

Abstract
The conformational preference of a peptide with three phenylalanine-glycine (FG) repeats from the intrinsically disordered domain of nucleoporin 159 (nup159) from the yeast nucleopore complex (NPC) is studied. Conformational states of this FG-peptide in dimethyl sulfoxide (DMSO), a non-native solvent are first studied. A solvent exchange scheme is designed and performed to understand how the conformational preferences of the peptide are altered as the solvent shifts from DMSO to water. An ensemble of structures of a 19-residue peptide is determined based on 13Cα, 1Hα, and 1HN chemical shifts and with inter-proton distances. An experimental model is then presented where chemical shifts and amide-proton temperature dependence is probed at changing DMSO to water ratios. These co-solvent experiments provide evidence of a conformational change as the fraction of water increases by the stark change in the behavior of amide protons under varied temperature. This investigation provides a NMR based experimental method in the field of intrinsically disordered proteins to realize conformational transitions from a non-native set of structures (in DMSO) to a native set of disordered conformers (in water).
Keywords: Intrinsically disordered protein (IDP), Nuclear Magnetic Resonance (NMR), Solvent Perturbation, Nucleoporins
Introduction
The concept that a macromolecule's function is determined by its structure was an early realization. However, a class of proteins, called Intrinsically Disordered Proteins (IDPs), are defined as fully or partially unfolded proteins at physiological conditions 1. These proteins have at least one domain which does not hold tertiary structure and has transient secondary structure. Though protein and protein domains classified as IDPs do not have these high level structural components, they have been associated with many important cellular functions, most commonly with protein-protein interactions 2. Dunker et al. showed, via proteomic techniques, that approximately 30% of eukaryote proteins contain a disordered section of at least 40 residues 1. While these proteins cannot be said to have distinct structure in the traditional sense, they can exist in an ensemble of structures, that is, there are a number of possible thermodynamically allowed conformational states at physiological temperature 3. When comparing IDPs to the free-energy landscape of protein folding, the energy distribution is very flat and broad with little energy difference between the many conformational states 4.
The nuclear pore complex is the central transmission point for nuclear cytoplasmic cross-membrane traffic and contains multiple IDPs in the pore region 5-7. Yeast NPC is a large protein-protein complex that contains approximately 30 proteins in multiple repeats 8. Proteins of smaller then 3-4 nm pass through the NPC through unmediated diffusion while larger proteins, up to 39 nm, are transported via mediation 9. This assisted diffusion for larger proteins involves complexing with multiple possible transport factors 10,11. One of the early structural studies involves the interaction of importin (classified as a karyopherin) with phenylalanine glycine (FG) repeat peptides 12,13. The inter-pore region is known to be highly concentrated with these FG-nucleoporins (FG-nups) and many of these FG repeat rich protein domains have been classified as IDPs 6.
Dimethyl sulfoxide (DMSO), though an organic solvent has been used to mimic the bilayer/water transition area (dielectric constant ε ∼ 40) 14. DMSO also has a high viscosity (η = 2.03 mPa·s) which may mimic molecular crowding due to a high concentration of IDPs 15. As the DMSO-water mixture is fully miscible, this combination of solvents allows a systematic transition of the experimental conditions from 100% DMSO to that of 100% water, including any other co-solvent conditions in between. This study, therefore will sample structural information in DMSO and DMSO-H2O co-solvent conditions. In this work we first determine a structural ensemble to gain insight into the possible conformational behavior of a 19 amino acid (AA) polypeptide containing three FG repeats. This peptide was chosen due to the relatively short distance between FG motifs leading to simplified spectra. Such a short peptide allows for relatively fast NMR experiments and reduced peak overlap while retaining three possible FG interactions.
The structure of a minimal FG-domain in 100% DMSO was obtained from NMR restraint based molecular modeling. With the solution structure at 100% DMSO-d6 provided a well-defined boundary condition to explore residue specific changes as the solvent conditions are perturbed. These co-solvent experiments were performed to both analyze the inherent solvent effects on the backbone chemical shifts as well as determine any conformational differences between the aqueous and organic solvents. A domain which becomes disordered after the introduction of water indicates a location of possible structure in the overall conformational space. Specifically, the backbone alpha position for each residue (both carbon and hydrogen) was sampled at varying ratios of water to DMSO along with the backbone amide hydrogen. The amide hydrogen was further be tested at each dilution by a variable temperature (VT) experiment to estimate possible loss or gain in intra or inter-molecular hydrogen bonding 16. This information should provide both a consistent reduction in structural stability as the concentration of water increases, as well as serve a fundamental data set for such co-solvent NMR experiments on IDPs in general.
Materials and Methods
FG Nucleoporin Peptide
A Minimal FG-peptide (MFGP) of Nup159 (P40477) residues 598-616 from Saccharomyces cerevisiae was used for these studies. MFGP contains two SAFG repeats near the N and C termini (residues 600-603 and 610-613) and a central PSFG motif (residues 605-608).
(598/1) SGSAFGKPSFGTSAFGTAS (616/19)
The sequence chosen includes two terminal residues buffering the four amino acid repeats in question (N terminus-SG, C terminus-AS) and all future numberings will consider Ser589≡Ser1 and Ser616≡Ser19. The peptide, 99.41% purity as determined by HPLC, was purchased from Biomatik Co. and was used without further purification. MFGP is soluble in 100% DMSO, 100% H2O and all attempted mixtures of the two solvents. NMR samples were prepared with 1.5 mg of MFGP dissolved in 600 μL DMSO-d6 with 0.03% TMS in a 5 mm NMR tube.
