

Abstract
Cu is an essential element with many biological roles, but its roles in the mammalian nervous system are poorly understood. Mice deficient in the cuproenzyme peptidylglycine α-amidating monooxygenase (PAM+/− mice) were initially generated to study neuropeptide amidation. PAM+/− mice exhibited profound deficits in a few behavioral tasks, including enhancements in innate fear along with deficits in acquired fear. Interestingly, several PAM+/− phenotypes were recapitulated in Cu restricted wildtype mice and rescued in Cu supplemented PAM+/− mice. These behaviors correspond to enhanced excitability and deficient synaptic plasticity in the amygdala of PAM+/− mice, which are also rescued by Cu supplementation. Cu and ATP7A are present at synapses, in key positions to respond to and influence synaptic activity. Further study demonstrated that extracellular Cu is necessary for wildtype synaptic plasticity and sufficient to rescue PAM+/− LTP. These experiments support roles for PAM in Cu homeostasis and for synaptic Cu in amygdalar function.
Keywords: peptidylglycine alpha-amidating monooxygenase, copper, amygdala, fear conditioning, synaptic plasticity, ATP7A
Cu and PAM
Biologically, Cu is best known as a cofactor in oxidation-reduction reactions1. Only a handful of enzymes that require Cu for their activity are encoded by the human genome, yet these “cuproenzymes” are diverse in their biological roles2–5. Cu deficiency can be caused by mutation in the gene encoding the intracellular Cu transporter ATP7A and results in Menkes disease6–8. Menkes disease patients have low levels of brain Cu, and typically present in infancy with failure to thrive and seizures. Symptoms of Menkes disease are thought to correspond to insufficient activity of specific cuproenzymes; however the pathophysiology leading to seizures is not well understood9.
Peptidylglycine α-amidating monooxygenase (PAM) is a vesicular transmembrane protein with two luminal enzymatic domains, one of which is a cuproenzyme10. Peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL) act sequentially to catalyze the cleavage of the C-terminal glycine of their peptidylglycine substrate to yield an amidated product peptide11,12. The PHM domain requires 2 Cu molecules for its activity13, while PAL uses Zn14. PAM is the only enzyme in mammals that can accomplish this amidation reaction15,16, which is essential to the biosynthesis of approximately half of all neuropeptides; α-amidation is the final step required to confer full biological activity to many neuropeptides12,17.
PAM function is critical for development and nervous system function 16,18. Aside from their role in multiple endocrine systems, peptides can also act as neurotransmitters and neuromodulators in the nervous system to regulate neuronal and synaptic physiology and function19. Amidation is impaired in animal models of Menkes disease20; levels of key neuropeptides are reduced with brain region-specificity21.
PAM+/− mice and fear
To study the function of PAM in animals, our group developed a mouse genetically deficient in the gene encoding PAM. Mice homozygous for the deleted Pam gene die in utero with profound edema16, a phenotype nearly identical to a knockout model of adrenomedullin, an amidated peptide22. By contrast, mice heterozygous for the gene deletion grow and reproduce normally, but exhibit a striking set of behavioral and physiological deficits when challenged (Table 1). PAM+/− mice and their wildtype littermates are bred from wildtype dams and PAM+/− sires for use in all experiments (Fig. 1A). PAM+/− mice cannot maintain their body temperature in the cold, secondary to impairments in vasoconstriction23. When injected with the GABAA receptor antagonist pentylenetetrazol, PAM+/− mice have more severe seizures at lower doses than their wildtype littermates24. When tested in fear-related tasks, PAM+/− mice display an interesting dichotomy of behaviors24,25.
Table 1.
PAM+/− behavioral and physiological phenotypes and their responses to dietary Cu condition.
| Phenotype | Cu restriction mimic | Cu supp rescue | Reference |
|---|---|---|---|
| Thermoregulation deficit | yes | yes | 23,24 |
| Vasoconstriction deficit | yes | 23 | |
| Seizure hypersensitivity | yes | no | 24 |
| Anxiety-like behaviors | yes | yes | 24 |
| Fear conditioning – contextual | no | 25Gaier et al, resubmitted | |
| Fear conditioning – cued | yes | 25Gaier et al, resubmitted | |
| Fear-potentiate startle | yes | 25Gaier et al, resubmitted | |
| Amygdalar LTP | yes | 25Gaier et al, resubmitted |
Fig 1. Innate and learned fear behavioral testing.

(A) PAM+/− mice are generated from wildtype (Wt) dams and PAM+/− sires, yielding 50% Wt and 50% PAM+/− offspring. Wt and PAM+/− littermates are used in all experiments. (B,C) Innate fear is tested in the elevated zero maze, and fear learning and memory is tested in fear conditioning. (B) The elevated zero maze is comprised of four quadrants, two of which are “open” (have low walls) and two of which are “closed” (have high walls). Mice are placed into the maze for a fixed length of time, and the amount of time spent in the open versus the closed arms of the maze is recorded. The percentage of time spent in the closed arm of the maze corresponds directly to the amount of anxiety-like behavior of the animal in this test. (C) Fear conditioning tests begin with training, during which a tone is paired with an aversive footshock. Subsequently, mice are either tested in contextual (left) or cued (right) fear conditioning. In contextual fear conditioning, mice are placed in the same chamber in which training occurred. In cued testing, mice are placed in a novel chamber and the same tone that was played during training is played. The amount of time the animal spends freezing (immobile except for breathing) while in the training context or during the tone is recorded. The percentage of time spent freezing is a direct measure of how well the animal learned and remembers the association between the conditioned stimulus (context or tone) and the unconditioned stimulus (footshock).
