

Abstract
The prometastatic protein NEDD9 (Neural precursor cell Expressed, Developmentally Down-regulated 9) is highly expressed in many cancers and is required for mesenchymal individual cell migration and progression to the invasive stage. Nevertheless, the molecular mechanisms of NEDD9-driven migration and the downstream targets effecting metastasis are not well defined. In the current study, knockdown of NEDD9 in highly metastatic tumor cells drastically reduces their migratory capacity due to disruption of actin dynamics at the leading edge. Specifically, NEDD9 deficiency leads to a decrease in the persistence and stability of lamellipodial protrusions similar to knockdown of cortactin (CTTN). Mechanistically, it was shown that NEDD9 binds to and regulates acetylation of CTTN in an Aurora A kinase (AURKA)/HDAC6-dependent manner. The knockdown of NEDD9 or AURKA results in an increase in the amount of acetylated CTTN and a decrease in the binding of CTTN to F-actin. Overexpression of the deacetylation mimicking (9KR) mutant of CTTN is sufficient to restore actin dynamics at the leading edge and migration proficiency of the tumor cells. Inhibition of AURKA and HDAC6 activity by Alisertib and Tubastatin A in xenograft models of breast cancer leads to a decrease in the number of pulmonary metastases. Collectively, these findings identify CTTN as the key downstream component of NEDD9-driven migration and metastatic phenotypes.
Implications
This study provides a mechanistic platform for therapeutic interventions based on AURKA and HDAC6 inhibition for metastatic breast cancer patients to prevent and/or eradicate metastases.
Keywords: NEDD9, migration, CTTN, metastasis, AURKA, HDAC6
Introduction
NEDD9 is a scaffolding protein that is up-regulated in multiple cancers, including invasive breast carcinomas (1, 2), metastatic melanomas (3), aggressive glioblastomas (4), and ovarian cancers (3, 5). There is a strong correlation between NEDD9 overexpression and an increase in metastasis in patients (6-8), suggesting that NEDD9 potentially affects cancer metastasis through the regulation of cancer cell migration. NEDD9 knockdown leads to impaired tumor cell migration in vitro and in vivo (9, 10). NEDD9 overexpression promotes mesenchymal-based cell movement, which is dependent on actin polymerization and matrix proteinase activity (3, 10-13). Several lines of evidence suggests that NEDD9 functions to promote cancer cell migration and invasion through the sequential phosphorylation of NEDD9 by FAK and Src (14), as well as activation of small GTPase Rac1 (4, 15, 16).
The formation of leading edge lamellipodia through dynamic cycles of regulated actin assembly is critical for the motility of cells (17). Lamellipodia formation requires actin nucleation and subsequent polymerization to generate filamentous (F)-actin networks utilized to propel the cell membrane forward (18). The role of NEDD9 in lamellipodia dynamics is unknown.
Cortactin (CTTN) is a lamellipodia protein that is essential for cancer cell migration (19). Cortactin localizes to lamellipodia, where it binds actin related Arp2/3 protein complex to activate actin nucleation and to stabilize resultant F-actin branch junctures (20). CTTN is acetylated by P300/CBP-associated factor (PCAF) at multiple lysine residues within the F-actin-binding region, preventing the association of CTTN with F-actin (21). Cortactin deacetylation by histone deacetylase 6 (HDAC6) restores the ability of cortactin to bind to actin filaments. Hyperacetylation or loss of CTTN expression in mesenchymal cells impairs cell migration (22) through decreased lamellipodia persistence and stability (23).
HDAC6 is involved in both tumor cell migration and invasion, and is postulated to play a role in facilitating cancer cell metastasis (21, 24, 25). We have previously shown that NEDD9 binds to and activates oncogenic serine/threonine kinase Aurora A (AURKA), which in turn phosphorylates HDAC6 to increase its deacetylase activity (26). While the role of AURKA in cell cycle regulation is well established (27), recent work suggests that AURKA functions to promote tumor cell motility through multiple mechanisms, including phospho-activation of the F-actin severing protein cofilin (28-29). However, additional mechanisms of regulation of the actin cytoskeleton by AURKA within lamellipodia are unknown.
In our current work, we report a new molecular mechanism underlining NEDD9-dependant migration through the regulation of CTTN. Our findings suggest that NEDD9 depletion significantly impedes the migration of breast cancer (BCa) cells due to the accumulation of hyperacetylated CTTN, destabilizing actin networks at the leading edge. Overexpression of a deacetylation mimicking CTTN point mutant (9KR) is sufficient to rescue actin dynamics at the leading edge. Depletion or inhibition of AURKA or HDAC6 recapitulates the phenotype observed in NEDD9 deficient cells. In support of these observations, inhibition of AURKA with the small molecule inhibitor MLN8237 (Alisertib) or HDAC6 with Tubastatin A decreases the metastatic capability of NEDD9-overexpressing BCa cells in orthotopic xenografts. Collectively, these results indicate that AURKA and HDAC6 are critical effectors of NEDD9-mediated BCa metastasis by increasing the pool of deacetylated cortactin required for lamellipodia stability.
Materials and Methods
Plasmids, reagents and cell culture
Authenticated cell lines MDA-MB-231, BT549, HEK293T were purchased from American Type Culture Collection, MDA-231-LN (Caliper Life Sci.) and grown based on the manufacturer's recommendations. NEDD9 wild type and knockout fibroblasts, vectors expressing human full-length NEDD9 or truncation mutants of NEDD9 were previously described (30). The shRNA/siRNAs expressing constructs against NEDD9, HDAC6, AURKA, CTTN and the control were purchased from ThermoFisher Scientific (sequences available upon request). Mouse wild type, C- and N-term CTTN truncation mutant constructs were previously described (31). The acetylation-null 9KR cortactin construct was generated by site-directed mutagenesis using full-length mouse cortactin. Tissue culture medium and supplements were purchased from ATCC. Cells were transfected with plasmid DNA and/or siRNA using nucleofection (Amaxa).
ECM coated Boyden chamber migration assays
Migration assays were performed using BD FluoroBlok™ insets (BD Biosciences) as previously described (7).
Kymography assay and analysis
Cells expressing mCherry-LifeAct (32) (Addgene, 40908) were plated on glass bottom dishes (Fisher Scientific) for 12h; live cell imaging was conducted using a Nikon LiveScan swept-field confocal microscope with a 37°C heated chamber in L15 medium. Images were captured every 1 min for 120 min. Kymograms were produced by extracting 1 pixel-width strips from each movie frame during lamellipodia extension, and then assembled using ImageJ (v1.40). At least 10 cells per treatment were analyzed; protrusion velocity, maximum extension, and persistence were calculated as described (Suppl.Fig.S1) (23, 33).
