

Abstract
Consumption and over-consumption of alcoholic beverages are well-recognized contributors to a variety of pulmonary disorders, even in the absence of intoxication. The mechanisms by which alcohol (ethanol) may produce disease include oxidative stress and prolonged endoplasmic reticulum (ER) stress. Many aspects of these processes remain incompletely understood due to a lack of a suitable animal model. Chronic alcohol over-consumption reduces hepatic alcohol dehydrogenase (ADH), the principal canonical metabolic pathway of ethanol oxidation. We therefore modeled this situation using hepatic ADH-deficient deer mice fed 3.5% ethanol daily for 3 months. Blood ethanol concentration was 180 mg% in ethanol fed mice, compared to <0.2% in the controls. Acetaldehyde (oxidative metabolite of ethanol) was minimally, but significantly increased in ethanol-fed vs. pair-fed control mice. Total fatty acid ethyl esters (FAEEs, nonoxidative metabolites of ethanol) were 47.6 μg/g in the lungs of ethanol-fed mice as compared to 1.5 μg/g in pair-fed controls. Histological and immunohistological evaluation showed perivascular and peribronchiolar lymphocytic infiltration, and significant oxidative injury, in the lungs of ethanol-fed mice compared to pair-fed controls. Several fold increases for cytochrome P450 2E1, caspase 8 and caspase 3 found in the lungs of ethanol-fed mice as compared to pair-fed controls suggest role of oxidative stress in ethanol-induced lung injury. ER stress and unfolded protein response signaling were also significantly increased in the lungs of ethanol-fed mice. Surprisingly, no significant activation of inositol-requiring enzyme-1α and spliced XBP1 were observed indicating a lack of activation of corrective mechanisms to reinstate ER homeostasis. The data suggest that oxidative stress and prolonged ER stress, coupled with formation and accumulation of cytotoxic FAEEs may contribute to the pathogenesis of alcoholic lung disease.
Introduction
Alcohol and alcoholic beverage consumption and over-consumption has long been recognized as significant risk factor for pulmonary infections, acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), and pneumonia (Kershaw and Guidot, 2008; Zhang et al., 2008; Siu et al., 2010). Although some of the increase in these key morbidities can be attributed to intoxication and its consequences, these acute toxicities do not inform the potential long-term effects of lower amounts of ethanol on cellular homeostasis. Heavy alcohol consumption, or abuse, increases morbidity among patients with community-acquired pneumonia. Others have suggested that both the delivery of ethanol to airways in the lungs during chronic alcohol abuse, and formation and accumulation of metabolites of ethanol may contribute to other lung and airway disorders, including asthma (Hlastala., 1998; Hlastala and Anderson, 2007; Siu et al., 2010; Kaphalia and Calhoun, 2013). Accordingly, the metabolic and immunologic effects of moderate consumption of alcohol are of considerable interest, both scientifically and clinically.
The majority of ingested alcohol is metabolized oxidatively by alcohol dehydrogenase (ADH) in the liver (Lieber, 2004). However, it is well known, and of potential clinical importance, that chronic alcohol over-consumption inhibits hepatic ADH and induces hepatic microsomal mono-oxygenases, particularly cytochrome P450 2E1 isozyme (Cederbaum, 2010). These biochemical changes alter the host environment away from oxidative detoxification of ethanol, and increase the metabolites that induce oxidative stress and evoke markers of stress of the endoplasmic reticulum (ER). Both, ADH- and CYP2E1 catalyzed oxidation of ethanol are shown to be associated with generation of acetaldehyde (a reactive aldehyde that binds to cellular proteins and DNA) and/or reactive oxygen species (ROS) causing peroxidation of unsaturated lipids and oxidation of proteins and DNA. Such reactions alter systemic redox balance by reducing antioxidative capacity [such as glutathione (GSH) depletion] resulting in enhanced oxidative stress (Joshi and Guidot, 2007; Kershaw and Guidot, 2008; Cederbaum, 2010). However, it is not well understood whether generation of acetaldehyde and ROS, and depletion of GSH, or both are responsible for oxidative stress in lung disease related to ethanol over consumption.
A significant amount of ingested ethanol reaches the airways by the bronchial circulation, where it can be metabolized via oxidative and/or non-oxidative pathways as reviewed by us recently (Kaphalia and Calhoun, 2013). Because ethanol overconsumption impairs hepatic ADH, some individuals may have an increased time-weighted exposure to ethanol, as detailed in our previous studies (Bhopale et al., 2006; Kaphalia et al., 2010). However, chronic ethanol feeding also induces hepatic cytochrome P450 2E1, another potential metabolic pathway, with a different profile of metabolites (Lieber, 2004; Cederbaum, 2010), which may have overlapping or distinct toxicities. It is clear that inhibition of hepatic ADH facilitates substantial formation of cytotoxic fatty acid ethyl esters (FAEEs) via non-oxidative metabolism of ethanol catalyzed by FAEE synthase, which occurs in target organs including lungs of rodents fed ethanol (Manautou and Carlson, 1991; Manautou et al., 1992).
