

Abstract
Breast cancer is a heterogeneous disease with six molecularly defined subtypes, the most aggressive of which are triple negative breast cancers that lack expression of estrogen receptor (ER) and progesterone receptor (PR) and do not exhibit amplification of the growth factor receptor HER2. Triple negative breast cancers often exhibit basal-like gene signatures and are enriched for CD44+ cancer stem cells. In this report we have characterized the molecular actions of the VDR in a model of triple negative breast cancer. Estrogen independent, invasive mammary tumor cell lines established from wild-type (WT) and VDR knockout (VDRKO) mice were used to demonstrate that VDR is necessary for 1,25-dihydroxyvitamin D3 (1,25D) mediated anti-cancer actions in vitro and to identify novel targets of this receptor. Western blotting confirmed differential VDR expression and demonstrated the lack of ER, PR and Her2 in these cell lines. Re-introduction of human VDR (hVDR) into VDRKO cells restored the anti-proliferative actions of 1,25D. Genomic profiling demonstrated that 1,25D failed to alter gene expression in KO240 cells whereas major changes were observed in WT145 cells and in KO clones stably expressing hVDR (KOhVDR cells). With a 2-fold cutoff, 117 transcripts in WT145 cells and 197 transcripts in the KOhVDR clones were significantly altered by 1,25D. Thirty-five genes were found to be commonly regulated by 1,25D in all VDR-positive cell lines. Of these, we identified a cohort of four genes (Plau, Hbegf, Postn, Has2) that are known to drive breast cancer invasion and metastasis whose expression was markedly down regulated by 1,25D. These data support a model whereby 1,25D coordinately suppresses multiple proteins that are required for survival of triple-negative/basal-like breast cancer cells. Since studies have demonstrated a high prevalence of vitamin D deficiency in women with basal-like breast cancer, correction of vitamin D deficiency in these women represents a reasonable, but as yet untested, strategy to delay recurrence and extend survival.
Introduction
The heterogeneity of breast cancer makes the identification of intervention strategies for this disease particularly challenging. Although many trials have assessed nutrient status in relation to breast cancer, rarely are results stratified based on molecular sub-type. Of the >200,000 women diagnosed with breast cancer annually, between 15–25% are diagnosed with “triple negative” breast cancers which lack estrogen and progesterone receptors and do not exhibit amplification of HER2. These tumors often have basal-like gene expression signatures and are enriched for CD44+ breast cancer stem cells [1]. Women with triple negative cancers have poor prognosis and few treatment options, therefore new therapeutic targets for these aggressive tumors are critically needed. Data from the Cancer Genome Atlas [2] indicates that more than 95% of basal-like breast tumors express the vitamin D receptor (VDR). Furthermore, 1,25-dihydroxyvitamin D3 (1,25D, the active form of vitamin D3) and other VDR agonists induce cell cycle arrest, trigger apoptosis and inhibit invasion in basal-like breast cancer cell lines in vitro and reduce metastasis in nude mouse models [3–10]. Despite VDR expression, a high prevalence of vitamin D deficiency has been noted in women with triple negative/basal-like breast cancer [11,12]. Sub-optimal serum vitamin D levels would reduce availability of vitamin D to tumor VDR, limiting its activity. Correction of vitamin D deficiency or provision of supplemental vitamin D in women living with this sub-type of breast cancer would be predicted to delay recurrence and extend survival.
Although population studies have often demonstrated inverse correlations between vitamin D status and breast cancer risk/progression, the data are inconsistent and rarely account for the known molecular heterogeneity of the disease. Mechanistic insight into vitamin D actions in triple negative/basal-like breast cancer cells is sorely lacking. To further evaluate the effects of 1,25D in triple negative/basal-like breast cancers, we developed an in vitro model of murine breast cancer cell lines with differential VDR expression. In this report we have characterized the 1,25D/VDR dependent genomic profiles of these cells and demonstrate that 1,25D down-regulates a cohort of genes that are frequently up-regulated in aggressive breast cancers. Our data defines novel pathways that are targeted by vitamin D in human breast cancer and provide insight into mechanisms by which VDR signaling reduces invasion and metastasis.
Methods
Generation and culture of stable cell lines
WT145 and KO240 cells were originally established in our lab from tumors induced by DMBA treatment of wild-type and VDRKO mice [13]. For the studies described here, WT145 and KO240 cells were adapted to growth in DMEM/F12 medium supplemented with 5% charcoal-stripped FBS to eliminate exposure to serum vitamin D metabolites. In previous work [14] we confirmed that stable expression of hVDR into KO240 cells using a plasmid vector (pSG5-hVDR-hygro) recapitulated 1,25D mediated anti-cancer signaling, however, sensitivity to 1,25D was lost over time in these clones. To overcome this problem, we created a retroviral hVDR vector (pBABE-puro-hVDR, Fig 1) and generated a new series of hVDR expressing cells which we characterized in the present studies. Briefly, KO240 cells were infected with pBABE-puro-hVDR or pBABE-puro empty vector (EV) retroviral particles and selected with 5μg/ml puromycin to generate mass cultures from which individual clones were isolated and characterized. Once selected, KOEV and all KOhVDR stable cell lines were maintained in DMEM/F12 medium supplemented with 5% charcoal-stripped FBS. The KOhVDR clones maintained sensitivity to 1,25D for extended passages in the absence of puromycin.
Figure 1. Expression of hVDR in murine VDRKO cells.
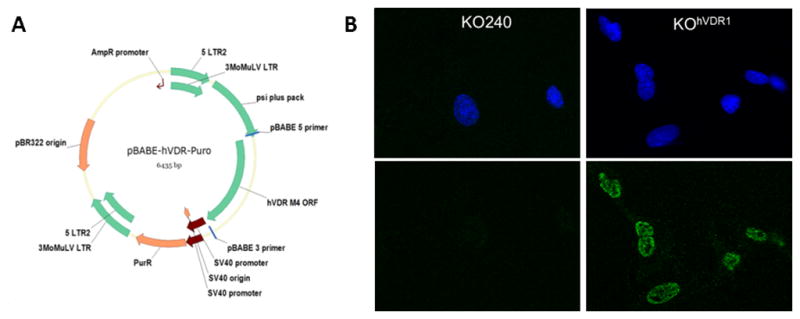
A, Retroviral expression vector for hVDR was created in pBABE-puro. B, Localization of VDR in KOhVDR cells. Cells were stained with DAPI for nuclear chromatin (top) and monoclonal VDR antibody Clone D6 (bottom) and visualized by confocal microscopy.
