

Abstract
Purpose
A randomized, phase III, placebo-controlled, partially blinded clinical trial (REGAL [Recentin in Glioblastoma Alone and With Lomustine]) was conducted to determine the efficacy of cediranib, an oral pan–vascular endothelial growth factor (VEGF) receptor tyrosine kinase inhibitor, either as monotherapy or in combination with lomustine versus lomustine in patients with recurrent glioblastoma.
Patients and Methods
Patients (N = 325) with recurrent glioblastoma who previously received radiation and temozolomide were randomly assigned 2:2:1 to receive (1) cediranib (30 mg) monotherapy; (2) cediranib (20 mg) plus lomustine (110 mg/m2); (3) lomustine (110 mg/m2) plus a placebo. The primary end point was progression-free survival based on blinded, independent radiographic assessment of postcontrast T1-weighted and noncontrast T2-weighted magnetic resonance imaging (MRI) brain scans.
Results
The primary end point of progression-free survival (PFS) was not significantly different for either cediranib alone (hazard ratio [HR] = 1.05; 95% CI, 0.74 to 1.50; two-sided P = .90) or cediranib in combination with lomustine (HR = 0.76; 95% CI, 0.53 to 1.08; two-sided P = .16) versus lomustine based on independent or local review of postcontrast T1-weighted MRI.
Conclusion
This study did not meet its primary end point of PFS prolongation with cediranib either as monotherapy or in combination with lomustine versus lomustine in patients with recurrent glioblastoma, although cediranib showed evidence of clinical activity on some secondary end points including time to deterioration in neurologic status and corticosteroid-sparing effects.
INTRODUCTION
Despite recent advances in radiation and neurosurgical techniques and the approval of new medical therapies, glioblastoma, the most common primary malignant brain tumor in adults, causes significant neurologic morbidity and is associated with survival of less than 2 years.1,2 Microvascular proliferation, a pathologic hallmark of glioblastoma, is due to the high expression of proangiogenic cytokines, particularly of vascular endothelial growth factor (VEGF) and signaling via its endothelial tyrosine kinase receptor VEGFR2.3–6 Levels of VEGF and its receptor are correlated with the histologic grade of gliomas, with the highest levels present in glioblastoma.7,8Thus glioblastoma has emerged as an attractive tumor in which to conduct clinical trials of novel anti-VEGF agents, such as monoclonal antibodies and tyrosine kinase inhibitors.9Bevacizumab, an anti-VEGF monoclonal antibody, was approved as monotherapy for recurrent glioblastoma by the US Food and Drug Administration in 2009 based on the radiographic response rates in two phase II trials.10–12
Cediranib is an orally available pan-VEGFR tyrosine kinase inhibitor with a half-life of 22 hours compatible with once daily dosing. Cediranib has a sub-nanomolar half maximal inhibitory concentration for VEGF receptors with additional activity against c-Kit and lower potency against platelet-derived growth factor β.13 In a prior phase II study of cediranib (45 mg/d) for patients with recurrent glioblastoma, eight (27%) of 30 subjects achieved a partial radiographic response based on consensus-based response criteria.14,15 Subsequently, this international, phase III, randomized, partially blinded, placebo-controlled study was conducted to investigate the efficacy of cediranib, as monotherapy and in combination with the synthetic alkylating agent lomustine (1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea), versus lomustine alone in patients with recurrent glioblastoma.
PATIENTS AND METHODS
Patients
Patients with recurrent glioblastoma were the target population for this study. Inclusion criteria included age ≥ 18 years, pathologic diagnosis of glioblastoma, prior treatment with a temozolomide-containing chemotherapy regimen, prior treatment with radiation, Karnofsky performance status (KPS) ≥ 70, Mini-Mental Status Examination score ≥ 15, and life expectancy ≥ 12 weeks. Exclusion criteria included any prior anti-VEGF therapy or cranial radiation within 3 months before study entry. All patients were required to sign an informed consent form approved by the institutional review board of the enrolling institution.