F-actin co-sedimentation assay
Cells were grown to confluence and lysed as described (34). Cell lysates were clarified by centrifugation at 10,000 g for 10 min. The monomeric rabbit skeletal muscle actin (Cytoskeleton) was polymerized to generate F-actin according to the manufacturer's protocol. F-actin co-sedimentation assays were carried out according to a previously published protocol (34). Briefly, cell lysates were incubated with 30μg of F-actin in 50 μl of F-actin binding buffer for 1 h at 4°C. Reaction mixtures were centrifuged at 100,000 g for 1 hour at 4°C. Supernatants were transferred to fresh tubes and the pellets were incubated in 50μl of G-buffer for 1 hour. Equal volumes of supernatant and pellet fractions were analyzed by Western blotting.
Western blotting and immunoprecipitation
Cells were disrupted by a PTY lysis buffer (35) containing the protease inhibitor cocktail (ThermoFisher Scientific). For immunoprecipitation (IP), primary antibodies were incubated with samples overnight at 4°C and followed by the addition of the protein A/G-sepharose (G&E Healthcare Life Sciences) for 2 h, precipitated by centrifugation, washed, and the immune complexes resolved by SDS-PAGE (35). The antibodies against Aurora A (AurA-N), NEDD9 (2G9), myc-tag (Santa Cruz), pan-acetyl lysine (Cell Signaling), or CTTN (4F11) were used for IP. Western blotting was conducted using standard procedures (7) with antibodies against: NEDD9 (2G9), alpha-tubulin, acetylated alpha tubulin, GAPDH, β-actin (Sigma), Aurora A (BD Biosciences), GFP (BD Bioscience), Myc-tag (Santa Cruz); cortactin (Novus Biologicals), phAurora A/T288 (Cell Signaling Technology), GFP (Abcam, ab2900) and a custom made anti-HDAC6 (#7) antibody raised against a recombinant fragment of human HDAC6 (890-1048aa) (Thermo Scientific). Secondary antibodies (Jackson ImmunoResearch) and quantification methods were previously described (7).
Immunofluorescence (IF)
IF was performed as previously described (35) using primary antibodies against cortactin (Novus), cortactin (4F11), phTyr166-NEDD9 (custom made against a peptide encompassing amino acids 159-172 of human NEDD9 by 21st Century Biochemicals), acetylated tubulin (Sigma), HDAC6 (#7) and AURKA (7). The secondary antibodies included: donkey anti-rabbit Alexa Fluor 405, 488, 555, and anti-mouse Alexa Fluor 647. F-actin was visualized by Rhodamine phalloidin staining (Life Technologies). Images were captured with a confocal microscope LSM510, 63x objective, NA1.43 at 0.2-0.3μm step (Carl Zeiss). Quantifications were performed on z stack projections and collected using the standard acquisition parameters within ImageJ (NIH) and LSM Image analysis software (Carl Zeiss).
Morphometric analysis of CTTN distribution
CTTN and F-actin IF images were analyzed in ImageJ by applying the Z-stack at the sum of 10 projections/cell. Signal intensities for CTTN and F-actin were calculated as outlined in the Suppl.Fig.S3.
Colorimetric HDAC activity assay
Cells were lysed in a PTY buffer and clarified lysates were used for the HDAC activity assessment using the colorimetric HDAC activity assay (Upstate) according to the manufacturer's recommendations. To differentiate the effect between class I&II and class III cytoplasmic deacetylases, lysates were treated with 4μM of the HDAC class I & II inhibitor Trichostatin A (Upstate). Assays were done in triplicate and read on a TECAN GENios plate reader.
Animal Bioluminescence Imaging (BLI) of mammary fat pad injections
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ/ (NSG) immunodeficient mice (Jackson Laboratory) were housed in the West Virginia University Animal Facility under pathogen-free conditions under an approved Institutional Animal Care and Use Committee protocol.
For orthotopic injections, luciferase-expressing MDA-MB-231LN were re-suspended in PBS (1×107 cells/ml), and 0.1 ml was injected into the 4th inguinal mammary gland of 6-8 weeks old female mice. Treatment groups consisted of at least six mice, and experiments were repeated three times. Mice were imaged weekly for quantitative evaluation of tumor growth and dissemination for a total of 4 weeks. Following the injection of 150 mg/kg of D-Luciferin (Perkin Elmer) into the peritoneum, tumors were visualized using the IVIS® Lumina-II Imaging System and Living Image-4.0 software (Perkin Elmer). The differences in tumor growth between the groups was determined by BLI and pathology measurements. All treatments (as described below) were initiated 1 week after the tumor cell injection.
Animal Bioluminescence Imaging (BLI) tail-vein injections
NSG males, 6-8 weeks old, were intravenously injected with 1×105 cells re-suspended in 0.05 ml of PBS and followed by BLI weekly for 2 weeks. The average lung radiance was estimated at each point in both the control and the treated animals. The lungs were collected at the end of the study and were fixed and pathologically analyzed for the number of pulmonary metastases by a pathologist as described below.
Pathology analysis of lung metastases
Collection, fixation and determination of pulmonary metastases were conducted as described (7) by a pathologist (RHL). The mean number of metastases per mm2 +/−S.E.M was determined from three independent experiments (n=6-10 animals/treatment group).
Treatment with AURKA Inhibitors. a) Cell lines studies
Cells were treated with 5-10nM of MLN8054 (Selleckchem) for 2-12h, disrupted in a PTY buffer (35), and processed for WB or IP. b) Xenograft studies. Compound administration began 1 week after the injection of the tumor cells into the mammary fat pad of the mice. MLN8237 was reconstituted in 10% 2-hydroxypropyl-b-cyclodextrin, 1% sodium bicarbonate in water. 20 mg/kg/dose was administered via oral gavage twice a day for 4 days/week for a total of 2 weeks. For the lung metastasis studies, MLN8237 was administered in the same manner as above starting 24 h after the tumor cell tail vein injections.
Treatment with HDAC6 Inhibitors. a) Cell lines studies
Cells were treated with 20μM Tubacin (Sigma), 10-20μM Tubastatin A (ChemScene), and 2μM ACY1215 (ChemieTek) or 1μg/ml Trichostatin A (Sigma) for 2-12h, and disrupted in a PTY buffer (35) and processed for WB or IP. b) Xenograft studies. Compound administration began 1 week after the injection of the tumor cells into the mammary fat pad of the mice. Tubastatin A was dissolved to 70mg/ml in DMSO and brought to 10mg/ml with PBS. A 40 mg/kg/dose was administered via intra peritoneal injection once a day for 5 days/week for a total of 2 weeks (36). For the lung metastasis studies, Tubastatin A was administered in the same manner as above, starting 24 h after the tumor cell tail vein injections. ACY-1215 was dissolved to 40mg/ml in DMSO and brought to 24mg/ml with saline. A 75 mg/kg/dose was administered via intra peritoneal injection once a day for 5 days/week for a total of 2 weeks (37).