NMR Spectroscopy and Structure Calculation
All the NMR experiments were performed in Agilent 400MHz (VNMR) system using an One-NMR probe equipped with a pulsed-field-gradient (PFG) along the Z-axis. In addition to one-dimensional NMR spectra, several 2D homonuclear and heteronuclear experiments were performed; TOtal Correlation SpectrosopY (TOCSY), NOESY (Nuclear Overhauser Effect SpectroscopY), DQFC (Double Quantum Filtered Correlation SpectroscopY) and 13C-HSQC (Heteronuclear Single Quantum Correlation Spectroscopy) 17-20. Primary TOCSY data sets were collected at 289 K with 64 transients per t1 point and a mixing time of 100, 80 or 60 ms, with Hartmann-Hahn matching during the mixing time achieved using the multi-pulse sequence DIPSI 21-23. Similarly the primary sequencing NOESY was run with a 150 ms mixing time, along with an 80 ms mixing time to identify direct and eliminate long-range NOE interactions 24,25. 13C Heteronuclear Single Quantum Correlation Spectroscopy with gradient based coherence selection (gHSQC) experiments were run over both the general 13C spectral range as well as specifically tuned to the alpha carbon region for increased resolution, using 2048 points in the direct dimension and 128 points in the indirect dimension and 256 transients per indirect point 20,26. NMR data processing was performed using nmrPipe 27, while spectral analyses were performed Sparky 3.0 28 on an Ubuntu 10.10-Linux quad core workstation.
Ensemble of structures were calculated using CYANA 3.0 using the NMR derived NOESY cross peak intensities 29. Backbone chemical shift information is also included in the structural ensemble calculations. NOE diagram and the distribution of constraints are given in supporting material (Fig, S4). Chemical shift difference of proline (Δδ = δCβ - δCγ) ratio predicts a trans-configuration. Torsion angle dynamics were performed 100000 steps with a random seed was used to generate 10000 random starting structures. These random structures are then allowed to come to an energy minimum within the given restraints. Lowest energy conformers (25 in total) had no constraint violations.
Solvent exchange experiments and NMR
A serial dilution was designed to capture a range of concentrations of DMSO to water with constant peptide concentrations while minimizing peptide waste. Three NMR tubes were used for the dilution experiment series. The first two tubes followed a serial dilution with DMSO- d6 and H2O. The third tube tested key dilution points at high water concentrations where the water solvent signal was not effectively attenuated and key alpha proton chemical shifts were obscured. This tube included DMSO-d6 and D2O with constant concentrations of peptide. The process of dilution required 1.5 mg of peptide dissolved in 600 μL DMSO-d6 in an NMR tube and 6.25 mg of peptide dissolved in 2.5 mL of H2O as stock solution. For each dilution the majority of sample in the NMR tube was removed and deposited in a clean Eppendorf tube where some sample was removed and an equal volume of water/peptide was added. This tube was then mixed and centrifuged for easy pipette collection, then deposited back in the same NMR tube. This process was used to make seven samples with molar ratio of DMSO to water (mol-DMSO/mol-H2O) at values: 0.83, 0.59, 0.43, 0.37, 0.50, 0.30, 0.20 along with a sample that has 100% DMSO-d6. Spectra were obtained along the same experimental parameters as described before. Collected spectra for all dilutions with H2O included HSQC centered on the Cα-Hα region, five TOCSY spectra were performed between 25-45 °C with increments of 5 °C.
All further references to mole fraction, χ, will be in units of mol DMSO/mol where only the DMSO and water are considered and the relatively small amount of peptide is not included. The temperature for the VT experiments was calibrated by the methanol hydroxyl hydrogen shift in a separate experiment 30. Pre-saturation was ineffective at the higher water concentrations where the water peak overlapped the alpha proton region. Starting most importantly with 0.43 χ some 13Cα- 1Hα peaks came directly from the D2O dilution scheme, later at 0.30 and 0.20 χ all 13Cα-1Hα peaks were derived from the DMSO-d6/D2O sample. Tetramethylsilane, included stock in the DMSO-d6, stayed in solution and at sufficient concentrations to be detected by TOCSY at all tested co-solvent dilutions. This was used to reference all TOCSY spectra, while the peak from residual 1H in DMSO-d6 was used to reference the gHSQC proton dimension with the corresponding TOCSY (at the same temperature and dilution). The carbon dimension of the HSQC experiments was referenced indirectly based on the proton chemical shifts 31. This indirect referencing should remain sufficient as the observation frequency was never changed, nor was the pulse power or pulse length throughout the entirety of the project and relative changes in chemical shifts were only considered for further analysis.
The peak locations measured were either the maximum intensity of the peak or under conditions of coupling, strong enough to show up on TOCSY, the average of the multiple peaks (center) is determined. These locations were then analyzed by LibreOffice. Two primary data sets were collected: the change in amide hydrogen chemical shift with respect to concentration and variation in temperature, second, the change in the hydrogen and carbon chemical shifts with respect to solvent concentration. The change in amide hydrogen CS with respect to temperature was determined individually for each residue and calculated by a linear least squares fit across all temperatures where data was available. The 1HN-1Hα peak was preferred for listing the 1HN chemical shift, though in cases where the alpha peak was buried under the water peak, the 1HN-1Hβ was substituted. The use of the 1HN-1Hβ peak should not affect the final value for the amide chemical shift because the choice between the coupled atoms in the indirect dimension does not affect the position of the amide proton in the directly detected dimension of the TOCSY.