The study was performed in accordance with the Declaration of Helsinki, the International Conference on Harmonization/Good Clinical Practice, applicable (national) regulatory requirements, and the AstraZeneca policy on bioethics.
Trial Design and Treatments
The study was a phase III, comparative, randomized, parallel group, multicenter trial with patients randomly assigned (stratified by age and resection status) in a 2:2:1 ratio to receive cediranib (30 mg) monotherapy, cediranib (20 mg) in combination with lomustine (110 mg/m2), or lomustine (110 mg/m2) in combination with a placebo. Cediranib doses (20 mg, 30 mg) were given once daily as oral tablets, lomustine doses (110 mg/m2) were given as oral capsules once every 6 weeks, and cediranib-matched placebo was given once daily as oral tablets. The primary end point of the study was to compare the efficacy of cediranib, as monotherapy and in combination with lomustine, with lomustine alone by independent, treatment arm–blinded, radiographic assessment of progression-free survival (PFS). Secondary objectives included assessments of the proportion of patients alive and progression-free at 6 months (APF6) after randomization, overall survival (OS), radiographic response rate, corticosteroid-sparing effects, time to deterioration in neurologic status (TTNS), safety, and tolerability. Exploratory end points included the time to deterioration in KPS and changes from baseline in the levels of soluble markers of angiogenesis and tumor growth.
Evaluations
The primary PFS end points were determined in patients on a stable dose of corticosteroids, based on postcontrast, T1-weighted brain magnetic resonance imaging (MRI) scans assessed by independent, centralized, treatment arm-blinded, radiographic review. The central PFS assessment took corticosteroid dose and timing of scans into account. The PFS end points were also assessed by local review of postcontrast, T1-weighted MRI brain scans by the treating physicians as well as independent, centralized, treatment-arm blinded, radiographic review of T2/fluid-attenuated inversion recovery MRI brain scans on the basis of published criteria.16,17 For patients with measurable disease at study entry (defined as contrast-enhancing tumor with a shortest diameter of ≥ 10 mm on two axial slices), progression was defined as either (1) ≥ 25% increase in the sum of the products of the largest perpendicular diameters for all lesions compared with the nadir scan (provided that the shortest diameter was ≥ 15 mm at the time of progression); (2) detection of a new lesion, with shortest diameter of ≥ 10 mm, outside the original tumor volume; (3) death from any cause. For postsurgical patients without measurable disease at study entry, progression was defined as either (1) detection of a new lesion; (2) a significant increase in lesion size (to a shortest diameter of ≥ 15 mm) for a small, baseline enhancing lesion at study entry; (3) death from any cause. The neurologic status and KPS were based on physical examination by the treating investigator before and during treatment. Corticosteroid doses were recorded for all subjects. Plasma samples obtained at baseline and during the study were used to determine levels of the soluble biomarkers VEGF, sVEGFR2, and basic fibroblast growth factor (bFGF) and to calculate changes from baseline levels. Adverse events (AEs) were recorded and graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
Statistical Analysis
The primary end point of the study was PFS based on centralized, radiographic review. Secondary end points were OS, response rate in patients with measurable disease, APF6, time to deterioration in neurologic status, mean change in average daily dosage of corticosteroids, and average number of progression- and corticosteroid-free days. Assuming a true hazard ratio (HR) of 0.55, with 300 patients randomly assigned 2:2:1 to cediranib, cediranib plus lomustine, and lomustine plus placebo, the trial had more than 80% power to demonstrate a statistically significant difference in PFS at a two-sided, 2.5% level. This would correspond to an 80% increase in PFS if median time to progression was 3 months in the lomustine-alone arm. The analysis for PFS was planned after 230 progression events had occurred. PFS was analyzed using the log-rank test, stratified by resection status and age, and the HR of progression and associated 95% CIs were estimated from a Cox proportional hazards model using stratification levels as covariates. Each cediranib-containing arm was independently compared with the lomustine plus placebo arm. Statistical significance would be declared if both comparisons were significant at the two-sided 5% level or if either comparison was statistically significant at the two-sided 2.77% level. OS was planned to be analyzed twice—at the time of PFS analysis and again when 270 deaths had occurred. To maintain an overall type I error rate of 5%, the significance level was prespecified at 0.00785 (two-sided) at interim for both comparisons and at 0.00417 (two-sided) at interim for either comparison. All efficacy assessments were performed on the intent-to-treat randomly assigned population.