Innate fear in rodents is typically tested in the elevated zero maze (Fig. 1B). In this task, animals are placed onto an elevated circular platform with four segments, half of which have tall walls (closed) and the other very low walls (open)26. Rodents instinctually spend more time in the closed regions due to an innate fear of open spaces, a behavior that likely evolved to avoid predation. The amount of time spent in the open regions can be used to quantify the degree of innate fear for comparison between different genetic and/or pharmacologic conditions. PAM+/− mice placed in the elevated zero maze spend significantly less time in the open areas than their wildtype littermates24,25. Anxiolytic medications increase the proportion of time animals spend in the open areas, owing to a reduction in innate fear. Interestingly, PAM+/− mice respond to low doses of diazepam, a GABAA receptor allosteric modulator and anxiolytic, by increasing the amount of time spent in the open regions while wildtype mice are unaffected25. Thus PAM+/− mice show enhanced innate fear behavior and sensitivity to a GABAergic anxiolytic in the elevated zero maze.
Acquired fear, or fear learning and memory, can be assessed in many ways. One commonly used method is fear conditioning, which tests an animal’s ability to associate a previously neutral stimulus (conditioned stimulus) with a footshock (unconditioned stimulus) (Fig. 1C)27. Two types of fear conditioning are typically tested. In contextual fear conditioning, the conditioned stimulus is the environment, consisting of the smell, shape and texture of the conditioning chamber. In cued fear conditioning, a single discrete auditory tone serves as the conditioned stimulus. Cued testing occurs in a different chamber where the exact same tone can be played. The conditioned and unconditioned stimuli are presented together during training, and the conditioned stimulus is presented alone during testing. Subsequent presentation of the conditioned stimulus causes the animal to outstretch the front paws and cease all motion aside from breathing, a readily quantifiable response referred to as “freezing”. The portion of time the animal spends freezing can be used as a direct measure of fear learning and memory.
PAM+/− mice freeze for less time than their wildtype littermates during both contextual and cued testing, indicating a deficiency in both forms of fear conditioning25. PAM+/− mice are also deficient in another fear learning and memory task, fear-potentiated startled. These results stand in stark contrast to the enhanced innate fear we observe in PAM+/− mice. Other paradigms aimed at manipulating neuropeptidergic signaling, including specific peptide knockouts, generally have similar effects on both innate and learned fear28–33. This paradox between PAM+/− innate and learned fear behaviors intrigued us and led us to perform physiological and biochemical experiments aimed at understanding amygdalar function in PAM+/− mice.
PAM+/− amygdalar physiology
The amygdala is a part of the brain’s limbic system situated in the medial temporal lobe in mammals and plays a role in both innate and learned fear behaviors (Fig. 2A)34. Manipulation of neuronal activity in the amygdala has direct effects on innate fear responses35–37. Amygdala function is required for both cued and contextual forms of fear conditioning; structural ablation or pharmacological blockade of synaptic transmission in the amygdala before or during, but not after, fear training abolishes learned fear responses27.
Fig 2. Amygdala structure, circuitry and recording methods.
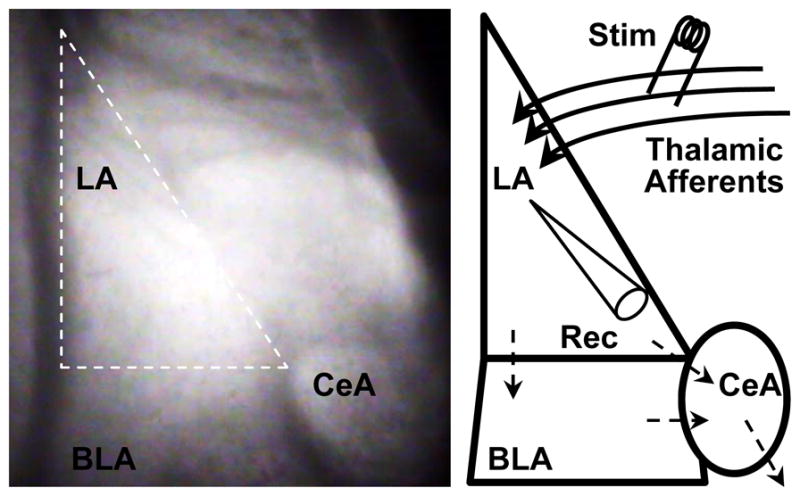
(Left) Infrared differential interference contrast image taken at 40× of a coronal mouse brain slice containing the amygdala. The amygdala is comprised of lateral (LA), basolateral (BLA) and central (CeA) nuclei. We performed our experiments in the LA (outlined in white dashes). (Right) Schematic depicting amygdala nuclei. Thalamic afferent axons enter the LA and synapse onto LA neurons. LA neurons send their projections to the BLA and CeA; the BLA sends its projections to the CeA as well. CeA neurons send their projections to midbrain and brainstem nuclei responsible for evoking the fight or flight response in mammals, which contributes to anxiety-like behaviors and freezing in fear conditioning. We stimulated (Stim) thalamic afferent synapses and recorded post-synaptic responses (Rec; whole cell patch clamp) in LA pyramidal neurons.