Clustering analysis
All the experimentally measured chemical shifts of amides as function of temperature and molar concentrations (18 amide resonances × 5 temperatures × 8 co-solvent concentrations) and chemical shifts of alpha proton and carbons as function of DMSO molar concentration (19 residues × 2 resonances × 8 co-solvent concentrations) are collectively categorized using cluster analysis. A correlation matrix was constructed across the AA residues using the Pearson squared distance metric. The calculations and the plots were generated by a combination of codes using Matlab 32 and SigmaPlot 33 programs. Correlation matrix was converted into heatmap using Matlab routines.
Results
Chemical Shift assignments and structure calculation in DMSO-d6
Chemical shift assignments were performed using the standard approach that combines DQFC/TOCSY for intra-residue assignments and NOESY for inter-residue correlations 34. Table 1 lists the complete chemical shift assignment of MFGP in DMSO-d6 and representative NMR spectra are included in the supporting information (Fig.S1, NOESY with sequential assignments and Fig.S2, Cα region 13C-HSQC). The alpha carbon and hydrogen chemical shifts, 3JHNα (HN to Hα) spin-spin coupling, and observable NOESY peaks were initially used to determine three dimensional structure 34. At 400 MHz, all expected protons were resolved with the exception of the F5 and F15 backbone atoms which were overlapped in the spectra and the aromatic atoms for all three F5, F10, and F15 which were all overlapped. Likewise, S1 and G2 1Hα and 1HN were overlapped. 13C-gHSQC provided carbon resonance assignments for all 13Cα and most backbone carbon atoms. The 1Hα for S1 and G2 was resolved in the carbon dimension, as were the overlapped F5 and F15 alpha protons, verifying the overlap and their chemical shift assignments. We are reassured that the analyzed peptide is not broken at the P8 position by strong NOESY signals between both K7 and S9 R-group protons. Overlap was noted on all resonances for F5 and F15 resulted in the same peaks (1Hα, 1Hβ, 1HN) being assigned to both, this was justified by an approximate double peak volume with respect to F10 in the NOESY (Table 1 and Fig.S1).
Table 1. Chemical shift assignment of MFGP in DMSO-d6.
| Residue | Backbone chemical shifts (13Cα, 1Hα, HN) in ppm | R-group shifts in ppm |
|---|---|---|
| Ser 1 | 54.15, 3.87, 8.59 | OH:5.46, Cβ:60.25, Hβ:3.72 |
| Gly 2 | 41.79, 3.86, 8.59 | |
| Ser 3 | 54.42, 4.37, 8.10 | OH:5.14, Cβ:61.68, Hβ:3.56 |
| Ala 4 | 48.39, 4.19, 8.17 | Cβ:17.57, Hβ:1.13 |
| Phe 5 | 53.78, 4.47, 7.99 | Cβ:37.32 Hβ:3.02 & 2.77 |
| Gly 6 | 41.56, 3.72 8.08 | |
| Lys 7 | 49.87, 4.50, 8.04 | Cβ:30.59, Hβ:1.65 & 1.528, Cγ:21.64, Hγ:1.36, Cδ:26.596, Hδ:1.53, Cε:38.53, Hε:2.75 |
| Pro 8 | 59.13, 4.36, N/A | Cβ:28.94, Hβ:1.98 & 1.81, Cγ:24.29, Hγ:1.87, δ:46.75, Hδ: 3.67 & 3.55 |
| Ser 9 | 54.89, 4.21, 7.91 | OH:4.969, Cβ:61.5, Hβ:3.52 |
| Phe 10 | 53.76, 4.47, 8.26 | Cβ:37.29, Hβ: 3.06 & 2.839 |
| Gly 11 | 41.86, 3.80, 8.16 | |
| Thr 12 | 57.63, 4.34, 7.82 | OH:4.98, Cβ:66.55, Hβ:3.98, Cγ:19.33, Hγ:1.04 |
| Ser 13 | 54.55, 4.35, 7.93 | OH:5.09, Cβ:61.53, Hβ:3.63 |
| Ala 14 | 48.39, 4.19, 8.06 | Cβ:1.13 |
| Phe 15 | 53.78, 4.47 7.93 | Cβ:37.32, Hβ:3.03 & 2.79 |
| Gly 16 | 41.86, 3.80, 8.15 | |
| Thr 17 | 57.80, 4.25, 7.77 | OH:4.89, Cβ:66.55, Hβ:3.98, Hγ:1.04 |
| Ala 18 | 47.74, 4.39, 7.95 | Cβ:18.19, Hβ:1.24 |
| Ser 19 | 54.54, 4.24, 7.99 | Cβ:61.13, Hβ:3.65 |
Three different data sets were examined towards the determination of MFGP's structure in DMSO: Backbone chemical shifts, coupling constants between 1HN and 1Hα, and NOESY through space cross peaks. Secondary 1Hα and 13Cα chemical shifts determined using both random coil chemical shifts in water and DMSO-d6 suggest and generally an extended conformation of MFGP (Fig. S3) 14,35-37. The ensemble of structures generated using the NOE constraints are shown in Figure 1. There is a distinct lack of NOESY backbone peaks for this unordered range and the few R-group based NOE peaks between the central residues are insufficient to fully confine the range (Fig. S4). It is worth mentioning NOESY experiments of MFGP in water (90% H2O/10% D2O) provided discernible cross peaks for analysis (data not shown).
Figure 1.