Statistical Analysis
Statistical comparisons were made using a one-way analysis of the variance (ANOVA). When more than two groups were analyzed, the two-way ANOVA was used. P≤0.05 was considered to be significant. Experimental values were reported as the mean +/−S.E.M. All calculations were made using GraphPad InStat software.
Results
NEDD9 depletion leads to a decrease in persistence and stability of the leading edge protrusions
The role of NEDD9 in the regulation of leading edge cortical actin dynamics during cell migration is currently unknown. To determine the role of NEDD9 on lamellar actin dynamics we performed a kymography of the leading edge of highly migratory BCa cells (MDA-MB-231) expressing mCherry-LifeAct along with siRNAs targeting NEDD9 (siN1 and siN2) or non-targeting control (siCon) (Fig.1A-B, Suppl.Fig.S1). Depletion of NEDD9 results in an 87% increase in the protrusions velocity and a 30% decrease in the persistence/stability of protrusions compared to the control cells. No changes in the maximum extension length were observed (Fig.1C). The decreased lamellipodia stability in NEDD9 knockdown cells corresponds with a 3-fold reduction in overall tumor cell migration (Suppl.Fig.S2A-B).
Figure 1. NEDD9 depletion leads to a decrease in stability of the leading edge protrusions.
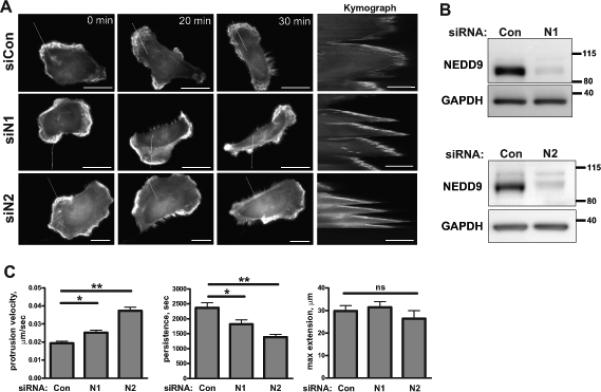
A. Representative images of the MDA-MB-231 cells co-trasfected with siControl (siCon) or siNEDD9 (siN1, siN2) and LifeAct-mCherry (white) were used to generate kymographs (inserts on the right, scale bar 10μm). White lines indicate the section across the cell membrane used to quantify the lamellipodia protrusion as described in the Supplemetary methods. The scale bar is 20μm. B. WB analysis of NEDD9 expression in siCon and siNEDD9 treated MDA-MB-231 cells with anti-NEDD9 antibodies, GAPDH is used as the loading control. C. Quantification of protrusion velocity, persistance/stability and maximum extension in cells as in A-B. Data are presented as mean values from three independent experiments, n=10 per treatment group, ± S.E.M. One-way ANOVA with pairwise comparison analysis of protrusion velocity: *p=0.0011 for siCon/siN1, **p<0.0001 siCon/siN2; protrusion persitance: *p=0.0103 for siCon/siN1, **p=0.0003 siCon/siN2; protrusion maximum extension: p is non-significant-(ns) for siCon/siN1/siN2.
NEDD9 interacts with cortactin in lamellipodia
The defects in lamellipodia dynamics observed in NEDD9 knockdown cells closely resembles the phenotype generated by depletion of the actin regulating protein cortactin (CTTN) (23). To evaluate if NEDD9-dependent changes in the lamellipodia involved CTTN, we determined the subcellular localization of both proteins. We found that NEDD9 co-localized with CTTN at the lamellar edge (Fig.2A). Interestingly, there is a high degree of co-localization between CTTN and the active Y166-phosphorylated form of NEDD9 (Fig.2B). To evaluate the possibility of an interaction between NEDD9 and CTTN, proteins were immunoprecipitated from MDA-MB-231 cells (Fig.2C) and mouse embryonic fibroblasts generated from NEDD9 knockout (KO) or wild type (WT) animals (Fig.2D). The NEDD9-CTTN complex was present in all cells except for NEDD9 KO fibroblasts, confirming the specificity of the NEDD9-CTTN association. Using the GFP-fusion truncation mutants of NEDD9 and full length CTTN, we identified that the serine-rich region (351-654aa) and the C-terminal (626-834aa) domain of NEDD9 were capable of binding to CTTN (Fig.2E) suggesting one or more cortactin binding sites exist between residues 351-834. Reciprocal experiments with CTTN truncation mutants determined that the N-terminal CTTN domain (1-350aa) is responsible for binding to NEDD9 (Suppl.Fig.S2C).
Figure 2. NEDD9 co-localizes to and interacts with CTTN at lamellipodia.
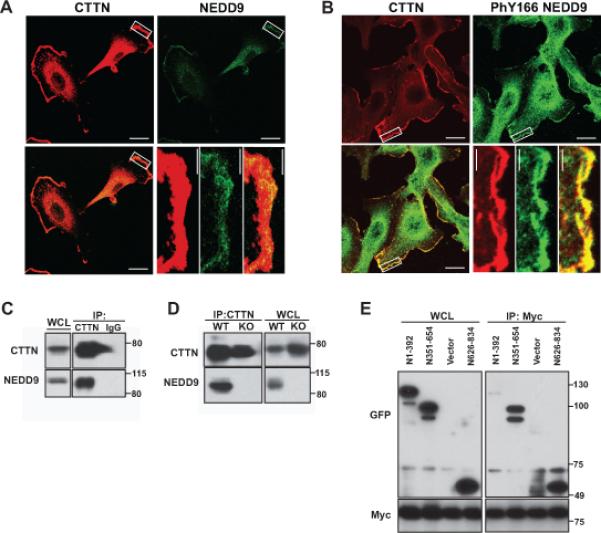
A. Representative images of immunofluorescent staining of the MDA-MB-231 cells with anti-CTTN (red) and anti-NEDD9 (green) antibodies. B. IF staining of cells with anti-CTTN (red) and anti-Y166-phNEDD9 (green) antibodies. Insets for A-B are enlarged areas at the leading cell edge indicated by a white rectangle. The scale bar is 20μm in the main panels and 4μm in the insets. C-D. Co-immunoprecipitation of CTTN and NEDD9 from the MDA-MB-231 cells (C) or from wild type (WT) or NEDD9 knock out (KO) mouse embryonic fibroblasts (MEFs) with anti-CTTN antibodies; nonspecific mouse IgG was used as the control. WB with anti-CTTN and anti-NEDD9 antibodies. Equal loading was assessed in whole cell lysates (WCL) and normalized to GAPDH (not shown). E. IP of CTTN using anti-myc tag antibodies from HEK293T cells cotrasfected with the indicated constructs: GFP-fusion truncation mutants of human NEDD9 (N1-392, N351-654, N626-834 aa) and full length myc-tagged human CTTN for 24h. WB with anti-GFP, -myc antibodies.