With the goals of elucidating the neurobiological underpinnings of the PAM+/− paradoxical fear behaviors, we performed whole cell patch clamp recordings in the basolateral complex of the amygdala. Baseline excitatory synaptic transmission is enhanced in the PAM+/− amygdala, and PAM+/− amygdalar neurons are hyperexcitable when compared to wildtype amygdalar neurons25. Interestingly, baseline inhibitory synaptic transmission is also increased, which may relate to the observed heightened sensitivity to benzodiazepines. These findings may relate to the increased innate fear of PAM+/− mice.
Long-term potentiation (LTP) at thalamic afferent synapses in the lateral nucleus of the amygdala is a well-accepted and extensively studied correlate to cued fear conditioning in mice (Fig. 3)38–40. We induce LTP in acute brain slices prepared from wildtype and PAM+/− mice by pairing thalamic afferent stimulations with post-synaptic depolarization to trigger action potentials in single neurons in the presence of the GABAA receptor blocker picrotoxin25,41 [as is necessary in these types of experiments42]. After induction, synaptic responses in PAM+/− neurons fail to increase above baseline, and are significantly lower than those recorded in wildtype neurons. This result corresponds to the deficit in cued fear memory25(Gaier et al, resubmitted). Notably, full blockade of GABAergic transmission with the addition of a GABAB receptor antagonist (CGP35348) restores PAM+/− LTP to wildtype levels (Fig. 3). It was initially hypothesized that these behavioral and physiological deficits arose from impairments in amidation of specific neuropeptides; however, for the few peptides examined, only modest reductions in amidation have been observed in PAM+/− mice16,43.
Fig 3. Amygdalar synaptic plasticity data summarized.

(Left) Schematic of neuronal/synaptic circuitry in the LA depicting methods for stimulating afferents to and recording from LA pyramidal neurons (PN). We stimulated thalamic afferents to the LA and recorded from PNs that receive those excitatory inputs. GABAergic interneurons (INs) also receive collateral thalamic afferent inputs and mediate feed-forward inhibition of PNs. Picrotoxin (PTX) is used in all experiments to block GABAA receptors to eliminate the fast component of feed-forward inhibition and permit LTP. Pairing of pre-synaptic stimulation with post-synaptic depolarization induces LTP (large arrow), resulting in potentiation of synaptic efficacy (right). LTP in this system under these conditions includes increased pre-synaptic neurotransmitter release. Several conditions are permissive to (+) or abolish (gray X) LTP. Pam heterozygosity (a low Cu condition) abolishes LTP, but not in the presence of GABAB receptor blockade (using CGP35348). Dietary Cu supplementation prior to slice preparation or inclusion of Cu in the perfusate (10 μM CuSO4) rescues PAM+/- LTP. On the other hand, inclusion of the membrane impermeant, Cu-specific chelator BCS abolishes LTP in both Wt and PAM+/− mice even when GABAB receptors are blocked. Combined, these results indicate that Cu is necessary for LTP and sufficient to rescue LTP in PAM+/− mice.
Cu rescue of PAM+/− phenotypes
Knowing that PAM requires Cu for its catalytic activity, we manipulated dietary Cu in wildtype and PAM+/− mice and performed the same behavioral and physiological experiments. Mild dietary Cu restriction (Cu depleted food for 4–6 weeks) in wildtype mice impairs thermoregulation, lowers seizure threshold and promotes anxiety-like behaviors very similar to PAM+/− mice at baseline (Table 1)24. Cu restriction has little to no effect on the PAM+/− phenotype. On the other hand, dietary Cu supplementation (300 ppm in the drinking water) in PAM+/− mice restores thermoregulation, ameliorates the anxiety-like behaviors, and rescues cued fear conditioning and fear potentiated startle 24(Gaier et al, resubmitted). Contextual fear conditioning and seizure susceptibility are the only behaviors examined that are not affected by dietary Cu supplementation in PAM+/− mice. Importantly, contextual fear conditioning tests an animal’s ability to integrate discrete sensory modalities, a task that requires the hippocampus, in addition to forming simple associations as in cued conditioning, which requires the amygdala, but not the hippocampus44. The difference in the ability of Cu to rescue only select fear memory tasks could reflect differences in the nature of the aberration in the PAM+/− amygdala and hippocampus. The hippocampus is also a very epileptogenic brain region and is likely involved in the seizure susceptibility of PAM+/− mice. That Cu supplementation distinguishes abnormal behaviors involving the hippocampus and amygdala strong suggests distinct aberrations in these brain regions and that PAM+/− amygdalar dysfunction is directly related to Cu.
We also tested the ability of Cu to rescue amygdalar LTP. Wildtype and PAM+/− mice were supplemented with dietary Cu, exactly as in the behavioral experiments, and recordings were performed on slices prepared from these mice. Dietary Cu supplementation restores PAM+/− LTP to normal levels and has no effect on wildtype LTP (Fig. 3; Table 1)(Gaier et al, resubmitted). These results draw a clear parallel between behavior and physiology in which the PAM+/− phenotype is rescued by dietary Cu. Interestingly, dietary Cu supplementation has no effect on peptide amidation, strongly arguing against our initial hypothesis that deficiencies of various amidated neuropeptides accounts for the PAM+/− phenotype24,43. Rather, these results suggest a role for Cu itself in mediating the effects of Pam heterozygosity and dietary rescue.