In humans, alcoholic lung disease develops after prolonged ethanol overconsumption (Kershaw and Guidot, 2008). A pathogenic role of ER stress in the pathogenesis of such lung diseases as COPD and idiopathic lung fibrosis has been suggested by several studies (Malhotra and Kaufman, 2007; Greene and McElvaney, 2010; Tanjore et al., 2012). However, the metabolic basis and pathogenic mechanisms of alcoholic lung disease are not well studied due to a lack of suitable animal model, and the perhaps insurmountable complexities of performing a properly designed human study of this question.
We hypothesize that chronic ethanol ingestion induces lung inflammation as a consequence of ethanol metabolism, oxidative stress, and endoplasmic reticulum stress, which are driven by the non-oxidative pathway generating FAEEs and CYP-2E1-catalyzed ethanol oxidation generating ROS rather than those by canonical metabolic pathways via ADH-catalyzed oxidation of ethanol. A conceptual framework for this work is shown in the Illustration (Fig. 1). To model situation, and unmask alternative metabolic pathways for ethanol, we adopted an animal model which is deficient in the canonical metabolic pathway, namely ADH. In the present study, we investigated ethanol metabolism and histological changes, oxidative stress, and ER stress responses in the lungs of hepatic ADH deficient deer mice after chronic ethanol feeding to understand the metabolic basis and mechanisms of alcoholic lung disease.
Fig. 1.

A conceptual schematic framework of ethanol metabolism and alcoholic lung injury.
Materials and Methods
Materials
Analytical grade fatty acid ethyl esters (FAEEs) standards, HPLC grade solvents, reagents, 200 proof ethanol and high purity grade acetaldehyde were obtained from Sigma-Aldrich (St. Louis, MO). Reagents for Western blot analysis were bought from BioRad. Antibodies against activating transcription factor (ATF)-6, cytochrome P450 (CYP) 2E1, 4-hydroxynonenal (4-HNE) and X binding protein (XBP) 1 were bought from IMGENEX, Millipore, Alpha Diagnostic Int. or Santa Cruz. All the other antibodies were from Cell Signaling (Boston, MA).
Animal experiments
All the animal experiments conducted in this study were in accordance to animal care protocols instituted by UTMB’s Institutional Animal Care and Use Committee. In our previous study, we found significant deaths in hepatic alcohol dehydrogenase (ADH)-deficient (ADH−) deer mice vs. hepatic ADH normal (ADH+) deer mice fed 4% ethanol daily for 2 months (Bhopale et al., 2006). Therefore, in these studies, we reduced ethanol exposure to 3.5% in the diet, as no significant deaths were found at this concentration of ethanol fed daily for 2 months (Kaphalia et al., 2010).
Hepatic ADH− deer mice are naturally deficient in hepatic class I ADH and more susceptible to ethanol toxicity as compared to ADH+ deer mice (Bhopale et al., 2006; Kaphalia et al., 2010). Multiple genes code for hepatic ADH in deer mice. However, hepatic class I ADH is the major isozyme responsible for ethanol metabolism to acetaldehyde in mammalian livers. The ADH− deer mouse, a natural variant ADHN/ADHN strain of Peromyscus maniculatis, is deficient in hepatic low-Km class I ADH, but has normal class II and class III ADH. The ADHF/ADHF strain or hepatic ADH-normal ADH+ have normal class I and class III ADH, but no significant class II ADH isozyme in the livers (Haseba et al., 1995). Therefore, a true wild type for ADH− deer mice, which are not knockout mice, is not available.
One year old male hepatic ADH negative phenotype (ADH−) deer mice (equivalent to human adult age) obtained from Peromyscus Stock Center, University of South Carolina, Columbia, SC, were housed in UTMB’s Animal Resource Center. After a mandatory 10 weeks quarantine as per institutional regulation, animals were divided into two groups. The experimental group was fed 3.5% ethanol daily for 3 months via Lieber–DeCarli liquid diet (Dyets Inc., Bethlehem, PA) (Kaphalia et al., 2010). Pair-fed ADH− deer mice were used as control in this study where liquid diet containing ethanol equivalent calories were substituted by maltose-dextrin. Comparison between ADH− deer mice fed ethanol vs. pair-fed controls (a true control group) should also account for any other metabolites, the levels of which theoretically might be altered by ADH deficiency.
