tomita Y, Fujita S, Nakamichi I, et al. (2005) Prognostic significance of BACH2 expression in diffuse large B-cell lymphoma: a study of the Osaka Lymphoma Study Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 23: 8012–8017. [DOI] [PubMed] [Google Scholar]
- 61. Vieira SA, Deininger MW, Sorour A, Sinclair P, Foroni L, et al. (2001) Transcription factor BACH2 is transcriptionally regulated by the BCR/ABL oncogene. Genes, chromosomes & cancer 32: 353–363. [DOI] [PubMed] [Google Scholar]
- 62. Muto A, Tashiro S, Tsuchiya H, Kume A, Kanno M, et al. (2002) Activation of Maf/AP-1 repressor Bach2 by oxidative stress promotes apoptosis and its interaction with promyelocytic leukemia nuclear bodies. The Journal of biological chemistry 277: 20724–20733. [DOI] [PubMed] [Google Scholar]
- 63. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, et al. (2010) Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 28: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumar R, Khan SP, Joshi DD, Shaw GR, Ketterling RP, et al. (2011) Pediatric histiocytic sarcoma clonally related to precursor B-cell acute lymphoblastic leukemia with homozygous deletion of CDKN2A encoding p16INK4A. Pediatr Blood Cancer 56: 307–310. [DOI] [PubMed] [Google Scholar]
- 65. Castro EC, Blazquez C, Boyd J, Correa H, de Chadarevian JP, et al. (2010) Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 13: 225–237. [DOI] [PubMed] [Google Scholar]
- 66. Wang E, Hutchinson CB, Huang Q, Sebastian S, Rehder C, et al. (2010) Histiocytic sarcoma arising in indolent small B-cell lymphoma: report of two cases with molecular/genetic evidence suggestive of a ‘transdifferentiation’ during the clonal evolution. Leuk Lymphoma 51: 802–812. [DOI] [PubMed] [Google Scholar]
- 67. Huang C, Geng H, Boss I, Wang L, Melnick A (2014) Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood 123: 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, et al. (2002) Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. Journal of the National Cancer Institute 94: 504–513. [DOI] [PubMed] [Google Scholar]
- 69. Berger M, Dirksen U, Braeuninger A, Koehler G, Juergens H, et al. (2013) Genomic EWS-FLI1 fusion sequences in Ewing sarcoma resemble breakpoint characteristics of immature lymphoid malignancies. PLoS One 8: e56408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 71. West S, Proudfoot NJ (2008) Human Pcf11 enhances degradation of RNA polymerase II-associated nascent RNA and transcriptional termination. Nucleic Acids Res 36: 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA (2010) Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol 191: 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang J, Chen S, Huang X, Han J, Wang Q, et al. (2010) Growth suppression of cervical carcinoma by pigment epithelium-derived factor via anti-angiogenesis. Cancer Biol Ther 9: 967–974. [DOI] [PubMed] [Google Scholar]
- 74. Bie L, Zhao G, Wang YP, Zhang B (2012) Kinesin family member 2C (KIF2C/MCAK) is a novel marker for prognosis in human gliomas. Clinical neurology and neurosurgery 114: 356–360. [DOI] [PubMed] [Google Scholar]
- 75. Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, et al. (2011) Raf family kinases: old dogs have learned new tricks. Genes Cancer 2: 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Karki S, Tokito MK, Holzbaur EL (2000) A dynactin subunit with a highly conserved cysteine-rich motif interacts directly with Arp1. J Biol Chem 275: 4834–4839. [DOI] [PubMed] [Google Scholar]
- 77. Xu J, O'Malley BW (2002) Molecular mechanisms and cellular biology of the steroid receptor coactivator (SRC) family in steroid receptor function. Reviews in endocrine & metabolic disorders 3: 185–192. [DOI] [PubMed] [Google Scholar]
- 78. Carpenter B, Hill KJ, Charalambous M, Wagner KJ, Lahiri D, et al. (2004) BASP1 is a transcriptional cosuppressor for the Wilms' tumor suppressor protein WT1. Mol Cell Biol 24: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Three elements for activating SB transposition in myeloid cells.
(TIF)
Breeding scheme for generating experimental animals and littermate controls.
(TIF)
Histiocytic neoplasms (HN) in multiple mice demonstrate involvement at multiple sites, local invasion/destruction, and aggressive morphology.
(TIF)
Change in mRNA levels of genes with transposon insertions comparing tumor to matched normal tissue.
(TIF)
Number and genotype of cohorts.
(XLSX)
IHC scoring for 5 markers in 8 tumors.
(XLSX)
List of tumors sequenced for transposon insertions.
(XLSX)
Non-redundant genomic regions containing transposon insertions (Excel version).
(XLSX)
Non-redundant genomic regions containing transposon insertions (BED formatted text version).
(BED)
Insertion distribution by donor chromosome.
(XLSX)
Clonality of multiple tumors from the same mouse based on insertion region overlap.
(XLSX)
Tumors with transposon insertions in/near MAPK pathway genes.
(XLSX)
CIS annotations.
(XLSX)
IPA Canonical Pathways.
(XLSX)
IPA Annotated Functions.
(XLSX)
Coordinately mutated genes based on frequent itemset mining.
(XLSX)