NEDD9 depletion results in disassociation of CTTN from the leading edge of the cell
Next we tested whether NEDD9 expression impacts CTTN localization at the leading edge of the migrating cells. The control and NEDD9 depleted cells were stained with anti-CTTN antibody and phalloidin to visualize the filamentous actin (Fig.3A). The distribution of the fluorescent signal corresponding to CTTN and the actin staining along the cell length was measured as outlined in the Suppl.Fig.S3. NEDD9 knockdown led to a significant reduction in the amount of CTTN localized to the leading edge of the cell without significant changes in the distribution of actin (Fig.3B). Thus, in the absence of NEDD9, CTTN has a limited ability to reside at the leading edge. The binding of CTTN to F-actin is crucial for its localization at the leading edge since deletion or post-translational modification of the F-actin binding region dislocates CTTN from the cell periphery and ablates cell migration (38, 39).
Figure 3. NEDD9 depletion results in CTTN disassociation from the leading edge of the cell.

A. Representative images of immunofluorescent staining of siNEDD9- and siCon-MDA-MB-231 cells incubated with anti-CTTN (red), actin (green) antibodies and DAPI (DNA; blue). White lines in the IF panels indicate the areas used for calculation of CTTN and actin distribution in relation to percent of the cell length. The scale bar is 10μm. B. Diagram of relative fluorescent intensities for CTTN (top graph) and actin (bottom graph) from staining shown in (A). Intensities across the cell was calculated as described in the material and methods and Supplementary Fig.S3. Data is represented as a sum of grey values for each pixel, n=10 per treatment group with ± S.E.M. Two-way ANOVA analysis, p<0.0001 for CTTN distribution; p=0.215 (non-significant, ns) for actin.
NEDD9 deficiency leads to decreased HDAC6 activity and an increase in acetylated CTTN
To determine the acetylation status of CTTN, we immunoprecipitated CTTN from the control and shNEDD9 cells. We found that depletion of NEDD9 led to an increase in acetylated CTTN, while total protein levels of CTTN were not affected (Fig.4A). A similar phenomenon was previously observed in cells with the depletion or inhibition of HDAC6, known to play a key role in the deacetylation of CTTN (21). Indeed, depletion of HDAC6 increased CTTN hyperacetylation (Fig.4B).
Figure 4. NEDD9 and AURKA deficiency leads to a decrease in HDAC6 activity and an increase in the acetylation of CTTN.
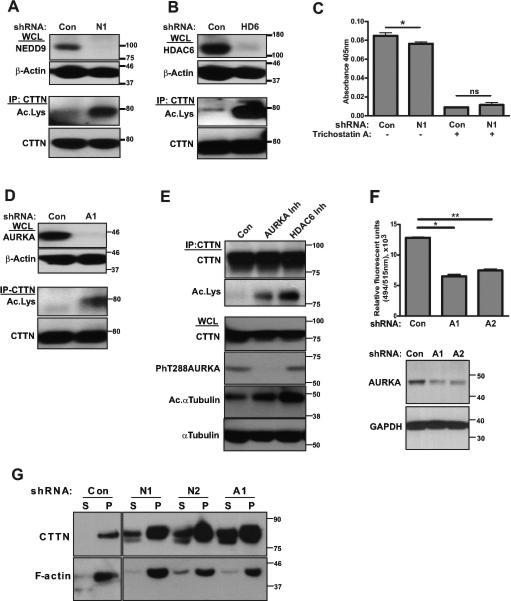
A. Top: Representative images of WB analysis of WCL for NEDD9 expression with anti-NEDD9 and -beta actin antibodies. Bottom: IP, WB analysis of total and acetylated CTTN using anti-CTTN and -acetylated lysine antibodies. B. Top: Representative images of WB analysis of HDAC6 (HD6) with anti-HDAC6 and -beta actin antibodies. Bottom: IP, WB analysis of total and acetylated CTTN with anti-CTTN and -acetylated lysine antibodies. C. Colorimetric HDAC activity assay with cytoplasmic extracts from shCon and shNEDD9 (N1) MDA-MB-231 cells with (+) or without (−) Trichostatin A. Graphs represent mean values ± S.E.M. from three independent experiments, n=3 per treatment group. One-way ANOVA with pairwise comparison *p=0.045 for shCon/shN1 (−) Trichostatin A, and ns for shCon/shN1 (+) Trichostatin A. D. Top: Representative images of WB analysis of AURKA expression with anti-AURKA and -beta actin antibodies. Bottom: IP, WB analysis of total and acetylated CTTN using anti-CTTN and -acetylated lysine antibodies. E. Top: IP of CTTN from lysates of MDA-MB-231 cells pretreated for 12h with: 1) Control 2) AURKA inhibitor, 3) HDAC6 inhibitor, and visualized by anti-CTTN and anti-acetylated lysine antibodies. Bottom: WB analysis of WCL, used for IP (top), with anti-CTTN, -phT288AURKA, -acetylated and -total alpha tubulin. F. Top: Fluoroblock Boyden Chamber migration assay for shCon and shAURKA (shA1, shA2) MDA-MB-231 cells. Data presented as mean values with ± S.E.M from three independent experiments, n=3 per treatment group. One-way ANOVA *p<0.0001 for shCon/shA1; **p<0.0001 for shCon/shA2. Bottom: WB analysis of cells used in migration assay with anti-AURKA and -GAPDH antibodies. G. F-actin pull-down assay using cell lysates shCon, shNEDD9 (N1, N2) and shAURKA (A1) as in (A, D). Representative images of WB analysis of F-actin precipitates (P) and supernatant (S) with anti-CTTN and -actin antibodies.
To measure the HDAC6 activity in NEDD9 depleted and control cells we utilized a colorimetric HDAC activity assay (Fig.4C). Cytoplasmic extracts from shNEDD9 cells had decreased deacetylase activity compared to the controls. To define whether this decrease was attributed to the decreased HDAC6 activity, both cell lysates were pre-treated with Trichostatin A (TSA). TSA inhibits HDACs of class I-II, including cytoplasmic HDAC6, but not cytoplasmic deacetylase of class III (SIRT-1/2), which potentially can also de-acetylate CTTN (40). The difference in de-acetylation activity between the control and shNEDD9 was abolished by treatment with TSA suggesting the involvement of HDAC6, but not SIRT-1/2, in the deacetylation of CTTN.