At the end of 3 months of ethanol feeding, animals were euthanized by intraperitoneal administration of Nembutal™ 80mg/kg, blood withdrawn directly from the heart and transferred into a heparinized tube. Immediately, 50 μl of the blood was transferred in to vial containing isopropanol (internal standard) and sealed with Teflon cap for head space gas chromatography for measuring ethanol and acetaldehyde levels. Lungs were excised for gross examination and a portion was fixed in 10% buffered formalin for 24 hr followed by preservation in 70% ethanol. The fixed tissue was properly embedded in paraffin blocks for histology and immunohistochemistry. Remaining portions of the lungs were frozen at −80°c for biochemical and ER stress studies.
Analysis of blood alcohol and acetaldehyde
Head space analysis was employed to determine blood alcohol and acetaldehyde levels using a 6980N Gas Chromatograph (GC) equipped with a flame ionization detector (FID) (Agilent Technologies) and 5975 GC-mass spectrometer (GC-MS, Agilent Technologies) (Wu et al., 2006)). Briefly, the vial containing 50 μl blood was heated at 100°C for 5 min and 50 μl from the head space was injected into the column (DB-ALC1, 30 m × 0.53 mm ID, J & W Scientific, Folsom, CA) preheated at 35°C. The column temperature was i ncreased to 150°C at 15°C/min after 2 min. The temperature of injector port and detector was maintained at 250 and 300°C, respectively. The GC was operated at a 50:1 spit ratio with 1 ml/min flow rate using nitrogen as carrier gas. Concentration of ethanol and acetaldehyde were further confirmed by GC-MS operated by Chemstation using single ion monitoring (SIM) with a dwell time of 150 ms for each ion using helium as carrier gas flowing at 1 ml/min. The temperatures were maintained at 250°C for transfer line to MS, 230°C for the source and 150°C for the quadrupole.
Extraction and analysis of FAEEs
Lung homogenate was prepared in 0.1 M sodium phosphate (pH 7.4), total lipids extracted, and the ester fraction separated and analyzed for FAEEs using GC-FID as described previously (Kaphalia et al., 2010). Individual FAEEs were further confirmed by GC-MS set at the transfer line temperature maintained at 230°C, source at 166°C and the quadrupole at 130°C using helium as carrier gas flowing at 1ml/min. SIM was used to identify individual FAEE with a dwell time of 150 ms.
Morphological and immunohistochemical studies
Sections (5 μm thick) of tissues embedded in paraffin blocks were cut and stained with Hematoxylin & Eosin (H & E) and for immunohistochemistry using antibodies against 4-HNE to assess histological changes and oxidative stress, respectively (Kaphalia et al., 2010). The images were captured with an Olympus IX71 microscope equipped with digital camera DP71 and OLYMPUS MICRO DP71, VER 03.01 software.
Western blot analysis for ethanol metabolizing, ER stress and cell death proteins
Lung homogenates were prepared in phosphate buffer (pH7.4) containing phosphatase and protease inhibitors, and the protein was measured using Pierce BCA protein assay. An equal amount of lung homogenate was mixed with sample buffer and boiled for 10 min, and equal amount of protein (45 μg) separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis as described previously (Kaphalia et al., 2010). The protein from the gels was transferred on to a nitrocellulose membrane for Western blot analysis using antibodies against ethanol metabolizing enzymes (ADH and CYP2E1), glucose regulated protein 78, ER stress response proteins, and caspases (cell death proteins). The blots were incubated with horseradish peroxidase-conjugated secondary antibodies, and the target proteins were visualized using a commercially available ECL detection system. The protein band intensity was assessed by NIH ImageJ Software and normalized by β actin values.
Statistical analysis
The data sets obtained from ADH-deficient vs. pair-fed ADH-deficient control mice were not normally distributed, and the variance between the groups was unequal. Therefore, Levene test was used to assess the equality of variances, and the data sets were analyzed using nonparametric Mann-Whitney U-tests for statistical significance, when two groups were being compared. The Kruskal-Wallis test, a nonparametric alternative to analysis of variance (ANOVA) is being used for comparing more than two groups. For post hoc tests, Mann-Whitney U tests were used with multiple hypothesis corrections. Two sided test adjusted p values of ≤ 0.05 were considered significant. The results are expressed as mean ± SD (standard deviations of mean) of ≥ 4 animals per group.
Results
Blood alcohol and acetaldehyde levels
The average blood alcohol concentration in ethanol-fed mice was 180 mg% vs. 0.65 mg% in pair-fed controls (Fig. 2). Although very low levels of blood acetaldehyde were detected in the ethanol-fed mice (<0.2 mg %), the values were significantly increased as compared to pair-fed controls. Such levels of blood alcohol and minimally elevated acetaldehyde are expected as a consequence of the ADH-deficient background in ADH− deer mouse model.
