

Abstract
The mature outflow tract (OFT) is, in basic terms, a short conduit. It is a simple, although vital, connection situated between contracting muscular heart chambers and a vast embryonic vascular network. Unfortunately, it is also a focal point underlying many multifactorial congenital heart defects (CHDs). Through the use of various animal models combined with human genetic investigations, we are beginning to comprehend the molecular and cellular framework that controls OFT morphogenesis. Clear roles of neural crest cells (NCC) and second heart field (SHF) derivatives have been established during OFT formation and remodeling. The challenge now is to determine how the SHF and cardiac NCC interact, the complex reciprocal signaling that appears to be occurring at various stages of OFT morphogenesis, and finally how endocardial progenitors and primary heart field (PHF) communicate with both these colonizing extra-cardiac lineages. Although we are beginning to understand that this dance of progenitor populations is wonderfully intricate, the underlying pathogenesis and the spatiotemporal cell lineage interactions remain to be fully elucidated. What is now clear is that OFT alignment and septation are independent processes, invested via separate SHF and cardiac neural crest (CNC) lineages. This review will focus on our current understanding of the respective contributions of the SHF and CNC lineage during OFT development and pathogenesis.
INTRODUCTION
Congenital heart defects (CHD)1 are the most common developmental anomaly and account for nearly one third of all major congenital anomalies.2,3 Within the scope of CHDs, cardiac outflow tract (OFT) defects and abnormalities of the great arteries that exit the heart are estimated to cause approximately 30% of these CHDs,4 causing significant morbidity and mortality in both children and adults. A variety of OFT defects result from disturbance of the morphogenetic patterning of the anterior pole of the heart, which is essential for the establishment of separate systemic and pulmonary circulations in higher vertebrates.5–7 Because proper formation of the OFT requires intricate coordination in space and time between multiple extra-cardiac progenitor populations, it is particularly susceptible to congenital anomalies. Specifically, this review will focus on the major OFT structural vascular defects including: persistent truncus arteriosus (PTA), in which there is only a single undivided OFT exiting the heart; double outlet right ventricle (DORV), in which the divided aorta and pulmonary trunks both exit the right ventricle (RV) only; transposition of the great arteries (TGA), in which the aorta and pulmonary trunks are fully septated but the aorta arises from the RV and the pulmonary trunk arises from the left ventricle (LV); and overriding aorta (OA), which is an OFT alignment defect usually associated with Tetratolgy of Fallot (TOF) [a spectrum that also includes pulmonary stenosis (PS), right ventricular hypertrophy and concomitant interventricular septal defects], summarized in Table 1 and illustrated in Figure 1. Other serious patient cardiovascular defects of the great arteries and veins exiting the heart not discussed here include5–7: coarctation/interruption of the aorta (CoARC); vascular ring; aberrant subclavian artery; PS; patent ductus arteriosus (PDA); persistent left superior vena cava; and anomalous pulmonary venous connection (APVC). In addition, there is a wide spectrum of OFT valvular defects including, aortic and pulmonary valve sub/supravalvular stenosis and bicuspid aortic valve that can often occur (reviewed expertly elsewhere8–10); and several of these CHDs can occur in various combinations. However, the underlying pathologies of these defects (i.e., CoARC, PS, APVC) remain unclear due to a lack of available experimental models that faithfully replicate great artery, vein, and valvular defects. In contrast, transgenic mouse mutants have been more instructive as to developmental etiologies of structural OFT defects, although many developmental processes remain opaque. It is not all doom-and-gloom, as remarkable surgical and medical advances now allow more than 90% of the children who survive surgery to live into adulthood, establishing the field of “adult CHDs” which can have ongoing consequences over decades.11 Moreover, advances in developmental biology have resulted in identification of numerous transcriptional factors, signaling molecules and structural genes that are now known to be critical for normal cardiac morphogenesis.11–14 This improved understanding of the molecular aspects of heart development has assisted the discovery of CHD-causing genes, identified using positional cloning and/or candidate gene screening approaches.13,15 Unfortunately the etiology of most CHDs, including the predominant structural OFT defects, remains unknown because of their multifactorial nature12,16 and the fact that several extra-cardiac progenitor lineages are required to combine seamlessly in order to form the mammalian OFT conduit. A greater understanding of possible genetic causes would provide insight into the pathogenesis perhaps even the prevention of CHDs as well as allow for better assessment of disease risk and prenatal diagnosis.
TABLE 1.
Major Cardiac Defects of the Mammalian Outflow Tract
| Outflow Tract Defect | Clinical Definition |
|---|---|
| Persistent truncus arteriosus (PTA) | An abnormal single artery resulting from failure of OFT septation exits the heart, resulting in mixing of systemic and pulmonary circulations |
| Double outlet right ventricle (DORV) | Both great arteries originate from the RV, frequently accompanied by a ventricular septal defect and sometimes pulmonary stenosis |
| Transposition of the great arteries (TGA) | The positioning of the septated aortic and pulmonary arteries is reversed, thus the aorta abnormally arises from the RV and the pulmonary trunk arises from the LV |
| Overriding aorta (OA) | The OFT and aortic valve is positioned directly over a ventral septal defect and receives blood from both the right and left ventricles |
FIGURE 1.