ATP7A and Cu at synapses
At this point we had gathered evidence that a deficiency in Cu in the PAM+/− amygdala could contribute to the synaptic impairments likely underlying the fear learning and memory deficits of these mice. Inherent to this hypothesis is the presumption that Cu and its handling machinery are present at synapses. We employed a well-established subcellular fractionation technique to directly study Cu biochemistry down to the synaptic level45. This technique requires amounts of tissue that exceed the volume of the mouse amygdala, so we used cortical tissue samples from individual animals. We focused on wildtype mice, knowing that changes in Cu and its chaperones/transporters are region specific in PAM+/− mice46.
We found that ATP7A is enriched in fractions containing synaptic vesicles and synaptosomal membranes (Fig. 4)46. The synaptic membrane fraction was further separated on a sucrose gradient and then solubilized using Triton X-100, allowing separation of insoluble proteins; neither cytoskeletal proteins nor the proteins concentrated in the post-synaptic density (PSD) are solubilized by Triton x-100. The gigadalton PSD complex forms the electron dense structure located at the tips of dendritic spines receiving excitatory inputs45. The majority of the ATP7A in the synaptosomal membrane fraction is Triton-soluble, but there is a clear and consistent portion of ATP7A that segregates with the Triton-insoluble PSD46. The C-terminal PDZ-binding motif of ATP7A may contribute to its localization in the PSD fraction47,48. The majority of Cu is in the cytosolic fraction, where Cu bound to chaperones and metallothioneins would be found46,49. Cu is also present in the synaptosomal membrane and cytosol fractions, totaling approximately 40% of the total Cu content. These data compellingly support the presence of ATP7A and Cu at synapses, but how ATP7A is distributed among synapses remained to be seen.
Fig 4. Cu and ATP7A are present at synapses.

Schematic summarizing biochemical and synaptic plasticity studies with respect to Cu and ATP7A. (Soma) Cu enters neurons and binds the chaperone Atox-1 in the cytosol, which transports Cu to ATP7A for delivery into the secretory pathway. PAM is also present in the secretory pathway and co-localizes with ATP7A in neurons. A cytosolic portion of PAM is cleaved and enters the nucleus, affecting transcription and up-regulation of Atox-1. Vesicles containing PAM, ATP7A and Cu are transported into the dendrite. (Synapse) Thalamic afferent glutamatergic terminals innervate pyramidal neuron (PN) dendritic spines. Interneuron (IN) terminals release GABA which inhibits excitatory transmission and excitation through activation of GABAA and GABAB receptors. Notably, INs express the highest levels of PAM and ATP7A. Extracellular Cu could come from any number of sources including the post-synaptic pyramidal neurons, pre-synaptic afferents, or INs. Inhibitory effects of Cu on synaptic receptors and ion channels including GABAA receptors, NMDA receptors and L-type voltage-gated Ca channels have been described (see text), but how Cu influences synaptic plasticity remains unknown. The role of Cu in modulating synaptic transmission and plasticity is complex and requires further study.
To address this issue, we simultaneously visualized ATP7A, VGlut1 (a presynaptic marker) and PSD95 (a post-synaptic marker) in cultured cortical neurons prepared from wildtype mice46. We found almost no pre-synaptic localization of ATP7A; ATP7A is primarily post-synaptic, partially overlapping with PSD95 (Fig. 4). ATP7A staining near the dendritic membrane typically either co-localizes or is juxtaposed with PSD95. It is possible that co-staining puncta represent ATP7A within the PSD that we observed in the subcellular fractionation studies. Thus, ATP7A is present only at select synapses, begging the question – what is the functional difference between ATP7A-positive and ATP7A-negative synapses? The developmental expression of ATP7A suggests a potential role in synaptogenesis50,51, so the presence of Cu in the adult animal may reflect an ongoing role in synaptic maintenance and plasticity. Nevertheless, these data provide compelling evidence for the presence of Cu and Cu handling machinery at synapses.
Cu is sufficient and necessary for PAM+/− LTP
Knowing that Cu and its handling machinery are present at synapses, we manipulated Cu in our in vitro slice recordings to study the functional role of Cu at amygdalar synapses (Gaier et al, resubmitted). If Cu is the key to rescuing PAM+/− amygdalar dysfunction, then acute application of Cu would be sufficient for LTP. Indeed Cu (10 μM) supplied in the bath solution is sufficient to potentiate amygdalar afferent synapse (Fig. 3). Previous studies in the hippocampus have found that 10 μM Cu inhibits LTP, but we found that the concentration of Cu has no effect on wildtype LTP in the amygdala. Again, this likely reflects differences in synaptic properties in these respective brain regions.