Fig. 2.
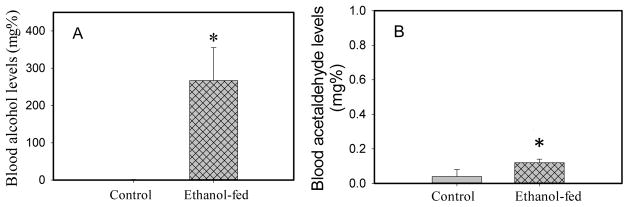
Blood ethanol (A) and acetaldehyde (B) concentrations in ADH− deer mice fed 3.5% ethanol daily for 3 months. Values are mean ± SD in each group (n = ≥ 5 in each). *- p value ≤ 0.05.
FAEE levels
Only palmitic (C16:0), stearic (C18:0), oleic (C18:1) and linoleic (C18:2) acid ethyl esters were detected in all the lung samples from the ethanol-fed mice analyzed in the present study; the average being 8.0, 12.1, 21.3 and 6.1 μg/g wet weight, respectively (Fig. 3). Total FAEEs were 47.6 μg/g wet weight in the lungs of the ethanol-fed mice. In contrast, only C16:0 and C18:0 FAEEs were detected in two control mice with average levels of 0.8 and 0.7 μg/g wet weights, respectively. Overall, total FAEEs in the lungs of the ethanol-fed mice were ~30 fold greater than those in pair-fed controls indicating a substantial formation and accumulation of FAEEs in the lungs of ADH-deficient mice fed ethanol.
Fig. 3.

Concentration of total FAEEs in the lungs of ethanol-fed and pair-fed control mice. Values are mean ± SD of five samples for ethanol-fed and four for control mice. *- p value ≤ 0.05.
Histological findings
Microscopic examination of H&E stained sections of the lungs of ethanol-fed mice showed multiple foci of perivascular and peribronchiolar lymphocytic infiltration. However, inflammation around the bronchioles was mild and not frequent (Fig. 4). All pair-fed control mice were free of pathologic changes; however, only one of 5 ethanol-fed mice was free of pathologic changes.
Fig. 4.

H&E stained lung tissues of control (A) ethanol-fed (B) mice (scanned microphotographs, 20X magnification). Arrows show perivascular (C) and bronchiolar (D) inflammatory infiltration (100X magnification).
Immunohistochemistry for oxidative stress
Oxidative stress as measured by an immmunohistochemical technique showed significantly greater 4-HNE immunostaining in the lungs of the ethanol-fed mice than the controls (Fig. 5). These results suggest increased oxidative stress in the lungs of the ethanol-fed mice as compared to pair-fed control mice.
Fig. 5.
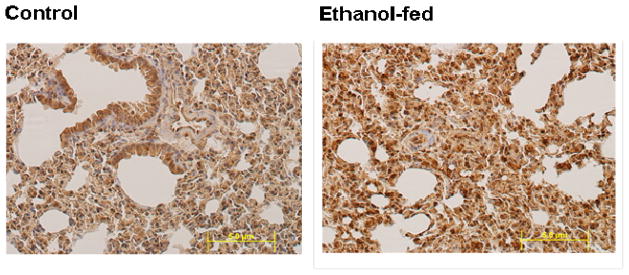
Oxidative stress evaluated by immunostaining using antibodies against 4-HNE in the lungs of ethanol-fed and control mice (20X magnification). Intense staining as seen in the lungs of ethanol-fed vs. pair-fed control mice indicates oxidative stress.
Enzymes involved in ethanol oxidative metabolism
Hepatic ADH and CYP2E1, important enzymes involved in oxidation of ethanol to acetaldehyde, were present in the lungs. Although ADH levels were not significantly different between the lungs of ethanol-fed mice and pair-fed controls (data not shown), CYP2E1 levels were significantly (~5 fold) greater in the lungs of ethanol-fed mice compared to pair-fed controls (Fig. 6).
Fig. 6.
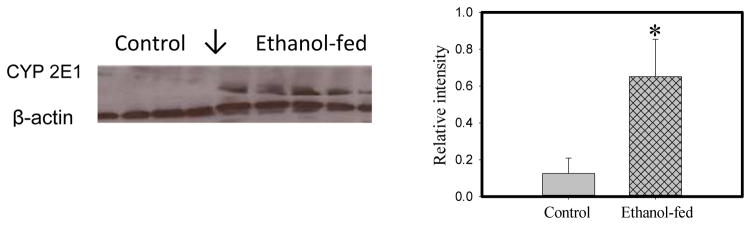
CYP2E1 levels in the lungs of ethanol-fed and pair-fed control mice; protein band and their relative intensities as determined by ImageJ Software are shown by Western blots and bar diagram, respectively. Values are mean ± SD of four samples in each group. *- p value ≤ 0.05.