AURKA activity is required for CTTN de-acetylation and binding to F-actin
We previously reported that HDAC6 is a direct substrate of Aurora A kinase (AURKA), and phosphorylation of the HDAC6 increases its activity (26). Similar to NEDD9 depletion, treatment with shRNAs against AURKA increases the acetylation of CTTN (Fig.4D). Importantly, inhibition of AURKA or HDAC6 with specific inhibitors led to an increase in the acetylation levels of CTTN (Fig.4E), indicating a potential clinical application. Hereby, expression and activity of AURKA is required for deacetylation of CTTN, even in the presence of HDAC6. The knockdown of AURKA also led to a 2-fold decrease in migration (Fig.4F) of the MDA-MB-231 cells. The increase in the amount of acetylated CTTN in shNEDD9, shHD6 and shAURKA expressing cells correlated with a decreased ability of CTTN to bind F-actin (Fig.4G, Suppl.Fig.S4A).
NEDD9 scaffolds an AURKA-CTTN-HDAC6 complex at the leading edge to promote CTTN de-acetylation
To determine where the AURKA-HDAC6 complex gets activated we have mapped the localization of HDAC6, CTTN and AURKA. We found that CTTN is co-localized with AURKA and HDAC6 at the leading edge (Fig.5A-B). HDAC6 was shown previously to co-localize with CTTN both at the cell membrane and cytoplasm (40). Here we show that AURKA and NEDD9 co-localize with CTTN at the leading edge (Fig.5A, Fig.2A-B), indicating the potential importance of this complex in lamellipodia maturation. NEDD9, AURKA, and CTTN were also found to form a complexes when co-immunoprecipitated from several different cell lines (Fig.5C); however, this is not the case in NEDD9 deficient cells, suggesting NEDD9 plays a critical role in scaffolding this complex at the leading edge.
Figure 5. NEDD9 scaffolds the AURKA-CTTN-HDAC6 complex at the leading edge.
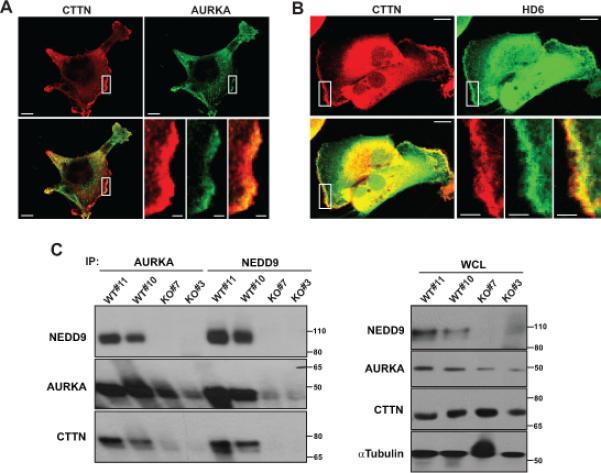
A. Representative images of immunofluorescent staining of the MDA-MB-231 cells with anti-CTTN (red) and anti-AURKA (green) antibodies. Images are reconstructed confocal projections. The scale bar is 10μm. B. Immunofluorescent staining of cells with anti-CTTN (red) and anti-HDAC6 (green) antibodies. The scale bar is 10μm. Insets for A-B are the enlarged areas at the leading edge indicated by white rectangular. The scale bar is 1.5 and 3μm in the insets A and B, respectively. C. Left: Representative images of WB analysis of co-immunoprecipitation of NEDD9, AURKA and CTTN from wild type (WT#11, #10) or NEDD9 knock out (KO#7, #3) MEFs by anti-AURKA and -NEDD9 antibodies; Right: WB analysis of WCL with anti-NEDD9, -AURKA, -CTTN and -alpha tubulin antibodies.
Re-expression of CTTN-9KR mutant rescues the NEDD9-driven deficiency in lamellipodia maturation
To test whether the decrease in the amount of de-acetylated/active CTTN was the cause of the shNEDD9-driven deficiency in lamellipodia persistence, and the increase in velocity, we overexpressed the de-acetylation mimicking mutant of CTTN-9KR or control vector in the shNEDD9 cells (Fig.6A, Suppl.Fig.S5). A kymography analyses of the MDA-MB-231 cells transfected with 9KR mutant, but not in the negative control, shows the rescue of protrusion velocity and persistence to the level observed in the control cells (Fig.6B-C), indicating that deacetylated CTTN is a direct and essential mediator of the NEDD9-AURKA-HDAC6 pathway of lamellipodia stabilization in the BCa cells.
Figure 6. Expression of the de-acetylation mimicking mutant of CTTN/9KR in shNEDD9 cells is sufficient to rescue lamellapodia stability.
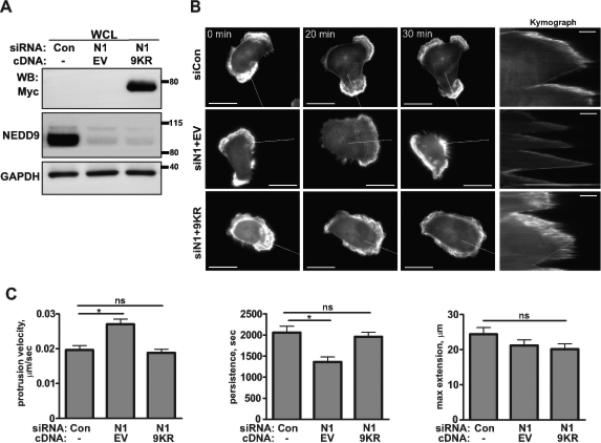
A. WB analysis of myc-CTTN/9KR mutant or empty vector (EV) expression in MDA-MB-231 cells transfected with siRNAs against NEDD9 (N1) or non-targeting control (Con) with anti-myc, -NEDD9 and -GAPDH antibodies. B. Representative images of the MDA-MB-231 cells co-trasfected with siControl (siCon); or siNEDD9 (siN1) and pRK5-myc-empty vector (siN1+EV); or siNEDD9 and pRK5-myc-CTTN/9KR (siN1+9KR) and LifeAct-mCherry (white) were used to generate composite kymographs (inserts on the right, scale bar 5μm). White lines indicate the section across the cell membrane used to quantify the velocity, persistance/stability and extension of protrusions as described in Supplemetary methods. The scale bar is 20μm. C. Quantification of protrusion velocity, persistance and maximum extension in cells as in A-B. Data presented as a mean values ± S.E.M from three independent experiments, n=10 per treatment group. One-way ANOVA with pairwise comparison for velocity: *p=0.0004 for siCon/siN1+EV, p is ns for siCon/siN1+9KR; persitance: *p=0.0003 for siCon/siN1+EV, p is ns for siCon/siN1+9KR; maximum extension: p is ns for siCon/siN1+EV or /siN1+9KR.