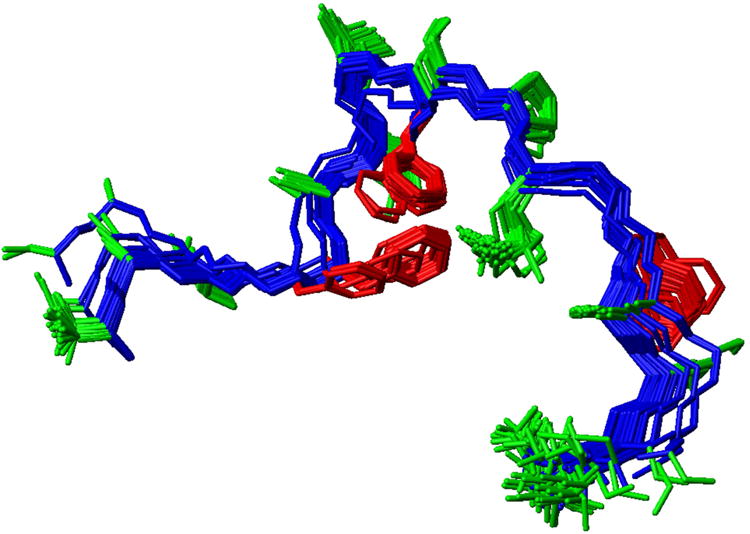
Ensemble of NMR determined structures of MFGP. The ensemble of structures also highlights the locations of the phenylalanines.
Systematic co-solvent experiments: residue specific contributions
Amide hydrogen temperature coefficients and solvent perturbation
The variable temperature data set is taken solely from the DMSO-d6/H2O dilution series. These TOCSY experiments were conducted at 1, 0.83, 0.59, 0.5, 0.43, 0.38, 0.3 and 0.2 χ mol DMSO/mol and at temperatures 299.03, 304.41, 309.80, 315.18, and 320.56 K. Figure 2 shows the relative change in the amide proton chemical shift as a function of χ mol DMSO/mol and temperature as a heat map for each residue. Supporting information (Fig. S5 and Tables S1) show the calculated ΔδHN/ΔT for each condition. The majority of amide hydrogen chemical shifts in the 19 residue peptide maintain a temperature dependence between -4 and -6 ppb/K (Fig. S5), a range considered congruent with weak intra-molecular hydrogen bonding. Error in the ΔδHN/ΔT values was found to range between 0.2 and 0.6 ppb. Seven of the residues deserve individual remark. S9 and F10 both make a large move from -4.4 ppb/K and -3.9 ppb/K to -5.4 ppb/K and -5.1 ppb/K respectively between 0.83 and 0.59 χ. This large step change is distinct and the higher values of chemical shift dependence on temperature at higher concentrations of water generally hold. Likewise, A4, F5, S13 A14 and A18 have a distinct change in ppm/K towards loss of non-solvent hydrogen bond, though at the break between 0.59 and 0.50 χ. This effect is particularly large for A4 and A14 where the 1-0.59 χ change in chemical shift with respect to temperature is between -4.9 to -5.2 ppb/K and -4.4 to -4.7 ppb/K respectively. This relatively low value moves at 0.50 χ to -7.0 ppb/K for A4 (with a range of-7.2 to -6.4 ppb/K between 0.50 and 0.20 χ) and -6.6 ppb/K for A14 (with a range of -7.1 to -6.4 ppb/K between 0.50 and 0.20 χ). These two residues have the highest dependence on temperature at the higher water concentrations. It is worthy of note that both of these alanines are part of the two SAFG motifs in MFGP.
Figure 2.
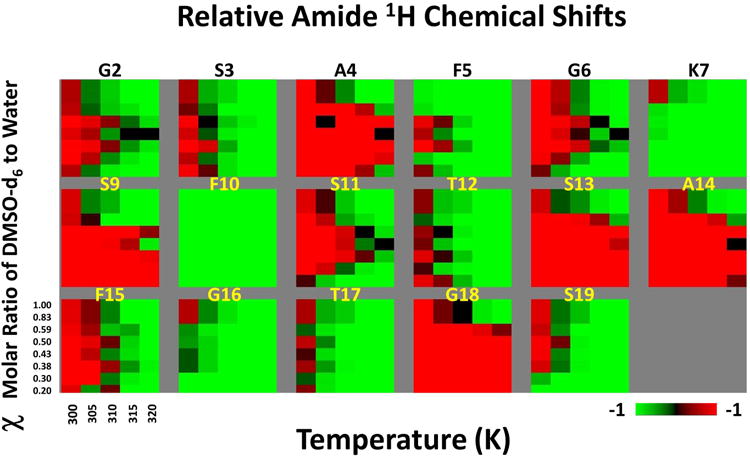
Heat map summarizing the solvent perturbation and temperature effects on the amide proton chemical shifts. Each panel represents a AA residue as labeled. The chemical shifts are normalized between -1 (green) to +1 (red) to highlight the relative effects.
In conjunction with the above data set, a non-linear change in the amide hydrogen chemical shift at 299 K occurs between 0.59 and 0.50 χ (Fig 3). Between 0.5 and 0.2 χ, a negative parabolic like fit is seen for many residues including G2, A4, F5, S9, F10, G11, S13, A14, F15, G16 and A18 with an apparent maximum near 0.37 χ. Lysine 7 is unique as it does not move downfield along with the bulk of residues between 0.5 and 0.37 χ but does make a large upfield shift between 0.37 and 0.2 χ. Two residues, T12 and T17, are unique in showing little change in chemical shift with respect to DMSO:H2O ratio while the S1 amide hydrogen was not resolvable under experimental conditions past 0.83 χ.