ER stress and UPR signaling
Glucose regulated protein (GRP) 78, a prominent marker of ER stress, was significantly increased (~150%) in the lungs of ethanol-fed group compared to pair-fed controls (Fig. 7). Significant increases in ATF6 (~140%), phosphorylated eukaryotic initiation factor (eIF) 2α (~175%), protein disulfide isomerase (PDI, ~210%), and XBP1 (~200%) were observed in the lungs of ethanol-fed vs. pair-fed mice. Inositol requiring enzyme (IRE) 1α, one of three signaling pathways of UPR, was not significantly induced in our studies (data not shown). Therefore, increased XBP 1 appears to be a consequence of significantly increased activation of ATF6. This observation is consistent with the lack of significant activation of spliced (s) XBP1 observed in the lungs of ethanol-fed mice, and further is consistent with normal IRE 1α levels. Increased C/EBP homologous protein [CHOP, also known as growth arrest- and DNA damage-inducible gene 153 (GADD153; an ER-stress apoptotic cell death mediator causing mitochondrial damage, cytochrome C release and caspase 3 activation)] was also observed (~150%) in ethanol-fed mice compared to pair-fed controls.
Fig. 7.


ER stress and UPR signaling in the lungs of ethanol-fed and pair-fed control mice as determined by Western blot analysis using antibodies against GRP 78 (A), ATF6 (B), XBP1 (C), PDI (D), CHOP (E) and p-eIF 2 (F) and their relative intensities, respectively. No significant changes were observed for sXBP1 and IRE-1α (data not shown). Values are mean ± SD of four samples in each group. *- p value ≤ 0.05.
Caspases
A statistically significant induction of caspase 8 and caspase 3 (>5 fold) was evident in the lungs of ethanol-fed mice (Fig. 8), compared to the pair-fed controls. Pro-caspase 12 (involved in ER stress related cell death) was not significantly altered in our studies (data not shown) by ethanol feeding. These results are consistent with the suggestion that oxidative stress-induced cell death is activated directly through caspase 3 via caspase 8, but not through the mitochondria-dependent caspase 9 or ER stress activation.
Fig. 8.
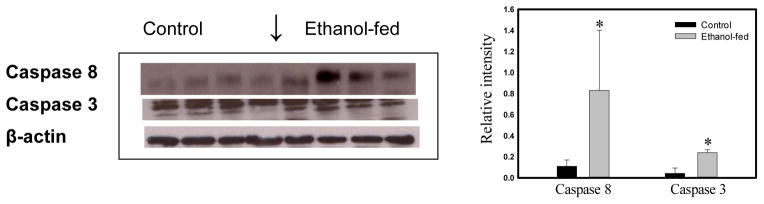
Western blot analysis of cell death proteins by using respective antibodies and their relative band intensities in the lungs of ethanol-fed and pair-fed control mice. No significant change was observed for caspase 12 and caspase 9 (data not shown). Values are mean ± SD of four samples in each group. *- p value ≤ 0.05.
Discussion
Being in direct contact with ambient air, the airways and lung parenchyma functionally is a potential target of environmental and infectious agents; this vulnerability is increased in the setting of impaired host defenses, or with abnormalities of innate or adaptive immunity in alcoholic patients. Chronic ethanol over-consumption has long been known to be a significant risk factor for lung infection (pneumonia), ARDS and COPD (Kershaw and Guidot, 2008; Zhang et al., 2008; Siu et al., 2010), even in the absence of acute intoxication. The effects of ethanol and its metabolites on redox homeostasis, antioxidants, and immune defense could lead to immunosuppression and predispose the lungs to infection (Joshi and Guidot, 2007; Kaphalia and Calhoun, 2013), and may be independent of intoxication or its consequences.
Binding of acetaldehyde to cellular macromolecules (proteins and nucleic acids) can result in oxidative stress (Cederbaum, 2010). We observed very low levels of acetaldehyde (a reactive aldehyde) in the blood of ethanol-fed mice (<0.2 mg%); accordingly, our findings regarding increased markers of oxidative stress in the lungs of ethanol-fed mice is remarkable, because it suggests that oxidative stress is likely induced by pathways other than the canonical ADH metabolic route. Our findings on significantly increased oxidative stress could be due to induction of hepatic CYP2E1 and related ethanol metabolites and ROS production (Cederbaum, 2010). However, the overall very low levels of blood acetaldehyde as found in our animal model reflect hepatic ADH-deficiency. Since hepatic ADH-deficiency attenuates acetaldehyde production, alternative ethanol metabolites such as FAEEs (non-oxidative metabolites of ethanol) may also contribute pathogenically to ethanol-induced oxidative stress due to their further metabolism. Although not investigated in this study, depletion of reduced glutathione as reported in the lungs after ethanol exposure might also contribute to the observed oxidative stress (Joshi and Guidot, 2007).