Logically, we next asked whether Cu is necessary for LTP. We used the membrane impermeant, Cu-selective chelator bathocuproine disulfonate (BCS; 50 μM). BCS is highly selective for Cu at this concentration, and application just prior to LTP induction minimized concerns about intracellular Cu depletion52–54. Inclusion of BCS in the perfusate eliminates LTP in wildtype mice and has no effect in PAM+/− mice (Table 1; Fig. 3)(Gaier et al, resubmitted). Addition of CGP35348 to block GABAB receptors in addition to picrotoxin to block GABAA receptors, permits LTP in both genotypes, but addition of BCS under these conditions still abolishes LTP in both genotypes. Together this suggests that while altered GABAergic signaling is involved in the PAM+/− LTP deficit, Cu is necessary for LTP in both genotypes. To our knowledge, this was the first set of synaptic plasticity experiments employing a Cu chelator. Previous studies have found that Cu chelation enhances NMDA receptor activity55, which is necessary for some forms of amygdalar LTP41. However, our induction protocol has been previously shown to be dependent on voltage-gated calcium channels rather than NMDA receptors41, possibly accounting for this discrepancy. Moreover, previous physiological studies employing Cu chelators were performed in hippocampal neurons, not amygdalar neurons55.
PAM and Cu: reciprocal influences
All of our data up to this point suggest that deficits in PAM reversibly affect Cu homeostasis, with behavioral and physiological consequences. How PAM affects Cu homeostasis is not clear. In vitro experiments using a well-studied corticotropic tumor cell line (AtT-20) have been conducted to study the bidirectional influences between PAM and Cu. Low Cu levels (achieved by BCS application) promote proteolytic cleavage of PAM and PHM secretion, whereas high Cu conditions promote retention of intact PAM on the membrane surface54. Along the same lines, dietary Cu deficiency promotes serum amidating activity24. Dietary Cu deficiency also affects proteolytic processing of membrane PAM. However, dietary Cu supplementation has no effect on serum amidating activity, likely due to the mild amount of Cu provided through this supplementation protocol. In the other direction, overexpression of PAM in the same cell line results in up-regulation of Atox-1, a chaperone that delivers Cu to the secretory pathway56. The cytosolic domain of PAM (sfCD) could be cleaved and signal to the nucleus to affect gene transcription (Fig. 4)57. In mice, Pam heterozygosity alters markers of Cu homeostasis in the liver and heart24. These interactions between PAM and Cu on the cellular lever reflect a role for PAM in the regulation of Cu homeostasis58.
Our behavioral and physiological data strongly suggest a deficiency of Cu at synapses in the PAM+/− amygdala. However, total brain Cu levels are unchanged in PAM+/− mice, and dietary Cu supplementation does not affect brain Cu levels24. Immunohistochemical staining of brain slices containing the amygdala reveal normal amygdala structure with similar staining patterns of both PAM and ATP7A25,46. Staining for both PAM and ATP7A was high in the lateral and basolateral nuclei of the amygdala. Levels of ATP7A protein and mRNA are reduced in the PAM+/− amygdala and normal in the hippocampus. The same pattern of expression was seen when we analyzed transcript levels of Atox-1, the cytosolic chaperone that delivers Cu to ATP7A in the secretory pathway. Levels of Cu itself are reduced in homogenized brain punches of the amygdala isolated from PAM+/− mice compared to wildtype(Gaier et al, resubmitted). This effect is also region-specific as there is no genotypic difference in Cu levels seen in the hippocampus. The deficit in amygdalar Cu has effects on other cuproenzyme functions, namely dopamine β-monooxygenase, that is also region-specific46. This finding reflects not only a consequence of Cu deficiency, but also implies that Cu functioning in the secretory pathway in particular is deficient in the PAM+/− amygdala. Thus, Pam heterozygosity affects Cu homeostasis specifically in the amygdala, perhaps explaining the Cu-sensitive behavioral and physiological dysfunction we observed in studying the PAM+/− amygdala.
Future directions and application
The PAM+/− mouse is a model connecting brain and behavior ideal for studying the role of Cu in synaptic function. Combined, the data we have summarized here support the suggestion that Cu is released with neuronal activation and signals to the pre-synaptic terminal to promote vesicle release. Future studies should determine the cells in which ATP7A and Cu are essential for synaptic plasticity and fear conditioning, namely the prevalent glutamatergic neurons or the interneurons with high PAM and ATP7A expression. Conditional knockout models for ATP7A are the key to conducting such studies, especially when combined with induction via viral vectors using cell-type specific promoters to target ATP7A knockout. Cultured neuronal systems are more easily manipulated and will serve as ideal models for studying the role of Cu in neuronal and synaptic function. Moreover, culture models can be combined with promising new methods to visualize Cu at subcellular levels in real time59,60. We anticipate a time when these techniques will greatly expand our ability to visualize the dynamics of Cu homeostasis at synapses.
We have shown that secreted Cu is necessary for amygdalar LTP, but exactly how Cu modulates synaptic transmission and plasticity remains unclear. Cu interacts with several different receptors, binding proteins and enzymes at synapses and could therefore affect synaptic function in a multitude of ways 61. We have studied how manipulation of Cu affects behavioral and physiology, but no experiments we have performed to date have gone in the other direction. It would be interesting to test whether neuronal ATP7A and Cu localization can be manipulated by neuronal stimulation in vivo, perhaps through seizure induction or fear conditioning training. We hypothesize that the ATP7A distribution between the Triton-soluble and PSD fractions will change with stimulation, but even more interesting will be to test whether and how those responses may be altered in PAM+/− mice. Investigating these dynamics of Cu homeostasis at synapses is critical to understanding the mechanism by which Cu regulates synaptic plasticity.