Growth assays and microscopy
To determine the effect of 1,25D on culture growth, cells were treated on the day after plating with 100nM 1,25D or vehicle. After 96 hours, adherent cells were fixed in glutaraldehyde, stained with crystal violet and dried overnight. Stain was resuspended in 0.2% Triton X-100 and absorbance was read at 590 nm as an indication of adherent cell density. To visualize cell morphology, cells were treated with vehicle or 10nM 1,25D for 96h and photographed live on a Nikon Eclipse TS100 phase contrast microscope. For VDR immunofluorescence, KOhVDR cells growing in chamber slides were fixed and permeabilized with ice-cold methanol, blocked overnight in PBS/1% BSA and incubated with antibody against VDR (clone D6, Santa Cruz Biotechnology, Santa Cruz, CA) followed by anti-mouse Alexa Fluor 488 secondary antibody (Life Technologies, Grand Island, NY). Coverslips were applied with ProLong Gold Antifade reagent with DAPI (Life Technologies, Grand Island, NY) and imaged on a Leica DMI6000 microscope with TCS SP5 confocal laser scanner using Leica Application Suite AF version 2.6.0.7266 software.
Western blotting
Whole cell lysates were prepared by scraping confluent monolayers in Laemmli buffer containing protease and phosphatase inhibitors. Lysates were sonicated and protein concentration was determined with the BCA protein assay (Pierce Biotechnology, Rockford, IL). Cell lysates (50μg per lane) were separated via SDS-PAGE, transferred to nitrocellulose or PVDF membranes, blocked in skim milk, and incubated overnight with primary antibodies including VDR clone D6 (Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal antibody against ER (6F11, Vector Laboratories, Burlingame, CA), mouse monoclonal antibody against PR (PR-1, Thermo Scientific, Fremont, CA), or rabbit monoclonal antibody against HER2 (Cell Signaling, Danvers, MA) followed by appropriate HRP-conjugated secondary antibodies. Detection was with ECL Plus (Pierce Biotechnology, Rockford, IL) on a Storm 860 Molecular Imaging system. Blots were then stripped using acetonitrile, 0.1M glycine and 1M Tris base (pH 7.5) and re-probed with mouse monoclonal antibodies against GAPDH or actin.
Microarray screening, pathway analysis and quantitative PCR
Three KOhVDR clones that were confirmed to express functional VDR (KOhVDR1, KOhVDR2, and KOhVDR4), along with WT145 and KO240 cells were treated with ethanol (EtOH) or 100nM 1,25D for 24h. RNA was isolated and hybridized to Affymetrix mouse gene 1.0 ST arrays comprising >28,000 annotated genes and datasets were analyzed with Genespring v10 software (Agilent, Santa Clara, CA). Briefly the data was filtered to remove probe sets with low signal across all samples. The resulting list was filtered to exclude genes with >25% CV across all replicates within a condition. The filtered entities were subjected to a moderated t-test (p<0.05) with a Benjamini-Hochberg false discovery rate to correct for multiple comparisons included. These were further filtered to identify entities that were differentially expressed in either WT145 or all three hVDR-expressing KO clones in the presence of 1,25D at a 2-fold or higher level. A venn diagram was used to identify a subset of 35 genes that were commonly altered by 1,25D in WT145 cells and in all three KOhVDR clones. Pathway analysis on this 35-gene set was performed using Babelomics 4 gene expression and functional profiling analysis suite (http://babelomics.bioinfo.cipf.es).
For real-time PCR, mRNA was harvested with RNEasy Mini Kits (Qiagen, Valencia, CA) according to manufacturer’s instructions, and two-three independent cDNA stocks were prepared from each sample using random hexamer primers (Applied Biosystems, Foster City, CA). cDNA preparations were independently analyzed in duplicate with mouse specific primers for Cyp24a1, Cib2, Enpp1, Prelp, Has2, Hbegf, Plau and Postn (Origene, Rockville, MD) using SYBR Green PCR Master Mix (ABgene, Thermo Scientific) on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Data were calculated by the ΔΔCt method, normalized against 18S and expressed relative to values obtained for vehicle treated cells.
Statistics
Data are expressed as mean±standard error, and statistical evaluation was by Students t test using GraphPad Prism software (La Jolla, CA). Comparisons were routinely made on the effect of treatment within (rather than between) each cell line. In all cases p < 0.05 was considered to be significant and is indicated by an asterisk above data bars.
Results
Development of mammary tumor cells with differential VDR expression
To study the VDR dependent effects of 1,25D on breast cancer, we previously established a series of cell lines from DMBA-induced mammary tumors that were generated in wild-type (WT) and VDR knockout (KO) mice [13]. Cells derived from WT mice (termed WT145 cells) abundantly expressed VDR protein which was transcriptionally activated by 1,25D as measured by a vitamin D responsive luciferase reporter assay [13]. Furthermore, we showed that growth of WT145 cells was dose and time dependently inhibited by 1,25D. In contrast to WT145 cells, KO240 cells derived from tumors induced in VDRKO mice did not express detectable VDR and failed to respond to 1,25D. We also demonstrated that treatment of mice with a VDR agonist (EB1089) retarded growth of xenografts composed of WT145 cells, but not those composed of KO240 cells [15].
Recently we provided proof of principle that expression of VDR in KO240 cells using a plasmid vector recapitulated 1,25D mediated anti-cancer signaling [14] however, these VDR expressing cells lost sensitivity over time. For these follow-up studies, we created a retroviral hVDR expression vector (pBABE-hVDR-Puro, Fig 1A) and selected multiple KO240 clonal cell lines with stable expression of hVDR protein. Immunofluorescent staining (Fig 1B) confirmed that KOhVDR cells expressed VDR protein in the nucleus as evident by co-localization with the nuclear stain DAPI. Four clones were originally characterized, three of which (clones 1, 2 and 4) expressed detectable VDR by western blotting (Fig 2A) and underwent growth arrest and apoptosis when treated with 1,25D (data from clone 4 is shown in Fig 2B and 2C). The magnitude of growth inhibition in response to 1,25D treatment in hVDR-expressing clones #1, #2 and #4 was similar to or greater than that observed in WT145 cells (Fig 2B and data not shown). Similar to KO240 cells, growth of in KOEV cells and KOhVDR clone #3 (which failed to express detectable VDR protein by western blotting) was unaffected by 1,25D (not shown).