Formation and accumulation of lipophilic FAEEs as we observed is of considerable interest and has great biological significance in human diseases, because these esters are cytotoxic and proinflammatory, and in addition, uncouple oxidative phosphorylation (Wu et al., 2006; Wu et al., 2008; Lange and Sobel, 1983). Metabolism of ethanol in lungs appears to be predominantly through nonoxidative pathways, with the metabolic fate of the ethanol carbons incorporated into FAEEs (via FAEE synthase-catalyzed reaction) rather than oxidation by ADH or CYP2E1 (Manautou and Carlson, 1991; Manautou et al., 1992). The significant oxidative stress in our model appears to be a consequence of synthesis of FAEEs or their further metabolism, including production of ROS related to CYP2E1-catalyzed oxidation of ethanol. Ethanol can also be produced in the gut via bacterial fermentation of dextrose (Mezey et al., 1975; Ostrovsky, 1986). Therefore, the small quantities of FAEEs detected in the lungs of pair-fed controls in this study are likely to be related to endogenous ethanol production (Bhopale et al., 2006). The differential ethanol-induced toxic effects in the lungs may be a consequence of greater synthesis and accumulation of FAEEs in the lungs as compared to the livers and other organs of rodents fed ethanol (Manautou and Carlson, 1991).
The mean values of FAEEs found in the lungs of the ethanol-fed mice in this study are similar to those reported previously (Manautou and Carlson, 1991), suggesting external validity of our methodology. The specific mechanisms by which FAEEs induce lung damage require additional studies, although the mechanisms of ethanol-induced toxicity could be analogous to those previously reported in pancreatic acinar cell toxicity (Werner et al., 1997; Gukovskaya et al., 2002; Wu et al., 2008).
Studies by the Guidot laboratory demonstrated a significant reduction of GSH levels in the lungs of experimental animals fed ethanol, which were restored by dietary supplements of GSH precursors or GSH (Joshi and Guidot, 2007). The role of GSH is worth noting because a majority of GSH is synthesized in the liver. Therefore, it is also possible that the increased oxidative stress observed in our studies was a consequence of decreased lung levels of GSH due to chronic ethanol feeding. Moreover, the low and relatively elevated blood acetaldehyde levels observed in the ethanol-fed vs. pair-fed control mice suggest a significant role of CYP2E1 in catabolism of ethanol in our animal model of chronic ethanol feeding. However, generation of ROS during CYP2E1-catalyzed oxidation of ethanol could be an underlying mechanism of ethanol-induced oxidative stress (Fig. 1).
Various diseases including neurodegenerative disorders, metabolic disorders, inflammatory diseases, and malignancies involve ER stress (Zhang and & Kaufman, 2008; Hosoi and & Ozawa, 2010; Hotamisligil, 2010). Besides being a major site for the synthesis of lipids and cellular proteins, primary role of the ER is to fold proteins correctly into their tertiary and quaternary structures. However, aggregation of misfolded proteins in the ER-lumen results in an unfolded protein response (UPR, a conserved and evolutionary recovery mechanism), whereby cells respond to stress conditions that target the ER membrane. GRP 78 (BiP), a prosurvival ER chaperone that controls stress signaling in the ER membrane, is activated under ER-stress conditions as an protective mechanism that limits ER-stress-induced cell damage (Reddy et al., 2003). The UPR initially activates intracellular signaling pathways mediated by three resident sensors that are naturally complexed with BiP: i) protein kinase-like ER kinase (PERK); ii) IRE-1α (down regulates protein synthesis); and iii) ATF6 [essential for XBP1 mRNA expression and introduction of the ER chaperone GRP78 and PDI (Kaplowitz and Ji, 2006)]. Activation of the UPR signaling network alleviates ER stress and restores ER homeostasis by promoting cell survival and adaptation mechanisms. We demonstrated ER stress in the lungs of ethanol-fed mice increases GRP78 levels followed by activation of UPR, particularly ATF-6 and downstream PERK pathways [phosphorylated eukaryotic initiation factor (eIF) 2] in this study. However, the observed activation of XBP1 and CHOP appears to be secondary to activation of ATF-6 and in part via activation of downstream signaling of PERK. Surprisingly, no activation of spliced (s) XBP1 as found in this study is likely a consequence of the lack of activation of IRE 1α. We believe the absence of activation of IRE 1α in the lungs of ethanol fed mice is a novel finding.