All together these findings suggest an essential role for Cu in amygdalar synaptic functions with behavioral relevance. The PAM+/− model therefore may be particularly relevant to studies of post-traumatic stress disorder (PTSD), in which the amygdala is known to play a critical role 62. Progressive strengthening of traumatic memories, which drives baseline anxiety and memory-related fear, is at the heart of PTSD63. Thus, identification of Cu signaling as a critical player in amygdalar synaptic plasticity and fear memory formation may have therapeutic potential as a pharmacological target in the treatment of PSTD. Expanding the study of PAM+/− mice and Cu signaling in amygdala function to the other half of fear conditioning, extinction (not discussed here), would have even higher relevance to novel treatments for PTSD64. Aside from anxiety disorders, disruptions in Cu homeostasis have been implicated in several neurodegenerative diseases65,66 including Alzheimer’s disease67,68, prion-related diseases69 and multiple sclerosis70.
References
- 1.Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63:797S–811S. doi: 10.1093/ajcn/63.5.797. [DOI] [PubMed] [Google Scholar]
- 2.Andreini C, Banci L, Bertini I, Rosato A. Occurrence of copper proteins through the three domains of life: a bioinformatic approach. J Proteome Res. 2008;7:209–216. doi: 10.1021/pr070480u. [DOI] [PubMed] [Google Scholar]
- 3.Klinman JP. The copper-enzyme family of dopamine beta-monooxygenase and peptidylglycine alpha-hydroxylating monooxygenase: resolving the chemical pathway for substrate hydroxylation. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 4.Olivares C, Solano F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009;22:750–760. doi: 10.1111/j.1755-148X.2009.00636.x. [DOI] [PubMed] [Google Scholar]
- 5.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MENKES JH, Aleter M, STEIGLEDER GK, WEAKLEY DR, SUNG JH. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764–779. [PubMed] [Google Scholar]
- 7.Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tumer Z, Moller LB. Menkes disease. Eur J Hum Genet. 2010;18:511–518. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasad AN, Levin S, Rupar CA, Prasad C. Menkes disease and infantile epilepsy. Brain Dev. 2011;33:866–876. doi: 10.1016/j.braindev.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: the structure of peptidylglycine a-hydroxylating monooxygenase. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 11.Perkins SN, Husten EJ, Eipper BA. The 108-kDA peptidylglycine alpha-amidating monooxygenase precursor contains two separable enzymatic activities involved in peptide amidation. Biochem Biophys Res Commun. 1990;171:926–932. doi: 10.1016/0006-291x(90)90772-f. [DOI] [PubMed] [Google Scholar]
- 12.Eipper BA, Stoffers DA, Mains RE. The biosynthesis of neuropeptides: peptide alpha-amidation. Annu Rev Neurosci. 1992;15:57–85. doi: 10.1146/annurev.ne.15.030192.000421. [DOI] [PubMed] [Google Scholar]
- 13.Siebert X, Eipper BA, Mains RE, Prigge ST, Blackburn NJ, Amzel LM. The catalytic copper of peptidylglycine alpha-hydroxylating monooxygenase also plays a critical structural role. Biophysical J. 2005;89:3312–3319. doi: 10.1529/biophysj.105.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chufan EE, De M, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: the structure of the lyase domain of the amidating enzyme. Structure. 2009;17:965–973. doi: 10.1016/j.str.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mains RE, Bloomquist BT, Eipper BA. Manipulation of neuropeptide biosynthesis through the expression of antisense RNA for peptidylglycine alpha-amidating monooxygenase. Mol Endocrinol. 1991;5:187–193. doi: 10.1210/mend-5-2-187. [DOI] [PubMed] [Google Scholar]
- 16.Czyzyk TA, Ning Y, Hsu MS, Peng B, Mains RE, Eipper BA, Pintar JE. Deletion of peptide amidation enzymatic activity leads to edema and embryonic lethality in the mouse. Dev Biol. 2005;287:301–313. doi: 10.1016/j.ydbio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Mains RE, Dickerson IM, May V, Stoffers DA, Perkins SN, Ouafik LH, Husten EJ, Eipper BA. Cellular and molecular aspects of peptide hormone biosynthesis Front. Neuroendocrinol. 1990;11:52–89. [Google Scholar]
- 18.Jiang N, Kolhekar AS, Jacobs PS, Mains RE, Eipper BA, Taghert PH. PHM is required for normal developmental transitions and for biosynthesis of secretory peptides in drosophila. Dev Biol. 2000;226:118–136. doi: 10.1006/dbio.2000.9832. [DOI] [PubMed] [Google Scholar]
- 19.Gozes I, Brenneman DE, Geppetti P, Kastin AJ, Mains RE, Moody TW, Seroogy K, Spier AD, Zimmerman M. Neuropeptides: brain messengers of many faces. Trends Neurosci. 2001;24:687–690. doi: 10.1016/s0166-2236(00)02001-4. [DOI] [PubMed] [Google Scholar]
- 20.Steveson TC, Ciccotosto GD, Ma XM, Mueller GP, Mains RE, Eipper BA. Menkes protein contributes to the function of peptidylglycine alpha-hydroxylating monooxygenase. Endocrinology. 2003;144:188–200. doi: 10.1210/en.2002-220716. [DOI] [PubMed] [Google Scholar]