Inhibition of HDAC6 decreases metastasis in the xenograft models of BCa
To evaluate the potential clinical application of the inhibition of NEDD9-downstream targets, such as HDAC6 or AURKA in suppressing migration and metastasis, we treated NOD-Scid-gamma (NSG) female mice injected with the human MDA-MB-231LN cells with either vehicle, HDAC6 inhibitor-Tubastatin A, AURKA inhibitor-MLN8237/Alisertib, or in combination. After the injection of the cancer cells, the tumors were allowed to grow for 1 week, then followed by two weeks of treatment. Bioluminescent images of the primary tumors within the mammary fat pads, which were acquired at the beginning (week 0) and at the end (week 2) of the treatment, showed no significant differences in the primary tumor growth among all treatment groups (Fig.7A-B). Nevertheless, the application of Tubastatin A as single agent led to a two-fold decrease in the number of pulmonary metastases when compared to the control (vehicle treated) animals. Next, we combined Tubastatin A and MLN8237 to test if this combination would increase the efficacy of Tubastatin A against metastasis. The addition of the MLN8237 improved the effect of Tubastatin A, indicating a potential additive effect (Fig.7C-D). A complementary analysis of the pulmonary metastases by ex vivo lungs BLI supports the pathology data (Suppl.Fig.S6A-B). We would also like to note that the application of the MLN8237 alone was very efficacious in comparison with the vehicle or other treatment groups, indicating that the combination of the anti-proliferative and anti-migrative action of the MLN8237 could be sufficient to prevent and treat metastasis, while the inhibition of HDAC6 could potentially only decrease migration of the tumor cells.
Figure 7. Inhibition of HDAC6 decreases metastasis in the xenograft models of BCa.

A. Representative images of bioluminescence (BLI) of mice orthotopically injected with MDA-MB-231LN cells; first day of treatment (week 0 of treatment (top), week 1 post injection) and after 2 weeks of treatment (bottom) with the vehicle, MLN8237, Tubastatin A and a combination. B. Quantification of the primary tumor growth based on BLI, n=6/group, mean total photon flux +/−S.E.M, two-way ANOVA, p values are non-significant (ns) p=0.6513 for week 2 in all treatment groups. C. Representative images of H&E staining used for quantification of metastatic foci in sections of the lungs from animals as in (A, bottom) (Vehicle, Tubastatin A). The scale bar is 100μm. D. Quantification of the number of metastases in the lungs of orthotopically injected animals at 2 weeks post treatment by a pathologist using serial sections of the lungs and H&E staining. Results are a mean number of metastases per mm2 +/−S.E.M, n=6 per treatment group, two-way ANOVA *p=0.0001 for Vehicle/MLN, **p=0.0094 Vehicle/TubA, ***p=0.0012 for Vehicle/MLN+TubA, ns for TubA/MLN+TubA. E. Representative images of BLI; mice intravenously injected with the MDA-MB-231LN cells; Top: Vehicle, ACY1215 or Tubastatin A (week 0, no treatment; 24h post injection). Bottom: 2weeks post treatment, end point. F. Representative images of H&E staining used for quantification of metastatic foci in sections of the lungs from animals as in (E) 2 weeks post treatment (Vehicle, Tubastatin A). The scale bar is 100μm. G. Quantification of the number of metastases in the lungs of intravenously injected animals at 2 weeks post treatment. Results are the mean number of metastases per mm2 +/−S.E.M, n=6 per treatment group, one-way ANOVA with pairwise comparison *p=0.0358 for Vehicle/ACY, **p=0.0318 Vehicle/TubA, ns for ACY/ TubA.
Next, we utilized intravenous injection of the MDA-MB-231LN cells to determine the impact of HDAC6 inhibitors on the lungs colonization by circulating the tumor cells (CTC). To inhibit HDAC6 we have used 2 specific inhibitors: 1) Tubastatin A, which is widely applied in in vitro and in vivo studies, and 2) ACY-1215, a small molecule inhibitor and currently in clinical trials for myeloma treatment (37). Based on the BLI data (Fig.7F) and the pathology reports on the dissected lungs (Fig.7E-G), tumor cells in circulation and the lung parenchyma were extremely sensitive to both Tubastatin A and ACY-1215 and had a limited capacity to colonize the lungs when compared with the vehicle-treated cells. The efficacy of the inhibitors against HDAC6 was confirmed by IF and WB analysis of the tumor cells looking for acetylation of tubulin and CTTN (Suppl.Fig.S6C-D). Application of Tubastatin A, which has minimal, if any, toxicity, decreased the ability of BCa to disseminate in xenograft models, suggesting a potentially successful clinical application of anti-HDAC6 compounds as an anti-metastatic agent. We also tested the efficacy of combination Tubastatin A and MLN8237 on the lungs colonization by CTCs. The addition of the MLN8237 improved by 2-fold the effect of Tubastatin A, indicating an additive effect (Suppl.Fig.6E-G).
Discussion
NEDD9 is a scaffolding protein highly expressed in many cancers and is associated with progression towards metastasis (1-9). However, little is known about the exact molecular mechanisms of NEDD9 driven metastasis. The ability of NEDD9 to regulate the migration was previously documented (10, 11, 13-16); however, the impact of NEDD9 in the regulation of cortical actin dynamics, particularly of lamellipodia-based protrusions, is unknown. In agreement with earlier published data (4, 41, 42), cells depleted of NEDD9 showed up to a 3-fold decrease in migration. The effect of NEDD9 depletion on migration is analogous to depletion of the well-known pro-migratory regulators CTTN and HDAC6 (21). Although the role of CTTN in cell migration has been well outlined, the critical components affecting its activity are just starting to emerge (19-21, 23, 43, 44). In our current study, we define a new signaling cascade responsible for the increased tumor cell migration via the regulation of CTTN de-acetylation and AURKA-dependent HDAC6 activation. We also provide evidence of the binding of CTTN to the NEDD9/AURKA complex at the leading edge, where CTTN is known to bind HDAC6, thereby bringing the key players of this novel pathway together.
Lamellipodia stability is greatly impaired upon NEDD9 depletion, resulting in a dramatic effect on the dynamics of the leading edge of the cell, with a 30% decrease in persistence of protrusions and an 87% increase in the velocity of membrane undulations. Similar effects were shown upon knockdown of the CTTN in a fibrosarcoma cell line (23).
We found that NEDD9 co-localizes and binds to CTTN within lamellipodia, where CTTN promotes actin nucleation and stabilization (45). Additionally, we determined that the carboxy-terminal region of NEDD9 (351-834aa) is responsible for the binding of NEDD9 to CTTN. CTTN was observed to be delocalized from the leading edge of the cell upon NEDD9 depletion, indicating the importance of NEDD9 expression for retaining CTTN at the cell periphery. Previously, dislocation of CTTN from the leading edge of the cell was described to be a result of CTTN being hyperacetylated (21, 46).