RESULTS
Patients
Between October 2008 and September 2009, 325 patients were randomly assigned in a ratio of 2:2:1 to receive cediranib (30 mg) monotherapy (n = 131), cediranib (20 mg) plus lomustine (110 mg/m2) n = 129), or lomustine (110 mg/m2) plus placebo (n = 65). A CONSORT diagram is included in Figure 1. The demographic and baseline clinical characteristics were generally well-balanced across the three arms, although fewer patients in the lomustine plus placebo arm had KPS less than 80 and used corticosteroids at baseline (Table 1).
Fig 1.
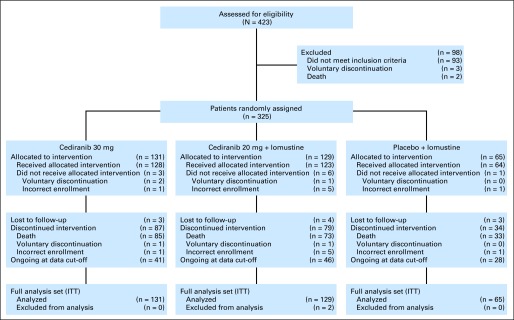
CONSORT diagram. ITT, intent to treat.
Table 1.
Baseline Characteristics
| Characteristic | Cediranib (n = 131) |
Cediranib + Lomustine (n = 129) |
Lomustine + Placebo (n = 65) |
|||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Median age, years | 54.0 | 54.0 | 54.0 | |||
| Resection for recurrent disease | ||||||
| Yes | 50 | 38.2 | 49 | 38.0 | 24 | 36.9 |
| No | 81 | 61.8 | 80 | 62.0 | 41 | 63.1 |
| Karnofsky performance status | ||||||
| < 70 | 0 | 1 | 0.8 | 1 | 1.6 | |
| 70-80 | 65 | 50.0 | 62 | 48.0 | 23 | 36.2 |
| 90-100 | 65 | 50.0 | 66 | 51.2 | 40 | 62.5 |
| Corticosteroid use at baseline | ||||||
| Yes | 64 | 48.9 | 71 | 55.0 | 26 | 40.0 |
| No | 67 | 51.1 | 58 | 45.0 | 39 | 60.0 |
| Time from last radiotherapy to randomization, months | ||||||
| 0-3 | 2 | 1.5 | 4 | 3.1 | 0 | |
| 3-6 | 32 | 24.4 | 29 | 22.5 | 16 | 24.6 |
| 6-12 | 52 | 39.7 | 42 | 32.6 | 27 | 41.5 |
| > 12 | 45 | 34.4 | 54 | 41.9 | 22 | 33.8 |
Efficacy Outcomes
The primary end point of PFS was not significantly different for either cediranib alone or in combination with lomustine versus lomustine alone. For the cediranib (30 mg) monotherapy arm, the HR versus lomustine was 1.05 (95% CI, 0.74 to 1.50; two-sided P = .90). For the cediranib (20 mg) plus lomustine arm, the HR versus lomustine was 0.76 (95% CI, 0.53 to 1.08; two-sided P = .16). The median PFS for the cediranib (30 mg) monotherapy arm, the cediranib (20 mg) plus lomustine arm, and the lomustine arm was 92 days (first quartile = 80 days, third quartile = 128 days), 125 days (first quartile = 83 days, third quartile = 201 days), and 82 days (first quartile = 42 days, third quartile = 168 days), respectively (Fig 2). Compared with the lomustine arm, neither cediranib-containing arm demonstrated a significant improvement in PFS based on either central review of postcontrast T1-weighted or noncontrast T2/ fluid-attenuated inversion recovery brain MRI scans or local review of postcontrast T1-weighted brain MRI scans. At the time of the primary PFS analysis, approximately 73% (197 of 270) of the death events prespecified for final analysis had occurred. At this time, there was no significant difference in the secondary end point of OS for either cediranib-containing arm versus the lomustine arm. For the cediranib (30 mg) monotherapy arm, the HR versus lomustine was 1.43 (95% CI, 0.96 to 2.13; two-sided P = .10). For the cediranib (20 mg) plus lomustine arm, the HR versus lomustine was 1.15 (95% CI, 0.77 to 1.72; two-sided P = .50). The median OS for the cediranib (30 mg) monotherapy arm, the cediranib (20 mg) plus lomustine arm, and the lomustine arm was 8.0 months, 9.4 months, and 9.8 months, respectively (Fig 3). On the basis of predictive power calculations, there was less than a 0.01% chance of concluding a positive outcome at the final OS analysis; thus it was considered of no value to continue to final analysis. A total of 136 patients received postprogression anticancer therapy, with the majority receiving bevacizumab either as monotherapy or in combination. Postprogression bevacizumab utilization was similar in the cediranib (20 mg) plus lomustine (43 of 84, 51%) and the lomustine plus placebo (20 of 39, 51%) arms but was less frequent in the cediranib (30 mg) open-label, monotherapy arm (22 of 81, 27%).
Fig 2.

Progression-free survival (PFS; based on central review postcontrast T1). No. at risk denotes the number of patients who were event free at the beginning of the period. Ced, cediranib; HR, hazard ratio; lom, lomustine; Pla, placebo.
Fig 3.

Overall survival (OS). No. at risk denotes the number of patients who were event free at the beginning of the period. Ced, cediranib; HR, hazard ratio; lom, lomustine; Pla, placebo.
There were no differences in the radiographic response rates between the two cediranib-containing arms (Table 2). The median reduction in contrast-enhanced tumor area was −36% and −28% in the cediranib (30 mg) monotherapy and cediranib (20 mg) plus lomustine arms, respectively, compared with a +14% increase in the lomustine plus placebo arm (Fig 4). The APF6 proportions in the cediranib (30 mg) monotherapy, cediranib (20 mg) plus lomustine, and lomustine arms were 16%, 35%, and 25%, respectively. Corticosteroid usage was reduced in the cediranib-containing arms compared with the lomustine arm; the mean change in corticosteroid use from baseline to progression was −26% for the cediranib (30 mg) monotherapy arm (P = .01 v lomustine), −23% for the cediranib (20 mg) plus lomustine arm (P = .01 v lomustine), and +5% for lomustine. The TTNS HRs for the cediranib (30 mg) monotherapy arm and the cediranib (20 mg) plus lomustine arm versus lomustine were 0.82 (95% CI, 0.55 to 1.22; P = .57) and 0.63 (95% CI, 0.42 to 0.95; P = .01), respectively. The time to deterioration of KPS HRs for the cediranib (30 mg) monotherapy arm and the cediranib (20 mg) plus lomustine arm versus lomustine were 1.03 (95% CI, 0.65 to 1.62) and 0.73 (95% CI, 0.45 to 1.17), respectively.
Table 2.
Best Overall Response (central review postcontrast T1)
| Best Overall Response | Cediranib (n = 118) |
Cediranib + Lomustine (n = 122) |
Lomustine + Placebo (n = 56) |
|||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Responders | 18 | 15.3 | 21 | 17.2 | 5 | 8.9 |
| CR | 1 | 0.8 | 2 | 1.6 | 0 | |
| PR | 17 | 14.4 | 19 | 15.6 | 5 | 8.9 |
| Stable disease | 76 | 64.4 | 67 | 54.9 | 23 | 41.1 |
| Unconfirmed CR | 0 | 1 | 0.8 | 0 | ||
| Unconfirmed PR | 32 | 27.1 | 9 | 7.4 | 2 | 3.6 |
| PD | 10 | 8.5 | 19 | 15.6 | 41.1 | |
| Nonevaluable | 14 | 11.9 | 14 | 11.5 | 5 | 8.9 |
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response.
Fig 4.