- 21.Niciu MJ, Ma XM, El MR, Pachter JS, Mains RE, Eipper BA. Altered ATP7A expression and other compensatory responses in a murine model of Menkes disease. Neurobiol Dis. 2007;27:278–291. doi: 10.1016/j.nbd.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci U S A. 2001;98:615–619. doi: 10.1073/pnas.021548898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bousquet-Moore D, Ma XM, Nillni EA, Czyzyk TA, Pintar JE, Eipper BA, Mains RE. Reversal of physiological deficits caused by diminished levels of peptidylglycine alpha-amidating monooxygenase by dietary copper. Endocrinology. 2009;150:1739–1747. doi: 10.1210/en.2008-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bousquet-Moore D, Prohaska JR, Nillni EA, Czyzyk T, Wetsel WC, Mains RE, Eipper BA. Interactions of peptide amidation and copper: novel biomarkers and mechanisms of neural dysfunction. Neurobiol Dis. 2010;37:130–140. doi: 10.1016/j.nbd.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaier ED, Rodriguiz RM, Ma XM, Sivaramakrishnan S, Bousquet-Moore D, Wetsel WC, Eipper BA, Mains RE. Haploinsufficiency in peptidylglycine alpha-amidating monooxygenase leads to altered synaptic transmission in the amygdala and impaired emotional responses. J Neurosci. 2010;30:13656–13669. doi: 10.1523/JNEUROSCI.2200-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd JK, Grewal SS, Fletcher A, Bill DJ, Dourish CT. Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology (Berl) 1994;116:56–64. doi: 10.1007/BF02244871. [DOI] [PubMed] [Google Scholar]
- 27.Davis M. Pharmacological and anatomical analysis of fear conditioning NIDA. Res Monogr. 1990;97:126–162. [PubMed] [Google Scholar]
- 28.Thompson BL, Rosen JB. Effects of TRH on acoustic startle, conditioned fear and active avoidance in rats. Neuropeptides. 2000;34:38–44. doi: 10.1054/npep.1999.0785. [DOI] [PubMed] [Google Scholar]
- 29.Karlsson RM, Holmes A, Heilig M, Crawley JN. Anxiolytic-like actions of centrally-administered neuropeptide Y, but not galanin, in C57BL/6J mice. Pharmacol Biochem Behav. 2005;80:427–436. doi: 10.1016/j.pbb.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Chen Q, Nakajima A, Meacham C, Tang YP. Elevated cholecystokininergic tone constitutes an important molecular/neuronal mechanism for the expression of anxiety in the mouse. Proc Natl Acad Sci U S A. 2006;103:3881–3886. doi: 10.1073/pnas.0505407103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bedard T, Mountney C, Kent P, Anisman H, Merali Z. Role of gastrin-releasing peptide and neuromedin B in anxiety and fear-related behavior. Behav Brain Res. 2007;179:133–140. doi: 10.1016/j.bbr.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 32.Pickens CL, ms-Deutsch T, Nair SG, Navarre BM, Heilig M, Shaham Y. Effect of pharmacological manipulations of neuropeptide Y and corticotropin-releasing factor neurotransmission on incubation of conditioned fear. Neuroscience. 2009;164:1398–1406. doi: 10.1016/j.neuroscience.2009.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pape HC, Jungling K, Seidenbecher T, Lesting J, Reinscheid RK. Neuropeptide S: a transmitter system in the brain regulating fear and anxiety. Neuropharmacology. 2010;58:29–34. doi: 10.1016/j.neuropharm.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sah P, Faber ES, Lopez De AM, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- 35.Rosen JB, Davis M. Enhancement of acoustic startle by electrical stimulation of the amygdala. Behav Neurosci. 1988;102:195–202. 324. doi: 10.1037//0735-7044.102.2.195. [DOI] [PubMed] [Google Scholar]
- 36.Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52:701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- 37.Truitt WA, Johnson PL, Dietrich AD, Fitz SD, Shekhar A. Anxiety-like behavior is modulated by a discrete subpopulation of interneurons in the basolateral amygdala. Neuroscience. 2009;160:284–294. doi: 10.1016/j.neuroscience.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sigurdsson T, Doyere V, Cain CK, Ledoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 40.Sah P, Westbrook RF, Luthi A. Fear conditioning and long-term potentiation in the amygdala: what really is the connection? Ann N Y Acad Sci. 2008;1129:88–95. doi: 10.1196/annals.1417.020. [DOI] [PubMed] [Google Scholar]
- 41.Bauer EP, Schafe GE, Ledoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin P, Bousquet-Moore D, Annangudi SP, Southey BR, Mains RE, Eipper BA, Sweedler JV. Probing the production of amidated peptides following genetic and dietary copper manipulations. PLoS One. 2011;6:e28679. doi: 10.1371/journal.pone.0028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips RG, Ledoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 45.Ma XM, Kiraly DD, Gaier ED, Wang Y, Kim EJ, Levine ES, Eipper BA, Mains RE. Kalirin-7 Is Required for Synaptic Structure and Function. J Neurosci. 2008;28:12368–12382. doi: 10.1523/JNEUROSCI.4269-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaier ED, Miller MB, Ralle M, Aryal D, Wetsel WC, Mains RE, Eipper BA. Pam (Peptidylglycine alpha-amidating monooxygenase) heterozygosity alters brain copper handling with region specificity. J Neurochem. 2013;127:605–619. doi: 10.1111/jnc.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linz R, Lutsenko S. Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J Bioenerg Biomembr. 2007;39:403–407. doi: 10.1007/s10863-007-9101-2. [DOI] [PubMed] [Google Scholar]