According to our findings, NEDD9 influences the acetylation status of CTTN. Upon NEDD9 depletion, CTTN is hyperacetylated. NEDD9, as shown previously, is required for the activation of its downstream target- Aurora A kinase (AURKA) (35), which is an essential effector promoting breast cancer cell invasion/migration (29). Depletion or inhibition of AURKA led to an increase in the acetylation of CTTN as well. The resulting phenotype in all cases resembled the effect of knockdown or the inhibition of HDAC6, which is responsible for deacetylating CTTN (21). The deacetylase activity of HDAC6 was previously shown by our group to be upregulated upon AURKA phosphorylation (26). The increase in CTTN acetylation upon either AURKA knockdown or inhibition suggests that, not only is the presence of AURKA necessary, but in addition, the AURKA activity is required for the deacetylation of CTTN; therefore supporting a role of AURKA phosphorylation in the regulation of HDAC6 activity towards CTTN.
The NEDD9 knockdown phenotype was rescued by a mutant variant of CTTN which mimics the deacetylated CTTN (9KR) (21). Re-expression of the 9KR mutant restored the normal actin dynamics at the leading edge of the NEDD9 depleted cells. Both the absence of CTTN (23) and, as our study indicates, hyperacetylation of CTTN triggers reduced persistence of lamellipodia protrusions. We suggest that an increase in the acetylation of CTTN, caused by NEDD9 depletion, leads to unstable protrusions formation, which is explained by the inability of the acetylated CTTN to bind F-actin in order to stabilize the actin branches (21). A reduction in the lamellipodial protrusion stability theoretically can be caused not only by an increased instability of the actin network, but also through a reduction of stable focal adhesions. It is known that the depletion of CTTN leads to a decrease in the rate of focal adhesion assembly (23); however, there is no indication that CTTN hyperacetylation can cause such an effect. To the contrary, there is evidence that an increase in the CTTN acetylation leads to stabilization of the focal adhesions (46).
NEDD9, a scaffold protein, forms a complex with CTTN and AURKA at the leading edge of the cell. Previous studies have shown that HDAC6 co-localizes and directly binds CTTN at the leading edge (21). Together, these findings suggests that NEDD9 brings CTTN, AURKA, and HDAC6 into a single complex at the cell's leading edge, where HDAC6 can be subjected to phosphorylation and activation by AURKA, a critical step allowing for HDAC6 to deacetylate CTTN (26). Collectively, our data suggests that NEDD9 is an upstream regulator of CTTN at the lamellar actin subdomain, which acts through the AURKA dependent activation of HDAC6.
Finally, we tested if targeting HDAC6 and AURKA would reduce breast cancer cell metastasis. AURKA inhibitors (MLN8237/Alisertib, PHA680632/Danusertib) are currently in Phase II-III clinical trials (47, 48). Since the increased expression of NEDD9 correlates highly with the overexpression of AURKA and metastatic disease in the human BCa (7), HDAC6 inhibitors could be utilized in patients with NEDD9 overexpressing tumors. Selective HDAC6 inhibitors are a promising group of anti-cancer compounds. One of which, ACY-1215, is currently in phase I-II clinical trials for the treatment of multiple myeloma by targeting the HDAC6 dependent proteasomal degradation pathway (37, 49). Additionally, it has been shown that the chemical inhibition of HDAC6 selectively reduces the amount of AURKA in the cell via degradation (50). Therefore, we hypothesized that simultaneous administration of HDAC6 and AURKA inhibitors might have a synergistic effect on metastasis. This approach was shown to be promising in the mouse orthotropic mammary fat pad xenograft experiments utilizing the BCa cell line MDA-MB-231LN, and representing a triple-negative BCa with high NEDD9 expression (7). HDAC6 inhibition, by the specific inhibitor Tubastatin A, in an orthotopic BCa model was able to decrease pulmonary metastasis, but not the primary tumor growth, similar to the AURKA inhibitor MLN8237. A combination of MLN8237 and Tubastatin A produced an intermediate phenotype, suggesting potential additive inhibitory effects of MLN8237. The application of the AURKA inhibitor alone was more efficient in eradicating metastases than the HDAC6 inhibitor. Further work is required to elucidate the lack of synergism in a combinatorial setting.
Utilizing tail-vein injection as a model for tumor colonization, comparable efficacy in reducing BCa cell metastasis in lungs was achieved for both HDAC6 inhibitors, ACY-1215 and Tubastatin A. Interestingly, combination of Tubastatin A and MLN8237 proved to be very efficient in eradication of metastases, improving the efficiency of Tubastatin A by 2 fold. Therefore, HDAC6 inhibitors may be utilized as a comparable alternative to AURKA inhibitor therapy for treatment of invasive BCa that may be resistant to AURKA inhibitors.
Supplementary Material
Acknowledgements
We thank Dr. Mikael Schaller (WVU, USA) and Mrs. Lana Yoho for their outstanding administrative support, in addition to the WVU Tissue Bank and Animal Care Facility. The Animal Models & Imaging Facility and the Microscope Imaging Facility was supported by the MBRCC and the NIH grants P20 RR016440, P30 RR032138/GM103488 and S10RR026378. This work was supported by a grant from the NIH-NCI (CA148671 to E.N.P), a Susan G. Komen for the Cure Foundation (KG100539 to E.N.P) and in part by a NIH/NCRR (5 P20 RR016440-09 to the Mary Babb Randolph Cancer Center) award.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest.
Disclaimers: This manuscript contains original work only and has not been published nor submitted elsewhere. All authors have directly participated in the planning, execution and analysis of this study and approved the submitted version of this manuscript.
Author Contributions. V.K.K, S.L.M, S.A.W, and E.N.P conceived the project and wrote the manuscript. Live cell imaging and analysis was done by V.K.K and L.C.K. Animal treatment, IVIS imaging, pathology and tumor analysis was done by V.K.K, S.L.M, R.H.L, and M.B.S. All remaining experiments and data analysis were performed by V.K.K, S.L.M, A.R, R.A.C, R.J.I, L.C.K, and E.N.P.