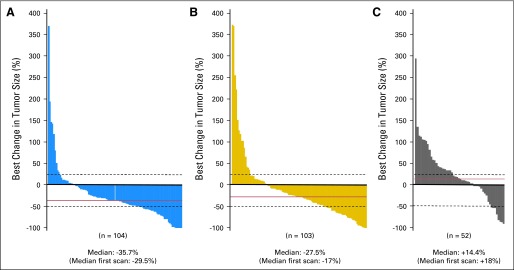
Best change from baseline in contrast-enhancing area. (A) Cediranib; (B) cediranib plus lomustine; (C) placebo plus lomustine.
Soluble Biomarkers
Median baseline levels of VEGF were 74 pg/mL (first quartile = 47, third quartile = 149), 76 pg/mL (first quartile = 46, third quartile = 165), and 66 pg/mL (first quartile = 34, third quartile = 93) in the cediranib 30 mg, cediranib 20 mg plus lomustine, and lomustine arms, respectively. Median VEGF levels in the cediranib 30-mg arm increased to 89%, 108%, and 106% above baseline at days 42, 84, and 126, respectively. By contrast, in the lomustine arm, median VEGF levels at day 42 were similar to those at baseline and decreased by 21% at day 84. An increase in VEGF levels was also recorded at each time point in the cediranib 20 mg plus lomustine arm (day 42, 46%; day 84, 41%; day 126, 61%), although these median increases were lower than those for the cediranib 30-mg arm. Subgroup analyses showed that baseline VEGF levels did not have a significant effect on either PFS or OS outcome. Median baseline levels of sVEGFR2 were similar in the three arms (range, 10,937 to 11,790 pg/mL). At day 42, decreases in median sVEGFR2 levels from baseline of 27% and 22% were recorded for the cediranib 30 mg and cediranib 20 mg plus lomustine arms, respectively; these decreases were maintained in both arms at days 84 and 126. Median sVEGFR2 levels in the lomustine arm remained similar to baseline levels throughout the trial. Median baseline levels of bFGF were 5 pg/mL, 4 pg/mL, and 3 pg/mL in the cediranib 30 mg, cediranib 20 mg plus lomustine, and lomustine arms, respectively. No consistent changes from baseline in bFGF levels were seen in any arm. At day 84, increases of 100% and 80% were observed in the lomustine arm and the cediranib 20 mg plus lomustine arm, respectively, but these increases were not maintained at day 126.
Dose-Intensity
The dose-intensity of cediranib was well maintained, and there was no evidence of early discontinuation before progression in either cediranib-containing arm. The mean dose-intensity was consistent (∼80%) during the first 3 months of therapy. There were more cediranib dose interruptions in the cediranib (20 mg) plus lomustine arm versus the cediranib (30 mg) monotherapy arm. However, there were more cediranib dose reductions in the cediranib (30 mg) monotherapy arm versus the cediranib (20 mg) plus lomustine arm, and the majority of reductions occurred after 3 months. The incidence of dose interruptions and reductions of lomustine was higher in combination with cediranib compared with placebo (although the number of cycles of lomustine received was generally greater in the combination arm, reflecting later progressions compared with the placebo arm). At least one dose interruption of lomustine occurred in 27% of patients in the placebo arm versus 40% in combination with cediranib. The dose of lomustine was reduced in 50% of subjects on the lomustine plus placebo arm versus 70% of subjects on the lomustine plus cediranib arm.
Safety and Tolerability
There were no unexpected AEs observed in any arm during the course of the study. There was no increased risk of intracranial bleeding observed in the cediranib-containing arms. The most common AE was diarrhea, experienced by 71%, 71%, and 19% of patients in the cediranib monotherapy, cediranib plus lomustine, and lomustine arms, respectively. AEs ≥ grade 3 were experienced by a higher proportion of patients in the cediranib plus lomustine arm (80%) compared with the cediranib monotherapy and lomustine arms (61% in each arm; Table 3). Grade 3 or 4 thrombocytopenia and neutropenia were observed in 38% and 20%, respectively, of patients in the cediranib combination arm compared with 2% and 1%, respectively, in the cediranib monotherapy arm and 22% and 3%, respectively, in the lomustine arm.