Figure 3.
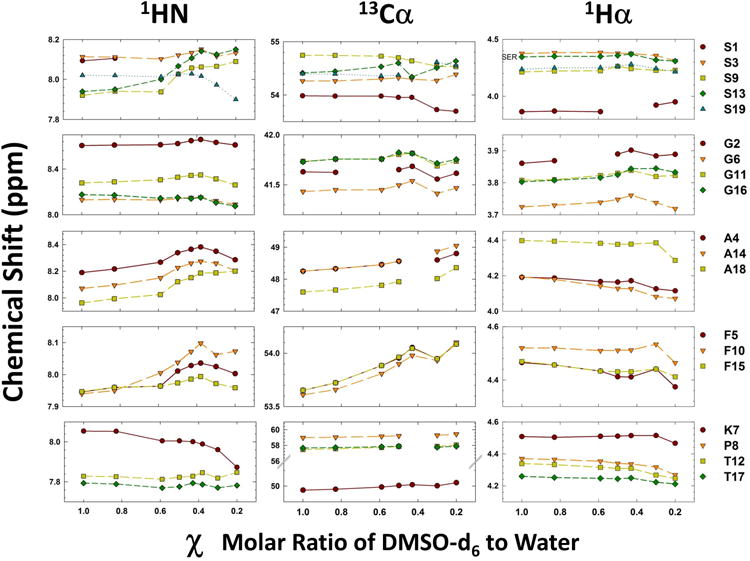
Chemical shift changes as a function of molar ratio of DMSO-water. Left (Amide proton), central (alpha carbon) and right (alpha proton) panels show the variation in the chemical shifts as function χ (molar ratio of DMSO-d6:water). Each sub-panels are grouped with residues as noted by the labels on the right
Alpha proton and carbon chemical shifts effects
Figure 3 (central and right panels) shows the variation in the 13Cα and 1Hα chemical shifts as a function of χ, while the left panel shows the amide proton chemical shift variation (see above) (Complete chemical shift data is given in the Supporting information Tables S2 and S4). The 1Hα data set (Fig.3 right panel), shows constant, if not linear, change in chemical shift with respect to mole fraction of the co-solvent for the majority of residues. This relatively constant change is broken for many residues between 0.43 and 0.20 χ with the majority of those residues breaking between 0.30 and 0.20 χ (as opposed to between 0.43 and 0.30 χ). The overall trend for the four glycines and the N-terminus serine is towards deshielding as the water concentration increases. The other 14 residues show a mild trend towards increased shielding as the water concentration increases. The 13Cα data set mirrors the 1Hα in that the general trend for non-glycine residues is towards shielding (opposite effect seen in the hydrogen set) yet the same jump in chemical shifts is seen between 0.43 and 0.2 χ (Fig. 3 central panel). This sudden change in the trend of chemical shifts with respect to DMSO:H2O ratio, while not the same for each residue, is seen in some manner for every residue.
In an attempt to elucidate the residues most susceptible to changes in solvent conditions a linear least squares fit was applied to both the αC and αH individually with a large slope taken as evidence of large general change in ppm/Δχ. Further, residues which made large changes in chemical shift over the course of different concentrations of solvent are shown by Figure 4. It should be noted that linear regression fits were used to approximate the data shown in Figure 4 though many residues follow a path which is far from linear. This data is best seen as a general indicator of direction and magnitude of movement relative to other residues. As well an absolute value of the movement in both carbon and proton dimension is shown by:
Figure 4.
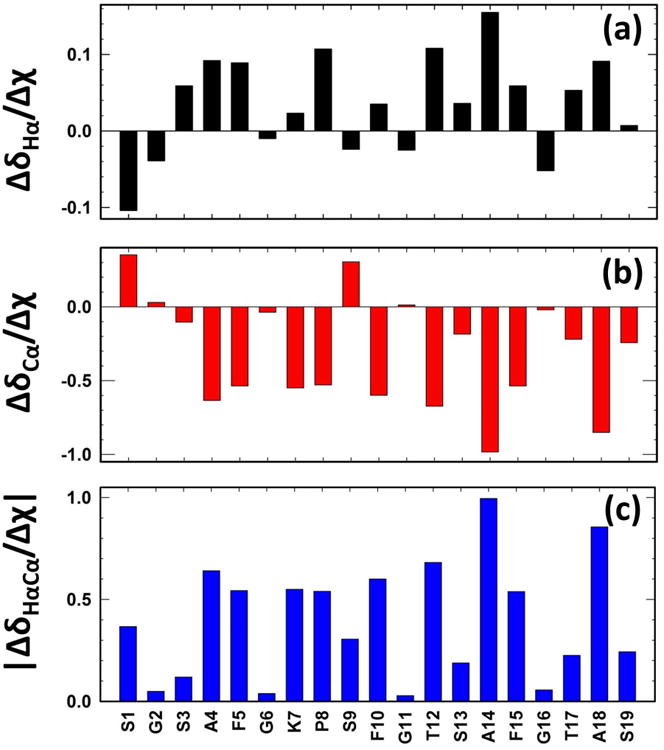
Chemical variation of the 13Cα-1Hα vector due to solvent perturbation. (a) ΔδHα/Δχ, (b) Δδcα/Δχ and (c) |ΔδHαCα/Δχ| plotted as a function of AA sequence. Note S9 which has an upfield move in the proton dimension and a downfield move in the carbon dimension while retaining a relatively large overall change as water increased.
| [1] |
This data set describes S1, S3, S9, S13, T17 and S19 as being susceptible to change with respect to DMSO:H2O concentration (0.4>|ΔδHαCα/Δχ| >0.1 ppm/Δχ) and A4, F5, K7, P8, F10, T12, A14, F15, and A18 being extremely susceptible to change |ΔδHαCα/Δχ|>0.2 ppm/Δχ. The Four glycine residues show very low effect overall on chemical shift at changing co-solvent concentration for the alpha position: G2, G6, G11, and G16 |ΔδHαCα/Δχ| <0.06 ppm/Δχ.