References
- 1.Minn AJ, Gupta GP, Padua D, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104(16):6740–5. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M, Gans JD, Nogueira C, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Natarajan M, Stewart JE, Golemis EA, et al. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene. 2006;25(12):1721–32. doi: 10.1038/sj.onc.1209199. [DOI] [PubMed] [Google Scholar]
- 5.Wang VW, Bell DA, Berkowitz RS, Mok SC. Whole genome amplification and high-throughput allelotyping identified five distinct deletion regions on chromosomes 5 and 6 in microdissected early-stage ovarian tumors. Cancer Res. 2001;61(10):4169–74. [PubMed] [Google Scholar]
- 6.Chang JX, Gao F, Zhao GQ, Zhang GJ. Role of NEDD9 in invasion and metastasis of lung adenocarcinoma. Exp Ther Med. 4(5):795–800. doi: 10.3892/etm.2012.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ice RJ, McLaughlin SL, Livengood RH, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 73(10):3168–80. doi: 10.1158/0008-5472.CAN-12-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WH, Lan HY, Huang CH, et al. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol. 14(4):366–74. doi: 10.1038/ncb2455. [DOI] [PubMed] [Google Scholar]
- 9.Izumchenko E, Singh MK, Plotnikova OV, et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res. 2009;69(18):7198–206. doi: 10.1158/0008-5472.CAN-09-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong C, Wang C, Wang L, et al. NEDD9 is a positive regulator of epithelialmesenchymal transition and promotes invasion in aggressive breast cancer. PLoS One. 6(7):e22666. doi: 10.1371/journal.pone.0022666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas JT, Jr., Salimath BP, Slomiany MG, Rosenzweig SA. Regulation of invasive behavior by vascular endothelial growth factor is HEF1-dependent. Oncogene. 29(31):4449–59. doi: 10.1038/onc.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 188(1):11–9. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law SF, Estojak J, Wang B, Mysliwiec T, Kruh G, Golemis EA. Human enhancer of filamentation 1, a novel p130cas-like docking protein, associates with focal adhesion kinase and induces pseudohyphal growth in Saccharomyces cerevisiae. Molecular and cellular biology. 1996;16(7):3327–37. doi: 10.1128/mcb.16.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh M, Cowell L, Seo S, O'Neill G, Golemis E. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys. 2007;48(1):54–72. doi: 10.1007/s12013-007-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–23. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 16.van Seventer GA, Salmen HJ, Law SF, et al. Focal adhesion kinase regulates beta1 integrin-dependent T cell migration through an HEF1 effector pathway. Eur J Immunol. 2001;31(5):1417–27. doi: 10.1002/1521-4141(200105)31:5<1417::AID-IMMU1417>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 17.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 18.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Tondravi M, Liu J, et al. Cortactin potentiates bone metastasis of breast cancer cells. Cancer Res. 2001;61(18):6906–11. [PubMed] [Google Scholar]
- 20.Weaver AM, Karginov AV, Kinley AW, et al. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11(5):370–4. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Molecular cell. 2007;27(2):197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 23.Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15(14):1276–85. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 24.Rey M, Irondelle M, Waharte F, Lizarraga F, Chavrier P. HDAC6 is required for invadopodia activity and invasion by breast tumor cells. Eur J Cell Biol. 90(2-3):128–35. doi: 10.1016/j.ejcb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011:875824. doi: 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agnese V, Bazan V, Fiorentino FP, et al. The role of Aurora-A inhibitors in cancer therapy. Ann Oncol. 2007;18(Suppl 6):vi47–52. doi: 10.1093/annonc/mdm224. [DOI] [PubMed] [Google Scholar]
- 28.Do TV, Xiao F, Bickel LE, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. doi: 10.1038/onc.2012.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang LH, Xiang J, Yan M, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 70(22):9118–28. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- 30.Zhong J, Baquiran JB, Bonakdar N, et al. NEDD9 stabilizes focal adhesions, increases binding to the extra-cellular matrix and differentially effects 2D versus 3D cell migration. PLoS One. 7(4):e35058. doi: 10.1371/journal.pone.0035058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans JV, Ammer AG, Jett JE, et al. Src binds cortactin through an SH2 domain cystine-mediated linkage. J Cell Sci. 2012;125(Pt 24):6185–97. doi: 10.1242/jcs.121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth JW, Vogan JM, Buch PJ, et al. Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ Res. 110(7):978–89. doi: 10.1161/CIRCRESAHA.111.257964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bear JE, Svitkina TM, Krause M, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109(4):509–21. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120(6):1417–26. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 2005;7(10):937–46. doi: 10.1038/ncb1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Zoeten EF, Wang L, Butler K, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Molecular and cellular biology. 31(10):2066–78. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 119(11):2579–89. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One. 5(11):e13847. doi: 10.1371/journal.pone.0013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weed SA, Karginov AV, Schafer DA, et al. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol. 2000;151(1):29–40. doi: 10.1083/jcb.151.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaluza D, Kroll J, Gesierich S, et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 30(20):4142–56. doi: 10.1038/emboj.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fashena SJ, Einarson MB, O'Neill GM, Patriotis C, Golemis EA. Dissection of HEF1-dependent functions in motility and transcriptional regulation. J Cell Sci. 2002;115(Pt 1):99–111. doi: 10.1242/jcs.115.1.99. [DOI] [PubMed] [Google Scholar]
- 42.Sima N, Cheng X, Ye F, Ma D, Xie X, Lu W. The Overexpression of Scaffolding Protein NEDD9 Promotes Migration and Invasion in Cervical Cancer via Tyrosine Phosphorylated FAK and SRC. PLoS One. 8(9):e74594. doi: 10.1371/journal.pone.0074594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell DH, deFazio A, Sutherland RL, Daly RJ. Expression and tyrosine phosphorylation of EMS1 in human breast cancer cell lines. International journal of cancer. 1996;68(4):485–92. doi: 10.1002/(SICI)1097-0215(19961115)68:4<485::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 44.Weaver AM. Cortactin in tumor invasiveness. Cancer letters. 2008;265(2):157–66. doi: 10.1016/j.canlet.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ammer AG, Weed SA. Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65(9):687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsunoda K, Oikawa H, Tada H, et al. Nucleus accumbens-associated 1 contributes to cortactin deacetylation and augments the migration of melanoma cells. J Invest Dermatol. 131(8):1710–9. doi: 10.1038/jid.2011.110. [DOI] [PubMed] [Google Scholar]
- 47.Carpinelli P, Ceruti R, Giorgini ML, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6(12 Pt 1):3158–68. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- 48.Huck JJ, Zhang M, McDonald A, et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res. 8(3):373–84. doi: 10.1158/1541-7786.MCR-09-0300. [DOI] [PubMed] [Google Scholar]
- 49.Mithraprabhu S, Khong T, Jones SS, Spencer A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br J Haematol. 162(4):559–62. doi: 10.1111/bjh.12388. [DOI] [PubMed] [Google Scholar]
- 50.Park JH, Jong HS, Kim SG, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. J Mol Med (Berl) 2008;86(1):117. doi: 10.1007/s00109-007-0260-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.