Table 3.
Grade 3 and 4 AEs (safety population)
| AE | Cediranib (n = 128) |
Cediranib + Lomustine (n = 123) |
Placebo + Lomustine (n = 64) |
|||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Any AE, ≥ grade 3 | 78 | 60.9 | 98 | 79.7 | 39 | 60.9 |
| Thrombocytopenia | 2 | 1.6 | 47 | 38.3 | 14 | 22 |
| Neutropenia | 1 | 0.8 | 25 | 20.3 | 2 | 3.1 |
| Fatigue | 21 | 16.4 | 19 | 15.4 | 6 | 9.4 |
| Leukopenia | 1 | 0.8 | 14 | 11.4 | 3 | 4.7 |
| Platelet count decreased | 0 | 10 | 8.1 | 1 | 1.6 | |
| Anemia | 0 | 8 | 6.5 | 0 | ||
| Hypertension | 18 | 14.1 | 8 | 6.5 | 0 | |
| ALT increased | 3 | 2.3 | 7 | 5.7 | 0 | |
| GGT increased | 1 | 0.8 | 7 | 5.7 | 2 | 3.1 |
| WBC decreased | 0 | 7 | 5.7 | 3 | 4.7 | |
| Diarrhea | 8 | 6.3 | 7 | 5.7 | 1 | 1.6 |
| Pulmonary embolism | 4 | 3.1 | 6 | 4.9 | 4 | 6.3 |
| Lymphopenia | 3 | 2.3 | 5 | 4.1 | 5 | 7.8 |
| Convulsion | 11 | 8.6 | 2 | 1.6 | 3 | 4.7 |
| Intracranial hemorrhage | 0 | 1 | 0.8 | 2 | 3.1 | |
Abbreviations: AE, adverse event; GGT, γ-glutamyltransferase.
Serious AEs were observed in 43%, 37%, and 41% of patients in the cediranib monotherapy, cediranib plus lomustine, and lomustine arms, respectively. AEs leading to permanent discontinuation of cediranib or placebo were observed in 15%, 18%, and 16% of patients in the cediranib monotherapy, cediranib plus lomustine, and lomustine arms, respectively.
DISCUSSION
The REGAL trial failed to meet its primary end point of demonstrating a benefit in PFS for either cediranib-containing arm versus lomustine in patients with recurrent glioblastoma. Cediranib demonstrated some clinical activity in certain secondary end points, including significant reduction of corticosteroid usage in both cediranib-containing arms, and there was a favorable delay in time to neurologic deterioration in the cediranib plus lomustine arm versus lomustine. There was no difference in OS between the three arms. Although OS may have been confounded by the use of postprogression bevacizumab therapy, bevacizumab use was similar between the cediranib 20 mg plus lomustine and placebo plus lomustine arms. Expected, dose-dependent increases in VEGF and decreases in sVEGFR2 were observed in the cediranib-containing arms but not in the lomustine arm. Baseline levels of these soluble biomarkers were not predictive of outcome in this trial. Dose-intensity of cediranib was well maintained in the cediranib-containing arms, although there were more lomustine reductions in the cediranib plus lomustine arm versus the lomustine arm despite the lower dose of cediranib (20 mg) in the lomustine plus cediranib arm. The AE profile observed in REGAL was consistent with prior cediranib trials, and no unexpected AEs were observed. However, patients in the cediranib plus lomustine arm experienced a higher incidence of toxicities, especially hematologic adverse effects, compared with the cediranib monotherapy and lomustine arms. This suggests that the combination of cediranib and lomustine exacerbates lomustine-induced hematologic toxicities. The increased incidence of toxicities did not result in an increase of cediranib discontinuations, suggesting that the toxicities were manageable. Importantly, no increased risk of intracranial bleeding was noted in patients receiving cediranib.