Figure 2. Sensitivity of murine cell lines to 1,25D.
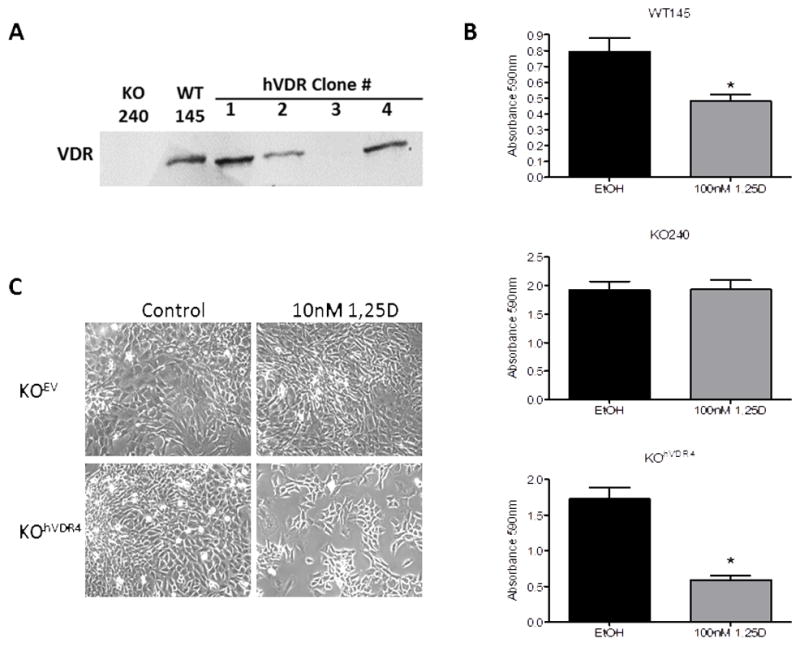
A, Western blot for VDR in WT145 cells, KO240 cells and four independent clones isolated from KOhVDR mass cultures. B, Adherent cell density was measured by crystal violet absorbance after 96h treatment with 100nM 1,25D or ethanol (EtOH) vehicle. Bars represent mean±standard error, n = 4. *p< .05, 1,25D vs control for each cell line, as evaluated by Student’s t test. C, Phase contrast images of KO240 cells expressing empty vector (KOEV) or hVDR (KOhVDR4) after 96h treatment with ethanol vehicle (Control) or 10nM 1,25D.
WT145 and KO240 cell lines are triple negative
Western blotting was used to assess expression of estrogen receptor α (ER), progesterone receptor (PR) and Her2 in WT145 and KO240 cells adapted to charcoal stripped FBS. As shown in Fig 3A, neither WT145 or KO240 cells grown in charcoal stripped FBS express ER, and there was no significant effect of the ER antagonist tamoxifen on growth of either cell line (Fig 3B). Since our previous data indicated that WT145 and KO240 cells grown in complete FBS express ER [13], adaptation to charcoal stripped FBS apparently selected for cells lacking this receptor. These results are consistent with our previous data indicating that xenografts derived from WT145 and KO240 cells grew robustly in ovariectomized mice indicating that the parental mass cultures contain estrogen independent cells [13]. No expression of either PR or HER2 was detected in WT145, KO240 or KOhVDR4 cell lysates (Fig 3C, 3D). These data indicate that the WT145 cells as well as the KO240 mass cultures and stable hVDR expressing clones can be categorized as triple negative breast cancer cell lines. Further studies are needed to determine whether these murine triple negative cells inherently express gene signatures similar to that of human basal-like breast cancers.
Figure 3. WT145 and KO240 cells are triple negative.
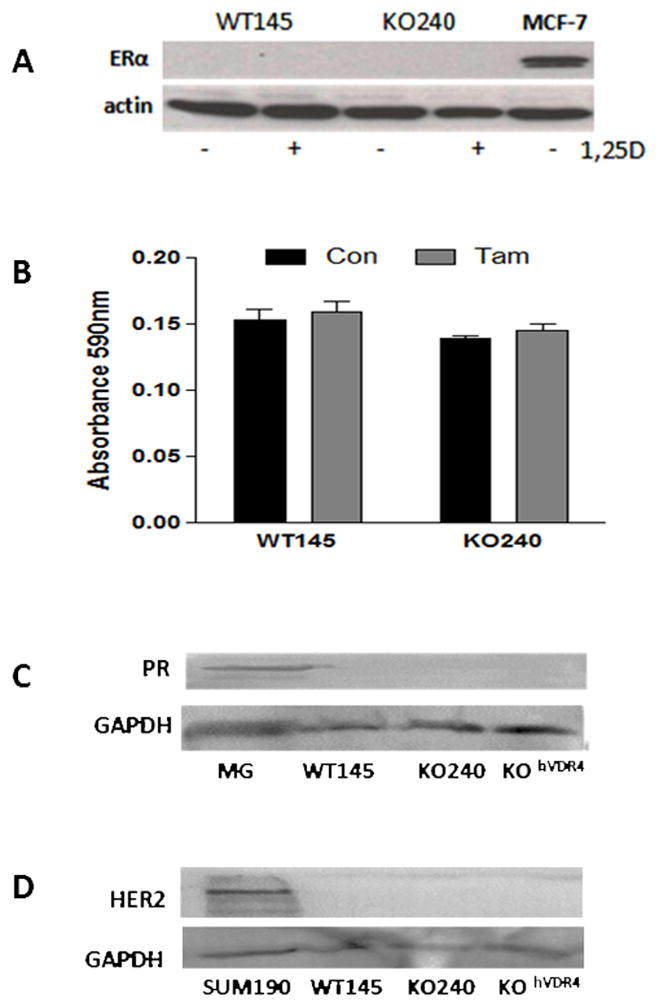
A, Western blot for ERα (top) in lysates from WT145 and KO240 cells. MCF7 breast cancer cell lysate was used as positive control. Blots were stripped and re-probed with actin antibody was used to verify protein loading. B. Effect of tamoxifen on WT145 and KO240 cell density. Cells were treated with ethanol vehicle control (Con) or 100nM tamoxifen (Tam) (bottom) for 96h and analyzed by crystal violet staining for adherent cell density. C. Western blot for progesterone receptor (PR) in WT145, KO240 and KOhVDR cell lysates; homogenate from pregnant mouse mammary gland (MG) was used as positive control. Blots were stripped and re-probed with Gapdh antibody to confirm protein loading. D. Western blot for Her2 in WT145, KO240 and KOhVDR cell lysates; lysate of known HER2+ human breast cancer cell line SUM190 was used as positive control. Blots were stripped and re-probed with Gapdh antibody to confirm protein loading.