Combined residue specific effects: non-native ordered to native disordered states
Chemical shift changes of amide proton of each residue were measured at five different temperatures and at eight different molar concentrations leading to 40 measurements per residue for most of the amino acids in MFGP (Figures 2 and 3). Similarly, 18 alpha proton/carbon chemical shifts were also measured (Figures 3 and 4) leading to ∼16 measurements/residue. On an average, we have measured ∼56 different parameters/residue that spans five different temperatures and eight different molar ratios. To develop a reductionist view of the data, we clustered these variables across the AA residues and the results are summarized as an interaction matrix in Figure 5. The heatmap color shows a spectral gradient from blue (no correlation) to red (high correlation). As Ser 1 and Pro8 do not have enough number of amide measurements, both these positions show no correlation with rest of the peptide. One of the hypotheses on the function of FG-nucleoporins in the NPC is the intramolecular cohesion elements that impart order to the FG domain and compact its ensemble of structures into native configurations 38,39. Of the three FG-motifs, the N-terminal motif (F5G6) is involved in a strong local correlation with K7 as well additional long range correlations with the central (F10G11) and C-terminal (F15G16) FG-motifs (shown by the grids in Fig.5).
Figure 5.
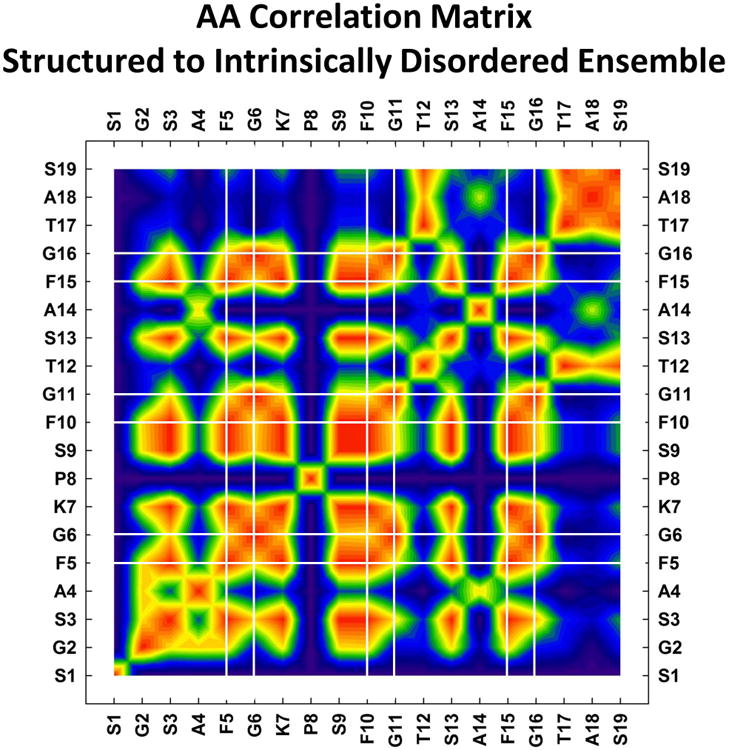
Correlation matrix summarizing the shift from non-native structured (in DMSO-d6) to natively disordered state (in water) of MFGP. Heatmap represents the Pearson squared correlation between the AA residues with no-correlation (0, blue) to high correlation (1, red). Vertical and horizontal lines identify the location of the FG-motifs.
There were some interesting points along the dilution path. Immediately on the addition of water the F5 and F10 Hα-HN and Hβ-HN TOCSY peaks became resolved. These were two sets of peaks, fully overlapped in the 100% DMSO data set, but moved apart and stayed apart throughout the rest of the dilutions. After the 0.83 χ data set, at the 0.59 χ set, the TOCSY spectra of the K7 Hα-HN peak drifted across the F5 and F10 Hα-HN peaks. This forced a NOESY to reconfirm which peak was which. This one point was the only time during the dilutions that a peak moved in a way where it became ambiguous as to its assignment extrapolated from previous spectra. In all other cases it was straight forward to copy the peaks from the previous dilution TOCSY and make minor adjustments to the peak locations (the relative location of peaks did not change significantly). This statement must be modified slightly as only the Hα, Cα, HN and some Hβ peaks were assigned during the dilution series and most R-group atoms were left unassigned.
Discussion
To address the question how the MFGP adopts the role of the IDP in water from a relatively ordered structure in DMSO, a comprehensive NMR based analysis was undertaken to determine how the chemical shifts of MFGP's backbone change as DMSO is replaced by water. To achieve this goal two different experiments will be performed and evaluated: (1) a variable temperature experiment to determine the change in the amide protons chemical shift with respect to temperature (ΔδHN/ΔT) at differing dilutions of water and DMSO (Fig. 2) and (2) the alpha position (1Hα & 13Cα) chemical shift at differing dilutions of the same co-solvent dilutions (Figures 3 and 4). The primary purpose of this research is to describe over one hundred NMR spectra taken in a systematic manner for further reference. As described before, the backbone chemical shifts are sensitive to the steric effects of secondary structure in a polypeptide. Equally, solvent dielectric constant and solvent peptide interactions such as hydrogen bonding and temperature can have an effect on the backbone chemical shifts. This data set is designed to point out the changes in chemical shift in relation to temperature and hydrogen bonding to the amide proton leaving only the steric effects of structure or dielectric effects of the solvent to be teased out.