The combination dose of cediranib (20 mg) was reduced from 30 mg after results from the National Cancer Institute of Canada BR24 study of metastatic lung carcinoma suggested a higher fatality rate in the cediranib-containing arms at this dose level.18 Whether selection of a lower dose of cediranib in the REGAL study may have contributed to the negative results of the study is impossible to determine.
Lomustine was approved in 1977 for use in patients with brain tumors.19,20 In a phase I study it was observed that lomustine 130 mg/m2 in combination with cediranib (20 mg) was insufficiently well tolerated in the population of patients with recurrent glioblastoma who had received prior temozolomide therapy. The dose-limiting toxicities were hematologic. It was determined that the optimal combination dose of lomustine for REGAL was 110 mg/m2, a dose also used in other studies in this patient population.21 The PFS and APF6 observed in the lomustine arm in the REGAL study were higher than the metrics typically used for comparative purposes from historical series of therapies for this patient population but consistent with that observed for lomustine in another control arm in a phase III study.22,23 However, it is noteworthy that patients were enrolled in REGAL at the time of first recurrence, whereas these historical series included patients treated at the first, second, or later recurrences. Nevertheless, lomustine monotherapy demonstrates efficacy in patients with recurrent glioblastoma previously treated with temozolomide and should be considered as a control arm in future comparative trials.
Although cediranib monotherapy or in combination with lomustine did not improve PFS compared with lomustine alone in REGAL, preclinical models suggest synergistic activity of anti-VEGF therapy in combination with radiation owing to the ability of these agents to normalize tumor vessels. On the basis of these observations, cediranib in combination with chemoradiotherapy is being studied in phase II trials in the newly diagnosed glioblastoma population24 (NCT00662506; NCT01062425).
Supplementary Material
Footnotes
Supported by AstraZeneca.
Presented in part at the 2010 Congress of the European Society of Medical Oncology, October 8-12, 2010, Milan, Italy, and the Society of Neuro-Oncology 15th Annual Scientific Meeting, November 18-21, 2010, Montreal, Quebec, Canada.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00777153.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Juliane M. Jürgensmeier, AstraZeneca (C); Rakesh K. Jain, XTuit Pharmaceuticals (C), Hambrecht & Quist Healthcare Investors (C), Hambrecht & Quist Life Sciences Investors (C); A. Gregory Sorensen, Siemens Medical Solutions (C); John Xu, AstraZeneca (C); Qi Liu, AstraZeneca (C), AstraZeneca (C) Consultant or Advisory Role: Tracy T. Batchelor, Roche (C), Millennium (C), Amgen (C), Novartis (C); Mario Campone, Novartis (C), Servier (C); Antje Wick, MSD (C), Roche (C); John DeGroot, Genentech (C), VBL Therapeutics (C), Deciphera Pharmaceuticals (C); Lawrence Cher, Roche (C), MSD (C); Rakesh K. Jain, Enlight Biosciences (C), SynDevRX (C), Noxxon Pharmaceuticals (C), Zyngenia (C); A. Gregory Sorensen, AstraZeneca (C); Martin van den Bent, AstraZeneca (C), Roche (C), MSD (C) Stock Ownership: Rakesh K. Jain, XTuit Pharmaceuticals, SynDevRx; A. Gregory Sorensen, Siemens AG; John Xu, AstraZeneca; Qi Liu, AstraZeneca, AstraZeneca Honoraria: Tracy T. Batchelor, Merck, UpToDate; Paul Mulholland, AstraZeneca; Mario Campone, Novartis; Antje Wick, MSD, Roche; Tom Mikkelsen, Roche, Merck; Surasak Phuphanich, Sigma Tau Pharmaceuticals; A. Gregory Sorensen, Astra Zeneca; Martin van den Bent, Roche, MSD Research Funding: Tracy T. Batchelor, AstraZeneca, Millennium, Pfizer; Paul Mulholland, AstraZeneca; Mario Campone, Novartis; Antje Wick, Boehringer Ingelheim, Apogenix, Eli Lilly; Tom Mikkelsen, Roche, Merck, Celgene; Surasak Phuphanich, AstraZeneca, Genetech Oncology, ImmunoCellular Therapeutics, Diffusion Pharmaceutical, Med-Immune, Boehringer Ingelheim, Myrexis; John DeGroot, Astra Zeneca, Sanofi-Aventis, EMD-Serono; Lawrence Cher, Astra Zeneca, Roche; Juliane M. Jürgensmeier, AstraZeneca; Rakesh K. Jain, MedImmune, Roche, Dyax; A. Gregory Sorensen, Astra Zeneca; Martin van den Bent, Roche Expert Testimony: None Patents: None Other Remuneration: John DeGroot, Merck; A. Gregory Sorensen, Astra Zeneca