Genomic Profiling
Microarray profiling was conducted in this model system to identify common 1,25D responsive gene targets in cells expressing mouse and human VDRs since both could trigger growth arrest in response to 1,25D. Cells were treated for 24h with 100nM 1,25D in an effort to identify gene changes that preceded growth inhibition (which is not evident for at least 48h after treatment). As shown in Fig 4A, and consistent with the lack of functional VDR, global gene expression was not significantly altered by 1,25D in KO240 cells. In contrast, major changes in gene expression were observed in WT145 cells and in all three KOhVDR clones (#1, #2, and #4). Interestingly, as evident from the heat map in Fig 4A, the genomic profiles of the three hVDR-expressing clones were similar to each other but quite distinct from the WT145 cells. After statistical analysis and 2-fold cutoff, 117 transcripts in WT145 cells and 197 transcripts in the KOhVDR clones were found to be significantly (p< 0.05) altered by 1,25D. In both cell lines approximately 70% of altered transcripts were up-regulated (85/117 in WT145 cells and 146/197 in KOhVDR cells).
Figure 4. Heat map of 1,25D regulated genomic profiles in murine cells with differential VDR expression.
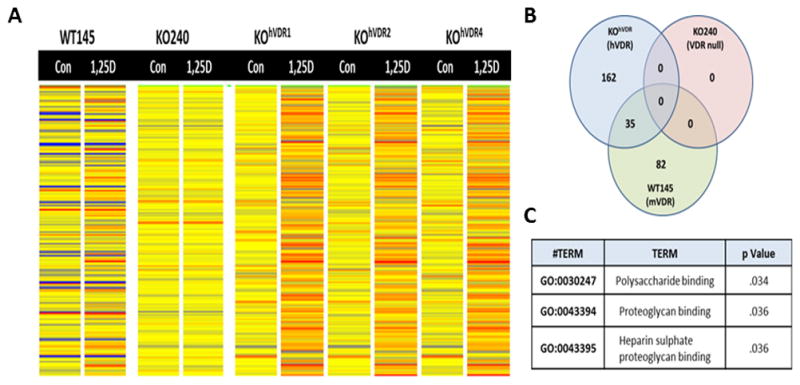
A, Heat maps of genomic profiles obtained from WT145 cells, KO240 cells and three independently derived KOhVDR clones (# 1, 2, 4) after 24h treatment with ethanol vehicle control (Con) or 100nM 1,25D. Cells were processed for Affymetrix microarray screening and analyzed by Genespring as described in Methods. B, Venn diagram of 1,25D regulated genes identified a cohort of 35 genes that were similarly regulated by 1,25D in both WT145 and KOhVDR cells. C, Pathway analysis of the 35 genes commonly regulated by 1,25D in WT145 and KOhVDR cells.
Through comparison of these distinct gene profiles, we identified a cohort of 35 genes that were similarly altered in WT145 cells and all three KOhVDR clones (Fig 4B). Gene ontology analysis (Fig 4C) indicated that these common transcripts are enriched in pathways related to polysaccharide and proteoglycan binding. The list of 1,25D responsive genes and the expression data as calculated from the microarray analysis for each cell line is shown in Table 1. Of these 35 commonly altered genes, the vast majority (31) were up-regulated, with only 4 genes down-regulated in WT145 cells and all three KOhVDR clones treated with 1,25D.
Table 1. Microarray data.
List of genes commonly regulated by 1,25D (100nM/24h) in all VDR positive cells (WT145 cells and three KOhVDR clones). None of these genes were significantly altered by 1,25D treatment in KO240 cells. Shown are fold change for each cell line (treated/control) calculated from the array dataset. Validation of a subset of these genes by real time PCR is shown in Figures 5 and 6.
| Fold Change (100nM 1,25D/Con) | |||||
|---|---|---|---|---|---|
| Gene | KO240 | WT145 | KOhVDR1 | KOhVDR2 | KOhVDR4 |
| A930018M24RK | 1.2 | 5.5 | 7.4 | 6.0 | 6.5 |
| Abcd2 | 1.0 | 2.2 | 2.1 | 1.8 | 2.2 |
| Apcdd1 | 1.1 | 3.8 | 2.1 | 2.6 | 1.9 |
| C3 | 1.2 | 2.1 | 2.7 | 2.0 | 1.7 |
| Cib2 | 1.0 | 4.6 | 9.9 | 8.0 | 9.5 |
| Ckb | 1.3 | 2.2 | 3.4 | 3.2 | 4.0 |
| Crabp2 | 1.0 | 4.0 | 2.6 | 2.6 | 4.3 |
| Cyp24a1 | 1.1 | 50.3 | 19.4 | 15.9 | 16.0 |
| Elovl7 | 1.0 | 4.3 | 4.5 | 3.4 | 4.4 |
| Enpp1 | 1.0 | 6.7 | 3.8 | 4.3 | 4.5 |
| Fam210b | 1.0 | 2.1 | 2.6 | 2.7 | 1.9 |
| Fbn1 | 1.0 | 2.2 | 2.0 | 2.0 | 2.1 |
| Fst | 1.0 | 2.6 | 2.2 | 1.9 | 2.1 |
| Gpc4 | 1.1 | 2.3 | 2.5 | 2.1 | 2.7 |
| Grk5 | 1.0 | 3.0 | 3.2 | 3.2 | 3.7 |
| Has2 | 0.9 | 0.5 | 0.3 | 0.4 | 0.3 |
| Hbegf | 1.0 | 0.5 | 0.4 | 0.5 | 0.4 |
| Igfbp4 | 1.0 | 2.9 | 3.1 | 3.2 | 6.4 |
| Islr | 0.8 | 4.3 | 5.2 | 2.2 | 3.5 |
| Kltl | 1.0 | 2.5 | 2.8 | 2.8 | 3.0 |
| Kl19 | 0.9 | 2.3 | 3.4 | 2.3 | 2.7 |
| Npr3 | 1.0 | 2.2 | 5.3 | 5.8 | 6.3 |
| Plau | 1.0 | 0.4 | 0.3 | 0.3 | 0.3 |
| Pnp1 | 1.0 | 2.1 | 2.3 | 2.0 | 2.4 |
| Postn | 1.0 | 0.2 | 0.3 | 0.3 | 0.4 |
| Prelp | 0.9 | 5.2 | 6.3 | 6.3 | 8.8 |
| Rassf2 | 1.0 | 3.4 | 5.0 | 3.3 | 4.1 |
| Relt | 1.0 | 2.4 | 2.5 | 2.5 | 2.6 |