The variable temperature experiment has been shown to provide key information towards the inter and intra-molecular hydrogen bonding of the amide proton 16,40. This proton is vital to many different secondary structures, and determining if it is solvent accessible or intra-molecularly hydrogen bonded provides powerful information for the existence of this structure. The literature describes two different ways of interpreting VT experiments, some simply state that a ΔδHN/ΔT greater than -6 ppb/K describes non-solvent hydrogen bonding 40. Other resources are more conservative and state that a ΔδHN/ΔT less than -4 ppb/K describe non-solvent hydrogen bonding, a ΔδHN/ΔT greater than -6 ppb/K describes solvent based hydrogen bonding, and the center range, -6<ΔδHN/ΔT<-4 ppb/K, is a gray area 16. In this study as we are adding the third dimension of co-solvent concentration to the set, we will be most interested in large changes in the ΔδHN/ΔT value between different dilutions; as opposed to discreet values, as such values are poorly defined in the literature in co-solvent concentrations of DMSO and water.
The importance of the alpha position towards structure was described (Figures 3 and 4). It can be very sensitive to secondary structure, but questions will always remain as to how significant the effects of solvent are to these values. There are two important goals of the data, first to determine radical changes between dilutions hinting towards a structural change, second to provide future researchers with a basis set for such co-solvent experiments with IDPs. While the primary goal of obtaining a data set of the basic behavior of an IDP under changing co-solvent conditions was quite successful, the true interest lies in the regular and clear critical points of the chemical shifts. One question which led to this set of experiments was, “Can we reliably use the alpha proton data set to restrict the backbone angles during structural calculations?” It is quite clear, where changes of Hα on the order of 0.1ppm are significant, that removing the data set from the 100% DMSO structure was valid. Not only was there a distinct change in the Hα and Cα chemical shifts as the concentration of water increased, the movement of the shifts could not have been modeled by a simple ‘systemic error’ addition or subtraction. Said another way, the change in the alpha position was complex and far from linear on most occasions with respect to the DMSO:H2O ratio. Yet, we expect that this approach might prove to be helpful to further explore of solvent based perturbation in the IDP research field.
A cumulative view of the solvent perturbation effects on the overall organizational changes on the ensemble of structures provides valuable clues on the transition from an ordered to disordered ensemble. The central region of MFGP, in particular the segment from K7 through F10 show a distinctive effect in terms of their organization within the ensemble compared to rest of the residues. Serine 9 is specifically interesting from the Δδ/Δχ data sets as it is unique (with the exceptions of the N-terminal S1) in showing a strong downfield shift in the carbon dimension as the water concentration increased. This is at the approximate location of the highly disordered region from the structure determined in the section above.
The parameters that signify the changes are not ‘traditional’ structural parameters, but a combination of protein dynamics, protein-solvent interactions as well as nature of the solvent itself. Even in this short peptide a consistent of FG-FG interactions that is considered important in longer FG-nups is self-evident 38 with the role of the central region of MFG and it particular S9 is notable. These experiments culminate in an approach towards identifying the structural habits of an IDP. We have found distinct similarities between two different experimental sets (structural ensemble in a non-native solvent and NMR structural parameters in native/non-native solvent concentrations) and can show MFGP made a discrete structural change along the path from DMSO to water. The use of DMSO to perturb a disordered peptide towards a smaller or different set of conformations is successful and under the assumptions made towards the dielectric constant and viscosity in the interior of the NPC may provide reliable information on the conformational preference of the in vivo FG-nucleoporin.
Supplementary Material
Supporting Figures: Figure S1: Finger print region of MFGP NOESY (150ms) spectrum in DMSO-d6.. Arrows represent the sequential assignment process.
Figure S2: Cα-Hα correlations in the HSQC spectrum of MFGP in DMSO-d6.
Figure S3: Variation of 13Cα and 1Hα chemical shifts with respect to random coil chemical shifts. Darker (red) and lighter (yellow) bars use random coil shifts derived in water 37 and DMSO 14, respectively.
Figure S4: Summary of structural calculation parameters under CYANA.
Figure S5: ΔδHN/ΔT for each residue. The left most bar for each residue represents 1 mol DMSO/mol and moving right .83, .59, 0.5, 0.43, 0.37, 0.30, 0.20. Note the large change for many residues between the third bar (0.59) and the fourth (0.50).
Acknowledgments
VVK acknowledges many discussions with Drs. E. Lau, M. Rexach and M. Colvin during the early part of this research. KAH was supported by a graduate fellowship from the National Science Foundation (NSF Award #1059994) and intramural funds from the office of the Provost and College of Science and Math, Fresno State. This research was in part supported by NIH grants P20 MD 002732, P20 CA 138025 and RO1 GM 077520.