Lack of activation of XBP1 splicing may also suggest lack of transcription of chaperones, genes for protein degradation and P58IPK. This ER stress-inducible protein P58IPK has been proposed to have independent function in protection from ER stress and a potential negative regulator of eIF 2 signaling, which attenuates UPR (VanHuizen et al., 2003; Reddy et al., 2003; Oyadomari Seiichi et al., 2006; Yoshida, 2007). A lack of activation of sXBP1 might also suggest failure of UPR signaling to a great extent and ER homeostasis. When such UPR adaptation to ER-stress is insufficient, particularly during chronic alcohol abuse and due to a prolonged UPR, the ER-stress response may unleash pathological consequences including necrosis and inflammation (Ji and Kaplowitz, 2006). The finding of scattered foci inflammation and lymphocytic infiltration in the lungs of our mouse model after chronic ethanol feeding is consistent with this hypothesis.
ER-stress-related cell death can be initiated by the activation of pro-caspase12 and CHOP (Kubisch and Logsdon, 2007). Since FAEEs are known to uncouple oxidative phosphorylation, their formation and accumulation in the ER membrane could hamper ATP production, resulting in delayed clearance of apoptotic bodies and even impaired cellular transport and trafficking. Instead, activation of caspase 8 and caspase 3 as found in the lungs of ethanol-fed mice suggest an oxidative stress response as an initiator of cell death mechanism. Therefore, a detailed study regarding various cell death mechanisms in this model, in conjunction with quantitation of formation and bioaccumulation of FAEEs, could help to disentangle the overlapping mechanisms that underlie ethanol-induced lung injury.
The mechanisms by which ethanol and its metabolites produce lung injury, inflammation, ER stress, and cell death remain enigmatic. Our study suggests that future work consider both oxidative and non-oxidative metabolism of ethanol as important contributors to the ultimate pathology. Oxidative mechanisms include increase oxidant species, and reduced anti-oxidant defenses (e.g. GSH depletion in the lungs), while non-oxidative metabolism of ethanol to FAEEs makes their cytotoxicity and inflammogenicity relevant to consider. It is likely that biologically conserved corrective processes such as UPR is not effective in the transcription of chaperones, P58IPK and other factors required for ER homeostasis mediated via splicing of XBP1 due to lack of activation of IRE 1α (VanHuizen et al., 2003). Therefore, the potential pathogenic role of FAEEs and abnormal UPR signaling could offer promising targets for the treatment of alcoholic lung diseases.
Highlights.
Alternative metabolism of ethanol was studied in hepatic-alcohol dehydrogenase (ADH)-deficient (ADH−) deer mice after chronic ethanol feeding, in order to model ADH-insufficiency in alcohol over consumption in humans.
Ethanol feeding produced pulmonary inflammation and markers of oxidative stress.
Fatty acid ethyl esters (FAEEs, nonoxidative metabolites of ethanol) were several folds higher in the lungs of ethanol-fed vs. pair-fed control mice; these FAEEs can be important mediators of the inflammatory response.
Endoplasmic reticulum (ER) stress and its downstream signaling consequences were significantly increased in the lungs of ethanol-fed mice irrespective of lung injury suggesting that prolonged ER stress may be an early marker of chronic ethanol feeding.
Acknowledgments
This study was conducted with the support of the Institute for Translational Sciences at the University of Texas Medical Branch, supported in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, National Institutes of Health. This work was also supported by grant AA19812 (BSK) from the National Institute of Alcohol Abuse and Alcoholism. Its contents are solely the responsibility of the authors, and do not necessarily represent the official views of the NIH or NIAAA. The authors also acknowledge the assistance of the UTMB’s Research Histopathology Core and Sealy Center for Environmental Health & Medicine, and Exposure Assessment & Biomarker Development Core supported through NIEHS Center grant P30ES06676.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GAS, Kaphalia BS. Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol. 2006;39:179–188. doi: 10.1016/j.alcohol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI. Role of CYP2E1 in Ethanol-Induced Oxidant Stress, Fatty Liver and Hepatotoxicity. Digestive Diseases. 2010;28:802–811. doi: 10.1159/000324289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CM, McElvaney NG. Z α-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J Gastrointest Pharmacol Ther. 2010;1:94–101. doi: 10.4292/wjgpt.v1.i5.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovskaya A, MM, IG, CNR, VNK, LDF, SJP Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology. 2002;122:106–118. doi: 10.1053/gast.2002.30302. [DOI] [PubMed] [Google Scholar]