| Rgl1 | 0.9 | 2.5 | 1.8 | 2.5 | 2.3 |
| Sepp1 | 1.0 | 3.4 | 5.7 | 4.0 | 6.1 |
| Serpinb9b | 0.9 | 2.0 | 2.3 | 3.0 | 3.0 |
| Slfn2 | 0.9 | 2.4 | 2.4 | 1.8 | 2.3 |
| Sulf2 | 1.0 | 2.9 | 3.3 | 2.5 | 2.6 |
| Tgm2 | 1.0 | 2.0 | 4.9 | 4.9 | 5.3 |
| Tspan2 | 1.0 | 2.3 | 4.9 | 3.6 | 4.1 |
Confirmation of selected VDR modulated genes by PCR
We chose to follow-up a subset of 8 genes identified in the microarray datasets by PCR analysis of independent samples. In addition to Cyp24a1 (a canonical VDR target gene [16]), three previously uncharacterized 1,25D up-regulated genes were selected: Cib2 (calcium and integrin binding protein2), Prelp (proline/arginine-rich end leucine-rich repeat protein) and Enpp1 (ectonucleotide pyrophosphate phosphodiesterase-1). All four genes were validated to be 1,25D responsive in a VDR dependent manner (Fig 5). Cyp24a1 was dramatically induced in all VDR positive cell lines, with the highest response in WT145 cells (this may indicate that the murine VDR is more effective at inducing the murine Cyp24a1 target gene than is hVDR). Cib2 was the second most highly induced gene, with significant increases of 50–100 fold above basal in WT145 cells and all KOhVDR clones in response to 1,25D. Under the same conditions the Prelp gene was up-regulated 10–20 fold and the Enpp1 gene was increased 5–15 fold in these cell lines. With the exception of Cyp24a1, there were no consistent differences in the magnitude of target gene induction by 1,25D in cells expressing mVDR or hVDR.
Figure 5. PCR analysis of select VDR up-regulated target genes in murine cell lines with differential VDR expression.

WT145 cells, KO240 cells and three clones of KO240 cells stably expressing hVDR (KOhVDR1, KOhVDR2, KOhVDR4) were treated with 100nM 1,25D or ethanol vehicle for 24h. RNA was isolated and used for real-time PCR evaluation of Cyp24, Cib2, Prelp and Enpp1. Data are normalized against 18S and expressed as fold change (1,25D treated vs control) for each cell line. Each bar represents mean ± standard error of 2–3 independent samples analyzed in duplicate. *p< .05, control vs 1,25D treated for each cell line.
All four genes that were found to be down-regulated upon 1,25D treatment of WT145 and KOhVDR cells in the microarray dataset were examined by PCR of independent samples (Fig 6). Plau (plasminogen activator, urokinase), Has2 (hyaluronan synthase 2) and Postn (periostin) were down-regulated by 1,25D from 50–75% in all VDR positive cell lines but unaffected in parental KO240 cells. Hbegf (heparin-binding EGF-like growth factor) was strongly down-regulated by 1,25D in KOhVDR cells but was not consistently altered in WT145 cells.
Figure 6. PCR analysis of select VDR down-regulated target genes in murine cell lines with differential VDR expression.
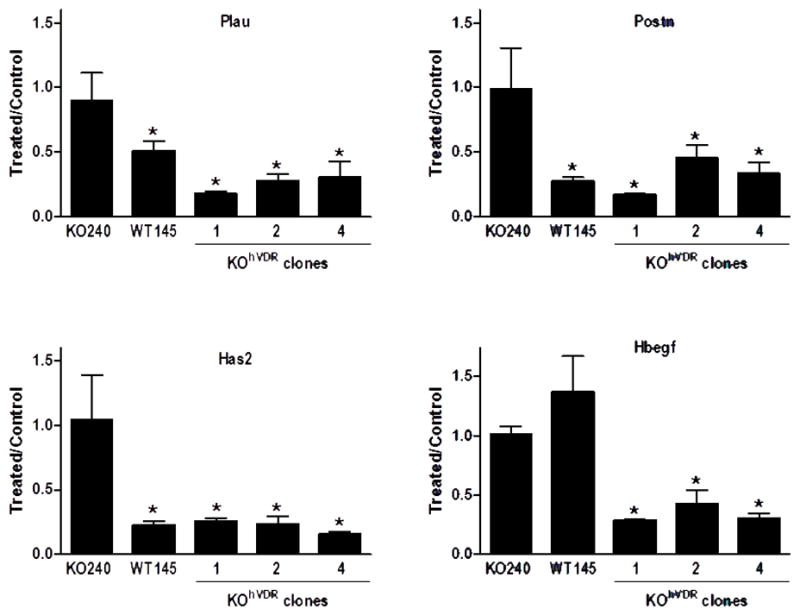
KO240 cells, WT145 cells, and three clones of KO240 cells stably expressing hVDR (KOhVDR1, KOhVDR2, KOhVDR4) were treated with 100nM 1,25D or ethanol vehicle for 24h. RNA was isolated and used for real-time PCR evaluation of Plau, Postn, Has2 and Hbegf. Data are normalized against 18S and expressed as 1,25D treated relative to control for each cell line. Each bar represents mean ± standard error of 2–3 independent samples analyzed in duplicate. *p< .05, control vs 1,25D treated for each cell line.
Discussion
In this report we have characterized a murine cellular model of triple negative breast cancer, an aggressive subtype of the disease which often exhibits the basal-like gene signature [2]. Similar to human triple negative breast tumors, the cell lines described here (which were derived from DMBA induced mammary tumors generated in C57Bl/6 mice) lack ER, PR and HER2 expression. This is not surprising since comparative genomic analysis of mouse tumor models and human breast cancers has demonstrated that most DMBA-induced mouse tumors show basal-like cell lineage features [17]. Since human triple negative breast cancers have been further sub-divided into six genomic clusters [18], further data mining will be needed to determine which subtype of triple negative human breast cancer these cells most resemble.