- 48.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 49.Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Affinity gradients drive copper to cellular destinations. Nature. 2010;465:645–648. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 50.El Meskini R, Cline LB, Eipper BA, Ronnett GV. The developmentally regulated expression of Menkes protein ATP7A suggest a role in axon extension and synaptogenesis. Dev Neurosci. 2005;27:333–348. doi: 10.1159/000086713. [DOI] [PubMed] [Google Scholar]
- 51.Niciu MJ, Ma XM, El Meskini R, Ronnett GV, Mains RE, Eipper BA. Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience. 2006;139:947–964. doi: 10.1016/j.neuroscience.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 52.Hawkins CJ, Perrin DD. Oxidation-reduction potentials of metal complexes in water. Part II. Copper complexes with 2,9-dimethyl- and 2-chloro-1,10-phenanthroline. Journal of the Chemical Society. 1963:2996–3002. [Google Scholar]
- 53.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 54.De M, Ciccotosto GD, Mains RE, Eipper BA. Trafficking of a secretory granule membrane protein is sensitive to copper. J Biol Chem. 2007;282:23362–23371. doi: 10.1074/jbc.M702891200. [DOI] [PubMed] [Google Scholar]
- 55.You H, Tsutsui S, Hameed S, Kannanayakal TJ, Len PE, bers JD, Lipton SA, Stys PK, Zamponi GW. ABeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Francone VP, Ifrim MF, Rajagopal C, Leddy CJ, Wang Y, Carson JH, Mains RE, Eipper BA. Signaling from the secretory granule to the nucleus: Uhmk1 and PAM Mol. Endocrinol. 2010;24:1543–1558. doi: 10.1210/me.2009-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajagopal C, Stone KL, Francone VP, Mains RE, Eipper BA. Secretory granule to the nucleus: Role of a multiply phosphorylated intrinsically unstructured domain. J Biol Chem. 2009;284:25723–25734. doi: 10.1074/jbc.M109.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bousquet-Moore D, Mains RE, Eipper BA. Peptidylgycine alpha-amidating monooxygenase and copper: a gene-nutrient interaction critical to nervous system function. J Neurosci Res. 2010;88:2535–2545. doi: 10.1002/jnr.22404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dodani SC, Domaille DW, Nam CI, Miller EW, Finney LA, Vogt S, Chang CJ. Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc Natl Acad Sci U S A. 2011;108:5980–5985. doi: 10.1073/pnas.1009932108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hirayama T, Van de Bittner GC, Gray LW, Lutsenko S, Chang CJ. Near-infrared fluorescent sensor for in vivo copper imaging in a murine Wilson disease model. Proc Natl Acad Sci U S A. 2012;109:2228–2233. doi: 10.1073/pnas.1113729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaier ED, Eipper BA, Mains RE. Copper signaling in the mammalian nervous system: Synaptic effects. J Neurosci Res. 2013;91:2–19. doi: 10.1002/jnr.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res. 2008;167:151–169. doi: 10.1016/S0079-6123(07)67011-3. [DOI] [PubMed] [Google Scholar]
- 63.Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 2012;35:24–35. doi: 10.1016/j.tins.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167:648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kapaki E, Segditsa J, Papageorgiou C. Zinc, copper and magnesium concentration in serum and CSF of patients with neurological disorders. Acta Neurol Scand. 1989;79:373–378. doi: 10.1111/j.1600-0404.1989.tb03803.x. [DOI] [PubMed] [Google Scholar]
- 66.Hozumi I, Hasegawa T, Honda A, Ozawa K, Hayashi Y, Hashimoto K, Yamada M, Koumura A, Sakurai T, Kimura A, Tanaka Y, Satoh M, Inuzuka T. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J Neurol Sci. 2011;303:95–99. doi: 10.1016/j.jns.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 67.Molina JA, Jimenez-Jimenez FJ, Aguilar MV, Meseguer I, Mateos-Vega CJ, Gonzalez-Munoz MJ, de BF, Porta J, Orti-Pareja M, Zurdo M, Barrios E, Martinez-Para MC. Cerebrospinal fluid levels of transition metals in patients with Alzheimer's disease. J Neural Transm. 1998;105:479–488. doi: 10.1007/s007020050071. [DOI] [PubMed] [Google Scholar]
- 68.Lin CJ, Huang HC, Jiang ZF. Cu(II) interaction with amyloid-beta peptide: a review of neuroactive mechanisms in AD brains. Brain Res Bull. 2010;82:235–242. doi: 10.1016/j.brainresbull.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 69.Zomosa-Signoret V, Arnaud JD, Fontes P, varez-Martinez MT, Liautard JP. Physiological role of the cellular prion protein. Vet Res. 2008;39:9. doi: 10.1051/vetres:2007048. [DOI] [PubMed] [Google Scholar]
- 70.Melo TM, Larsen C, White LR, Aasly J, Sjobakk TE, Flaten TP, Sonnewald U, Syversen T. Manganese, copper, and zinc in cerebrospinal fluid from patients with multiple sclerosis. Biol Trace Elem Res. 2003;93:1–8. doi: 10.1385/BTER:93:1-3:1. [DOI] [PubMed] [Google Scholar]