- Haseba T, Yamamoto I, HK, YO, Watanabe T. Alcohol dehydrogenase (ADH) isozymes in the AdhN/AdhN strain of Peromyscus maniculatus (ADH-deermouse) and a possible role of class III ADH in alcohol metabolism. Biochemical genetics. 1995;33:349–362. doi: 10.1007/BF02399933. [DOI] [PubMed] [Google Scholar]
- Hlastala MP, Anderson AJC. The Impact of Breathing Pattern and Lung Size on the Alcohol Breath test. Annals of Biomedical Engineering. 2007;35:264–272. doi: 10.1007/s10439-006-9216-3. [DOI] [PubMed] [Google Scholar]
- Hlastala M. The alcohol breath test--a review. J Appl Physiol. 1998;84:401–408. doi: 10.1152/jappl.1998.84.2.401. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Ozawa K. Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 2010;118:19–29. doi: 10.1042/CS20080680. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. ER stress: Can the liver cope? Journal of Hepatology. 2006;45:321–333. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Guidot DM. The alcoholic lung: epidemiology, pathophysiology, and potential therapies. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2007;292:L813–L823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- Kaphalia B, Bhopale KK, Kondraganti S, Wu H, Boor PJ. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol Appl Pharmacol. 2010;246:154–162. doi: 10.1016/j.taap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphalia L, Calhoun WJ. Alcoholic Lung Injury: Metabolic, Biochemical and Immunological Aspects. Tox letter. 2013;222(2):171–9. doi: 10.1016/j.toxlet.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplowitz N, Ji C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. Journal of Gastroenterology and Hepatology. 2006;10:1440–1746. doi: 10.1111/j.1440-1746.2006.04581.x. [DOI] [PubMed] [Google Scholar]
- Kershaw CD, Guidot DM. Alcoholic lung disease. Alcohol Research & Health. 2008;31:66–75. [PMC free article] [PubMed] [Google Scholar]
- Kubisch C, Logsdon c. Secretagogues differentially activate endoplasmic reticulum stress responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):1804–1812. doi: 10.1152/ajpgi.00078.2007. [DOI] [PubMed] [Google Scholar]
- Lange L, Sobel B. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. J Clin Invest. 1983;72:724–731. doi: 10.1172/JCI111022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxidants & Redox Signaling. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Manautou J, Buss N, Carlson G. Oxidative and non-oxidative metabolism of ethanol by the rabbit lung. Toxicol letter. 1992;62:93–99. doi: 10.1016/0378-4274(92)90082-u. [DOI] [PubMed] [Google Scholar]
- Manautou JE, Carlson GP. Ethanol-induced fatty acid ethyl ester formation in vivo and in vitro in rat lung. Toxicology. 1991;70:303–312. doi: 10.1016/0300-483x(91)90005-l. [DOI] [PubMed] [Google Scholar]
- Mezey E, Imbembo AL, Potter JJ, Rent KC, LLR, Hot PR. Endogenous ethanol production and hepatic disease following jejunoileal bypass for morbid obesity. Am J Clin Nutr. 1975;28:1277–1283. doi: 10.1093/ajcn/28.11.1277. [DOI] [PubMed] [Google Scholar]
- Ostrovsky YM. Endogenous ethanoldits metabolic, behavioral and biomedical significance. Alcohol. 1986;3:239–247. doi: 10.1016/0741-8329(86)90032-7. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Yun C, Fisher EA, et al. Cotranslocational Degradation Protects the Stressed Endoplasmic Reticulum from Protein Overload. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- Reddy R, CM, Baumeister P, Austin RCRJK, ASL Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. Biol Chem. 2003;278:20915–20912. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- Siu ST, Udaltsova Natalia, Iribarren Carlos, Klatsky aAL, Stanton T, Siu M. Alcohol and Lung Airways Function. The Permanente Journal. 2010;14:11–18. doi: 10.7812/tpp/09-089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2012;302:L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanHuizen R, JLMMG P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem. 2003;278:15558–15564. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- Werner J, Laposata M, FernandezDelCastillo C, Saghir M, Iozzo RV, Lewandrowski KB, Warshaw AL. Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology. 1997;113:286–294. doi: 10.1016/s0016-5085(97)70106-9. [DOI] [PubMed] [Google Scholar]
- Wu H, Cai P, Clemens DL, Jerrells TR, Ansari GAS, Kaphalia BS. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: Role of nonoxidative metabolism. Toxicology and Applied Pharmacology. 2006;216:238–247. doi: 10.1016/j.taap.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Wu H, Bhopale KK, Aansari GAS, Kaphalia BS. Ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells: role of fatty acid ethyl esters. Alcohol Alcohol. 2008;43:1–8. doi: 10.1093/alcalc/agm044. [DOI] [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS Journal. 2007;10.111:1742–4658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Bagby GJ, Happel KI, Raasch CE, Nelson S. Alcohol abuse, immunosuppression, and pulmonary infection. Curr Drug Abuse Rev. 2008;1:56–67. doi: 10.2174/1874473710801010056. [DOI] [PubMed] [Google Scholar]