To characterize the actions of 1,25D, we isolated triple-negative cell lines from mammary tumors generated in wild-type and VDRKO mice and demonstrated that 1,25D mediates growth inhibition only in VDR expressing cells. We also engineered cells from VDRKO mice that stably expressed human VDR and showed that 1,25D exerted similar anti-proliferative and genomic effects in cells expressing murine or human receptors. Our data are thus consistent with reports that 1,25D exerts anti-proliferative effects in cell lines derived from human triple negative breast tumors such as Hs578T, SUM159PT and DCIS.com cells [4–6,19]. The expression of VDR in human triple negative breast cancers suggests that optimizing the activity of this receptor (either through maintenance of adequate vitamin D status or via administration of synthetic agonists) may be useful therapeutically. Our data also raise concerns about the high prevalence of vitamin D deficiency reported in patients with triple negative/basal-like breast cancers [11,12].
We have utilized our model system to comprehensively profile the genomic actions of 1,25D with the goal of identifying specific pathways targeted by VDR signaling that contribute to growth inhibition. Microarray screening was used to compare genomic changes induced by 24h treatment with 100nM 1,25D in cells expressing either murine VDR (WT145 cells) or human VDR (KOhVDR cells). Despite similar origins of the WT145 and KOhVDR cells and identical side-by-side experimental protocols for the microarray, genetic background (and possibly differences between human and mouse VDR) markedly affect the cellular responses to 1,25D. Therefore we chose to focus on the 35 transcripts (Table 1) that were commonly altered by 1,25D in all VDR expressing cell lines reasoning that these would be most relevant to the common endpoint of growth inhibition. Since arrays were conducted after 24h of treatment, the cohort of genes we identified likely include both primary and secondary targets of the 1,25D-VDR complex.
Of the 35 altered genes we identified in all VDR positive cell lines, 13 (Apcdd1, C3, Cib2, Ckb, Crabp2, Cyp24a1, Enpp1, Has2, Prelp, Rassf2, Serpinb9b, Sulf2 and Tgm2) overlap with an array dataset of 1,25D regulated genes associated with induction of cell-cycle arrest, senescence, and differentiation in progenitor cells isolated from the murine prostate gland [20]. Four of the 35 common genes (Ckb, Rassf2, Sulf2 and Hbegf) contain VDR consensus binding sites as identified by in silico screening for DR3 or ER6 sequences within −10 to +5 kb of transcription start sites in the mouse genome [21] suggesting that these might represent direct VDR targets. However, since it is clear that VDR binding is not limited to proximal promoter regions or known consensus binding sites [22–25], and that indirect mechanisms involving other transcription factors or regulation of micro RNAs contribute to 1,25D actions [26–29], future mechanistic studies will likely uncover novel mechanisms for 1,25D regulation of these genes.
In this study, the majority (31 out of 35) of genes that were altered upon 1,25D treatment were up-regulated. Using real time PCR, we confirmed that Cyp24a1, Cib2, Enpp1 and Prelp were up-regulated, whereas Has2, Plau and Postn were down-regulated, in mammary tumor cells expressing either murine or human VDR. Hbegf was strongly repressed in all three KOhVDR clones but, despite concordance in the array dataset, PCR follow-up showed inconsistent down-regulation of Hbegf in WT145 cells. Further studies will be needed to determine if there are differences between human and mouse VDR signaling that account for these discrepancies, or whether Hbegf expression is deregulated in WT145 cells. PCR analysis did confirm that 1,25D had no significant effects on the expression of these genes in VDR negative KO240 cells (Figs 5, 6) or KOEV cells (data not shown).
Although further studies will be necessary to determine the significance of these genes to the anti-tumor actions of 1,25D, several of them have known roles in mammary gland biology or cancer progression. The Cib2 gene codes for a 22kDa, EF hand containing calcium binding protein with sequence similarity to calcineurin B and calmodulin. We chose to follow-up Cib2 since it was the second most highly up-regulated gene in both WT and KOhVDR cells (after Cyp24a1) and it is highly expressed in normal human breast tissue (http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.129867). In addition, the function of Cib2 in regulation of calcium signaling may be particularly relevant since 1,25D mediated growth inhibition of MCF-7 breast cancer cells has been linked to changes in intracellular calcium [30]. The Enpp1 gene may also contribute to the anti-proliferative actions of 1,25D since it codes for a 105kDa type II transmembrane glycoprotein that inhibits the tyrosine kinase activity of the insulin receptor. Insulin signaling, which promotes breast tumorigenesis and likely contributes to the link between obesity and breast cancer risk, is attenuated by 1,25D in breast cancer cells [19,31]. In addition, high Enpp1 protein expression was associated with increased progression-free survival [32], suggesting its importance in human breast cancer. In contrast to these findings, however, a recent report demonstrated that forced expression of Enpp1 enhanced the ability of triple negative MDA-MB-231 cells to form bone metastasis, suggesting a complex relationship between Enpp1 and breast cancer progression [33]. The fourth protein confirmed to be up-regulated by 1,25D, Prelp, codes for a heparin binding extracellular matrix protein that has been shown to reduce growth and spread of breast cancer to bone in xenograft models. Collectively, our data demonstrating up-regulation of Cib2, Enpp1 and Prelp by 1,25D identifies additional candidate mediators and mechanisms by which VDR signaling may act to inhibit breast cancer.
With respect to the functional significance of the genes that were down regulated by 1,25D in our model system, all four are known to be elevated during cancer progression. Plau codes for urokinase-type plasminogen activator (uPA), a serine protease that has consistently been linked to tumor cell migration and the invasive phenotype [34]. Binding of uPA to its receptor uPAR triggers the cleavage of plasminogen to plasmin with subsequent activation of metalloproteinases, favoring tumor cell invasion and metastasis. The Postn gene codes for periostin, a secreted heparin binding protein normally expressed in bone where it mediates cell adhesion and mineralization, however, it too has consistently been identified as a marker of invasion and metastasis [35–39]. Periostin is increased in breast cancer compared with benign and normal breast tissue and positively correlates with clinical stage by immunohistochemistry [36,40]. Mechanistically, periostin interacts with multiple cell-surface receptors to promote cancer cell survival, epithelial-mesenchymal transition, invasion, and metastasis. Has2 codes for hyaluronan synthase, the enzyme that synthesizes the extracellular proteoglycan hyaluronic acid (HA). HA binds to the breast cancer stem cell receptor CD44 and fosters complexes between CD44 and growth factor receptors/ligands, promoting survival [41,42]. Forced expression of Has2 in triple negative breast cancer cells drives bone metastasis while inhibition or silencing of Has2 reduces the malignant phenotype [41,43–45]. We have confirmed that 1,25D reduces Has2 expression and HA secretion in WT145 and KOhVDR cells and that exogenous HA partially rescues 1,25D mediated growth inhibition (data not shown), thus functionally linking Has2 to the anti-tumor actions of 1,25D. These newly discovered links between 1,25D, Has2 and HA complement data showing down-regulation of CD44 mediated survival signaling by 1,25D in the human DCIS.com cell line, a distinct model of triple-negative breast cancer [4,5]. The fourth 1,25D-repressed gene we identified, Hbegf, was one of three genes found to promote metastasis of breast cancer to brain [46]. Interestingly, Hbegf has been shown to signal through the EGF receptor, HA and CD44 in the epidermis during wound healing, epidermal hyperplasia and hair follicle morphogenesis [47–50], processes which are known to be modulated by VDR signaling. Thus, the four 1,25D repressed genes identified in this model system all function in the extracellular matrix to suppress distinct stages in cancer progression: proliferation, epithelial-mesenchymal transition, invasion and metastasis.