AUTHOR CONTRIBUTIONS
Conception and design: Tracy T. Batchelor, Warren Mason, Rao Gattamaneni, Lawrence Cher, Juliane M. Jürgensmeier, A. Gregory Sorensen, John Xu, Qi Liu
Financial support: A. Gregory Sorensen
Administrative support: Paul Mulholland, A. Gregory Sorensen
Provision of study materials or patients: Tracy T. Batchelor, Paul Mulholland, Bart Neyns, L. Burt Nabors, Mario Campone, Antje Wick, Warren Mason, Tom Mikkelsen, Lynn S. Ashby, John DeGroot, Rao Gattamaneni, Lawrence Cher, Mark Rosenthal, Franz Payer, Martin van den Bent
Collection and assembly of data: Tracy T. Batchelor, Paul Mulholland, L. Burt Nabors, Antje Wick, Tom Mikkelsen, Surasak Phuphanich, John DeGroot, Mark Rosenthal, Franz Payer, A. Gregory Sorensen, John Xu, Qi Liu, Martin van den Bent
Data analysis and interpretation: Tracy T. Batchelor, Bart Neyns, L. Burt Nabors, Mario Campone, Warren Mason, Surasak Phuphanich, Lynn S. Ashby, Rao Gattamaneni, Lawrence Cher, Mark Rosenthal, Juliane M. Jürgensmeier, Rakesh K. Jain, A. Gregory Sorensen, John Xu, Qi Liu, Martin van den Bent
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK, di Tomaso E, Duda DG, et al. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 4.Holash J, Maisonpierre PC, Compton D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 5.Shweiki D, Itin A, Soffer D, et al. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 6.Millauer B, Shawver LK, Plate KH, et al. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 7.Samoto K, Ikezaki K, Ono M, et al. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res. 1995;55:1189–1193. [PubMed] [Google Scholar]
- 8.Schmidt NO, Westphal M, Hagel C, et al. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84:10–18. doi: 10.1002/(sici)1097-0215(19990219)84:1<10::aid-ijc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 9.Jain RK, Duda DG, Clark JW, et al. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 11.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen MH, Shen YL, Keegan P, et al. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 13.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 14.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald DR, Cascino TL, Schold SC, Jr, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 18.Goss GD, Arnold A, Shepherd FA, et al. Randomized, double-blind trial of carboplatin and paclitaxel with either daily oral cediranib or placebo in advanced non-small cell lung cancer: NCIC Clinical Trials Group BR24 Study. J Clin Oncol. 2010;28:49–55. doi: 10.1200/JCO.2009.22.9427. [DOI] [PubMed] [Google Scholar]
- 19.Levin VA, Wilson CB. Nitrosourea chemotherapy for primary malignant gliomas. Cancer Treat Rep. 1976;60:719–724. [PubMed] [Google Scholar]
- 20.Shapiro WR, Young DF. Chemotherapy of malignant glioma with CCNU alone and CCNU combined with vincristine sulfate and procarbazine hydrochloride. Trans Am Neurol Assoc. 1976;101:217–220. [PubMed] [Google Scholar]
- 21.Batchelor TT, de Bono JS, Wen PY, et al. A phase I, open-label, multicenter study to assess the safety and tolerability of cediranib (Recentin) in combination with lomustine chemotherapy for patients with recurrent glioblastoma. Neuro-oncol. 2009;11:905. (abstr 0126) [Google Scholar]
- 22.Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17:2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 23.Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168–1174. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.