References
- 1.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Journal of Molecular Graphics and Modelling. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK, Oldfield C, Meng J, Romero P, Yang J, Chen J, Vacic V, Obradovic Z, Uversky V. BMC genomics. 2008;9:S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky VN. Protein science. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turoverov KK, Kuznetsova IM, Uversky VN. Progress in Biophysics and Molecular Biology. 2010;102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel SS, Belmont BJ, Sante JM, Rexach MF. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 6.Denning DP, Patel SS, Uversky V, Fink AL, Rexach M. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2450. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogan A, Stewart M, Giles E. Cochlear implants international. 2002;3:54–67. doi: 10.1179/cim.2002.3.1.54. [DOI] [PubMed] [Google Scholar]
- 8.Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT. The Journal of Cell Biology. 2000;148:635. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panté N, Kann M. Molecular biology of the cell. 2002;13:425. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moroianu J, Blobel G, Radu A. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:2008. doi: 10.1073/pnas.92.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enenkel C, Blobel G, Rexach M. Journal of Biological Chemistry. 1995;270:16499. doi: 10.1074/jbc.270.28.16499. [DOI] [PubMed] [Google Scholar]
- 12.Bayliss R, Littlewood T, Strawn LA, Wente SR, Stewart M. The Journal of biological chemistry. 2002;277:50597–50606. doi: 10.1074/jbc.M209037200. [DOI] [PubMed] [Google Scholar]
- 13.Bayliss R, Littlewood T, Stewart M. Cell. 2000;102:99–108. doi: 10.1016/s0092-8674(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 14.Tremblay ML, Banks AW, Rainey JK. Journal of Biomolecular NMR. 2010;46:257–270. doi: 10.1007/s10858-010-9400-5. [DOI] [PubMed] [Google Scholar]
- 15.Sacco A, Matteoli E. Journal of Solution Chemistry. 1997;26:527–535. [Google Scholar]
- 16.Iqbal M, Balaram P. Biochemistry. 1981;20:7278–7284. doi: 10.1021/bi00528a035. [DOI] [PubMed] [Google Scholar]
- 17.Rance M, Sørensen OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K. Biochemical and Biophysical Research Communications. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 18.Aue W, Karhan J, Ernst R. The Journal of Chemical Physics. 1976;64:4226–4227. [Google Scholar]
- 19.Piantini U, Sorensen O, Ernst RR. Journal of the American Chemical Society. 1982;104:6800–6801. [Google Scholar]
- 20.Hurd RE. Journal of Magnetic Resonance. 1990;87:422–428. [Google Scholar]
- 21.Braunschweiler L, Ernst R. Journal of Magnetic Resonance (1969) 1983;53:521–528. [Google Scholar]
- 22.Shaka A, Lee C, PINES A. Journal of Magnetic Resonance. 1988;77:274–293. [Google Scholar]
- 23.Rucker S, Shaka AJ. Molecular Physics. 1989;68:509–517. [Google Scholar]
- 24.Macura S, Ernst RR. Molecular Physics. 1980;41:95–117. [Google Scholar]
- 25.Thrippleton MJ, Keeler J. Angewandte Chemie International Edition. 2003;42:3938–3941. doi: 10.1002/anie.200351947. [DOI] [PubMed] [Google Scholar]
- 26.Bodenhausen G, Ruben DJ. Chemical Physics Letters. 1980;69:185–189. [Google Scholar]
- 27.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Journal of Biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Goddard T, Kneller D. University of California, San Francisco. 2006 [Google Scholar]
- 29.Guntert P. Methods in Molecular Biology-Clifton then Totowa. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 30.Van Geet AL. Analytical Chemistry. 1970;42:679–680. [Google Scholar]
- 31.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. Journal of Biomolecular NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 32.Mathworks Inc.; Mathworks Inc.,: Natick, MA 01760, USA, 2005.
- 33.Systat Software, I.; Systat Software, Inc.: San Jose, CA, USA, 2005.
- 34.Wuthrich K. NMR of proteins and nucleic acids. Wiley; 1986. [Google Scholar]
- 35.Mielke SP, Krishnan VV. Journal of Biomolecular NMR. 2004;30:143–153. doi: 10.1023/b:jnmr.0000048940.51331.49. [DOI] [PubMed] [Google Scholar]
- 36.Mielke SP, Krishnan VV. Progress in nuclear magnetic resonance spectroscopy. 2009;54:141–165. doi: 10.1016/j.pnmrs.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ. Journal of American Chemical Society. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 38.Krishnan V, Lau EY, Yamada J, Denning DP, Patel SS, Colvin ME, Rexach MF. PLoS Computational Biology. 2008;4 doi: 10.1371/journal.pcbi.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada J, Phillips JL, Patel S, Goldfien G, Calestagne-Morelli A, Huang H, Reza R, Acheson J, Krishnan VV, Newsam S, Gopinathan A, Lau EY, Colvin ME, Uversky VN, Rexach MF. Molecular & cellular proteomics: MCP. 2010;9:2205–2224. doi: 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothemund S, Weißhoff H, Beyermann M, Krause E, Bienert M, Mügge C, Sykes BD, Sönnichsen FD. Journal of Biomolecular NMR. 1996;8:93–97. doi: 10.1007/BF00198143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figures: Figure S1: Finger print region of MFGP NOESY (150ms) spectrum in DMSO-d6.. Arrows represent the sequential assignment process.
Figure S2: Cα-Hα correlations in the HSQC spectrum of MFGP in DMSO-d6.
Figure S3: Variation of 13Cα and 1Hα chemical shifts with respect to random coil chemical shifts. Darker (red) and lighter (yellow) bars use random coil shifts derived in water 37 and DMSO 14, respectively.
Figure S4: Summary of structural calculation parameters under CYANA.
Figure S5: ΔδHN/ΔT for each residue. The left most bar for each residue represents 1 mol DMSO/mol and moving right .83, .59, 0.5, 0.43, 0.37, 0.30, 0.20. Note the large change for many residues between the third bar (0.59) and the fourth (0.50).