In summary, our studies have identified a number of novel target genes that are modulated by the 1,25D/VDR complex in aggressive triple negative breast cancer cells. Challenges for the future will be to complete the validation of the 35 gene signature common to all VDR positive cell lines and to functionally characterize their role(s) in breast cancer invasion and metastasis. Such studies may identify new opportunities for targeting the vitamin D pathway in the treatment of human breast cancer.
Highlights.
A model of triple negative breast cancer with differential VDR expression was created
The growth regulatory and genomic effects of 1,25-dihydroxyvitamin D were profiled
1,25D failed to alter growth or gene expression in cells lacking VDR
1,25D regulated genes included Cib2, Enpp1, Prelp, Has2, Plau, Postn and Hbegf
Acknowledgments
We greatly appreciate the assistance of Meggan Keith, PhD, Gennifer Goode and Teresa Lloyd-Coronado with initial cell line characterization and James Keith, PhD with creation of the hVDR retroviral expression vector.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Idowu MO, Kmieciak M, Dumur C, et al. CD44(+)/CD24(−/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Human pathology. 2012;43(3):364–373. doi: 10.1016/j.humpath.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh J. Vitamin D and cancer: integration of cellular biology, molecular mechanisms and animal models. Scandinavian journal of clinical and laboratory investigation Supplementum. 2012;243:103–111. doi: 10.3109/00365513.2012.682870. [DOI] [PubMed] [Google Scholar]
- 4.So JY, Smolarek AK, Salerno DM, et al. Targeting CD44-STAT3 signaling by Gemini vitamin D analog leads to inhibition of invasion in basal-like breast cancer. PloS one. 2013;8(1):e54020. doi: 10.1371/journal.pone.0054020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.So JY, Lee HJ, Smolarek AK, et al. A novel Gemini vitamin D analog represses the expression of a stem cell marker CD44 in breast cancer. Molecular pharmacology. 2011;79(3):360–367. doi: 10.1124/mol.110.068403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanagan L, Packman K, Juba B, O’Neill S, Tenniswood M, Welsh J. Efficacy of Vitamin D compounds to modulate estrogen receptor negative breast cancer growth and invasion. The Journal of steroid biochemistry and molecular biology. 2003;84(2–3):181–192. doi: 10.1016/s0960-0760(03)00028-1. [DOI] [PubMed] [Google Scholar]
- 7.Mineva ND, Wang X, Yang S, et al. Inhibition of RelB by 1,25-dihydroxyvitamin D3 promotes sensitivity of breast cancer cells to radiation. Journal of cellular physiology. 2009;220(3):593–599. doi: 10.1002/jcp.21765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grotsky DA, Gonzalez-Suarez I, Novell A, et al. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. The Journal of cell biology. 2013;200(2):187–202. doi: 10.1083/jcb.201204053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopes N, Carvalho J, Duraes C, et al. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer research. 2012;32(1):249–257. [PubMed] [Google Scholar]
- 10.Ooi LL, Zhou H, Kalak R, et al. Vitamin D deficiency promotes human breast cancer growth in a murine model of bone metastasis. Cancer research. 2010;70(5):1835–1844. doi: 10.1158/0008-5472.CAN-09-3194. [DOI] [PubMed] [Google Scholar]
- 11.Peppone LJ, Rickles AS, Janelsins MC, Insalaco MR, Skinner KA. The association between breast cancer prognostic indicators and serum 25-OH vitamin D levels. Annals of surgical oncology. 2012;19(8):2590–2599. doi: 10.1245/s10434-012-2297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rainville C, Khan Y, Tisman G. Triple negative breast cancer patients presenting with low serum vitamin D levels: a case series. Cases journal. 2009;2:8390. doi: 10.4076/1757-1626-2-8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zinser GM, McEleney K, Welsh J. Characterization of mammary tumor cell lines from wild type and vitamin D3 receptor knockout mice. Mol Cell Endocrinol. 2003;200(1–2):67–80. doi: 10.1016/s0303-7207(02)00416-1. [DOI] [PubMed] [Google Scholar]
- 14.Keith ME, Laporta E, Welsh J. Stable expression of human VDR in murine VDR-null cells recapitulates vitamin D mediated anti-cancer signaling. Molecular carcinogenesis. 2013 doi: 10.1002/mc.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valrance ME, Brunet AH, Welsh J. Vitamin D receptor-dependent inhibition of mammary tumor growth by EB1089 and ultraviolet radiation in vivo. Endocrinology. 2007;148(10):4887–4894. doi: 10.1210/en.2007-0267. [DOI] [PubMed] [Google Scholar]
- 16.Matilainen JM, Malinen M, Turunen MM, Carlberg C, Vaisanen S. The number of vitamin D receptor binding sites defines the different vitamin D responsiveness of the CYP24 gene in malignant and normal mammary cells. The Journal of biological chemistry. 2010;285(31):24174–24183. doi: 10.1074/jbc.M110.124073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome biology. 2007;8(5):R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie SP, Pirianov G, Colston KW. Vitamin D analogues suppress IGF-I signalling and promote apoptosis in breast cancer cells. European journal of cancer (Oxford, England: 1990) 1999;35(12):1717–1723. doi: 10.1016/s0959-8049(99)00200-2. [DOI] [PubMed] [Google Scholar]
- 20.Maund SL, Barclay WW, Hover LD, et al. Interleukin-1alpha mediates the antiproliferative effects of 1,25-dihydroxyvitamin D3 in prostate progenitor/stem cells. Cancer research. 2011;71(15):5276–5286. doi: 10.1158/0008-5472.CAN-10-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang TT, Tavera-Mendoza LE, Laperriere D, et al. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Molecular endocrinology (Baltimore, Md) 2005;19(11):2685–2695. doi: 10.1210/me.2005-0106. [DOI] [PubMed] [Google Scholar]
- 22.Handel AE, Sandve GK, Disanto G, et al. Vitamin D receptor ChIP-seq in primary CD4+ cells: relationship to serum 25-hydroxyvitamin D levels and autoimmune disease. BMC medicine. 2013;11:163. doi: 10.1186/1741-7015-11-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satoh J, Tabunoki H. Molecular network of chromatin immunoprecipitation followed by deep sequencing-based vitamin D receptor target genes. Multiple sclerosis (Houndmills, Basingstoke, England) 2013;19(8):1035–1045. doi: 10.1177/1352458512471873. [DOI] [PubMed] [Google Scholar]
- 24.Heikkinen S, Vaisanen S, Pehkonen P, Seuter S, Benes V, Carlberg C. Nuclear hormone 1alpha,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic acids research. 2011;39(21):9181–9193. doi: 10.1093/nar/gkr654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. The Journal of biological chemistry. 2010;285(20):15599–15610. doi: 10.1074/jbc.M110.119958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang WL, Chatterjee N, Chittur SV, Welsh J, Tenniswood MP. Effects of 1alpha,25 dihydroxyvitamin D3 and testosterone on miRNA and mRNA expression in LNCaP cells. Molecular cancer. 2011;10:58. doi: 10.1186/1476-4598-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasiappan R, Shen Z, Tse AK, et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. The Journal of biological chemistry. 2012;287(49):41297–41309. doi: 10.1074/jbc.M112.407189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lisse TS, Adams JS, Hewison M. Vitamin D and MicroRNAs in Bone. Critical reviews in eukaryotic gene expression. 2013;23(3):195–214. doi: 10.1615/critreveukaryotgeneexpr.2013007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dimitrov V, Salehi-Tabar R, An BS, White JH. Non-classical mechanisms of transcriptional regulation by the vitamin D receptor: Insights into calcium homeostasis, immune system regulation and cancer chemoprevention. The Journal of steroid biochemistry and molecular biology. 2013 doi: 10.1016/j.jsbmb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Mathiasen IS, Sergeev IN, Bastholm L, Elling F, Norman AW, Jaattela M. Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. The Journal of biological chemistry. 2002;277(34):30738–30745. doi: 10.1074/jbc.M201558200. [DOI] [PubMed] [Google Scholar]
- 31.Gallagher EJ, Alikhani N, Tobin-Hess A, et al. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-1 receptor. Diabetes. 2013 doi: 10.2337/db13-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Umar A, Kang H, Timmermans AM, et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Molecular & cellular proteomics: MCP. 2009;8(6):1278–1294. doi: 10.1074/mcp.M800493-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau WM, Doucet M, Stadel R, Huang D, Weber KL, Kominsky SL. Enpp1: a potential facilitator of breast cancer bone metastasis. PloS one. 2013;8(7):e66752. doi: 10.1371/journal.pone.0066752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noh H, Hong S, Huang S. Role of urokinase receptor in tumor progression and development. Theranostics. 2013;3(7):487–495. doi: 10.7150/thno.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nature cell biology. 2013;15(7):807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D, Xu H, Ren Y, et al. Cancer stem cell-related gene periostin: a novel prognostic marker for breast cancer. PloS one. 2012;7(10):e46670. doi: 10.1371/journal.pone.0046670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Ouyang G. Periostin: a bridge between cancer stem cells and their metastatic niche. Cell stem cell. 2012;10(2):111–112. doi: 10.1016/j.stem.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Malanchi I, Santamaria-Martinez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481(7379):85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 39.Morra L, Moch H. Periostin expression and epithelial-mesenchymal transition in cancer: a review and an update. Virchows Archiv: an international journal of pathology. 2011;459(5):465–475. doi: 10.1007/s00428-011-1151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuetz CS, Bonin M, Clare SE, et al. Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer research. 2006;66(10):5278–5286. doi: 10.1158/0008-5472.CAN-05-4610. [DOI] [PubMed] [Google Scholar]
- 41.Okuda H, Kobayashi A, Xia B, et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer research. 2012;72(2):537–547. doi: 10.1158/0008-5472.CAN-11-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Udabage L, Brownlee GR, Nilsson SK, Brown TJ. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Experimental cell research. 2005;310(1):205–217. doi: 10.1016/j.yexcr.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 43.Urakawa H, Nishida Y, Wasa J, et al. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. International journal of cancer Journal international du cancer. 2012;130(2):454–466. doi: 10.1002/ijc.26014. [DOI] [PubMed] [Google Scholar]
- 44.Bernert B, Porsch H, Heldin P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1) The Journal of biological chemistry. 2011;286(49):42349–42359. doi: 10.1074/jbc.M111.278598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Li L, Brown TJ, Heldin P. Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. International journal of cancer Journal international du cancer. 2007;120(12):2557–2567. doi: 10.1002/ijc.22550. [DOI] [PubMed] [Google Scholar]
- 46.Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monslow J, Sato N, Mack JA, Maytin EV. Wounding-induced synthesis of hyaluronic acid in organotypic epidermal cultures requires the release of heparin-binding egf and activation of the EGFR. The Journal of investigative dermatology. 2009;129(8):2046–2058. doi: 10.1038/jid.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaya G, Tran C, Sorg O, et al. Hyaluronate fragments reverse skin atrophy by a CD44-dependent mechanism. PLoS medicine. 2006;3(12):e493. doi: 10.1371/journal.pmed.0030493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnes L, Tran C, Sorg O, et al. Synergistic effect of hyaluronate fragments in retinaldehyde-induced skin hyperplasia which is a Cd44-dependent phenomenon. PloS one. 2010;5(12):e14372. doi: 10.1371/journal.pone.0014372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson GD, Bazzi H, Fantauzzo KA, et al. KGF and EGF signalling block hair follicle induction and promote interfollicular epidermal fate in developing mouse skin. Development (Cambridge, England) 2009;136(13):2153–2164. doi: 10.1242/dev.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]