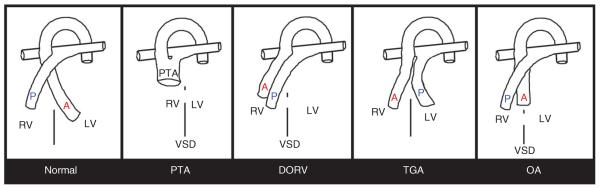
Schematics of the four major structural outflow tract (OFT) defects. Simplified illustration of a normal heart and OFT, including separate aorta (A) and pulmonary trunks (P), that exit the fully divided left (LV) and right ventricles (RV) respectively. Persistent truncus arteriosus (PTA), in which there is only a single undivided OFT exiting the RV of the heart. The blood exits the LV via an interventricular septal defect (VSD), indicated via broken line between RV and LV. Double outlet right ventricle (DORV), in which the divided aorta (A) and pulmonary trunks (P) both exit the RV only and the interventricular septum fails to close resulting in a VSD. Transposition of the great arteries (TGA), in which the aorta (A) and pulmonary trunks (P) are fully septated, but the aorta arises from the RV and the pulmonary trunk arises from the LV. Overriding aorta (OA) where the aorta is positioned directly over a VSD, instead of over the LV.
The most severe structural OFT defect in patients is thought to be PTA, as it is often associated with an unfavorable prognosis and complete surgical repair is not always possible.17 PTA is principally caused by defective septation of the initial single embryonic OFT into separate aortic and pulmonary trunks.5,7 On the basis of transgenic mutant mice and chick embryo surgical ablation studies, abnormal development of cardiac neural crest (CNC) cells, an ectoderm-derived extra-cardiac cell lineage, contributes significantly to the pathology of OFT defects including PTA.18–24 During early embryogenesis, CNC cells arise from the dorsal neural tube and migrate ventrally as mesenchymal cells to populate the OFT, where they merge with other cell lineages to form the aorticopulmonary septum. This septum divides the initially single embryonic OFT (also called truncus arteriosus) into separate aortic and pulmonary arteries, resulting in the establishment of separate systemic and pulmonary circulations.18 Transgenic mis-expression or deletion of genes in mice has revealed critical pathways, signaling cascades, and cell lineages essential for CNC morphogenesis and, thus, OFT formation (listed in Table 2). Beyond the extensive role of CNC cells, other lineages are emerging as important contributors to OFT formation. More recent studies have shown that reciprocal signaling between the CNC and cells derived from the pharyngeal mesoderm (or second heart field, SHF), which subsequently give rise to OFT/subpulmonary myocardium, is also required for OFT and heart development25,26 and the surgical removal of SHF progenitors27 results in other OFT defects (listed in Table 2). Additionally, endothelial cells that line the interior of the OFT vessels and a sub-region of the endothelial cells within the OFT (termed `endocardial cells' in the heart) undergo epithelial-mesenchymal transformation (EMT), to give rise to the endocardial cushions, that together with the invading CNC, form the OFT septum. The OFT endocardial cushions then undergo remodeling to form the semi-lunar valves that help maintain unidirectional blood flow.28–30 A range of transgenic mouse mutant studies have revealed that abnormal EMT (resulting in either deficient OFT cushion formation31 or excessive cushion formation32,33) can also give rise to structural OFT defects, particularly alignment defects (listed in Table 2). Although CNC cells are known to contribute a significant proportion of the endocardial cushion mass (likely important in early prevention of retrograde blood flow), CNC are not typically considered to have a direct role in semi-lunar valve formation.34 However, exciting recent data suggest that the CNC lineage is in close apposition to SHF precursors within the ventral pharynx during migration to the OFT endocardial cushions, and together they may provide instructive signals to orchestrate apoptosis and changes in extracellular matrix (ECM) production during valve remodeling.35 Hence, during OFT morphogenesis and creation of the distinct pulmonary and systemic conduits, multiple cell types arise from very distinct progenitor populations to undergo remodeling and accommodate ever-changing requirements of the developing embryo. Thus, CNC must interact with SHF myocardium and endocardial cushion mesenchyme for normal heart development to occur. As the role of the endocardial cushions/valves in CHDs has been expertly reviewed elsewhere,7,36–39 this review will therefore focus principally on the CNC lineage and also the SHF during OFT defect pathogenesis.
TABLE 2.
Examples of Transgenic Mouse Models Exhibiting Some of The Principal OFT Defects Observed in Patients
| Primary Defect | Primary Lineage Affected | Additional Cardiac Defects | Gene | Modification |
|---|---|---|---|---|
| PTA | NC migration | Arch artery | Acvr1 40 | Wnt1-Cre targeted deletion |
| NC differentiation in OFT | Disorganized CNC-derived mesenchyme and thinned ventricular myocardium | Cdh2 41 | Wnt1-Cre targeted deletion | |
| NC proliferation | PA hypoplasia | Ctnnb1 2 | Wnt1-Cre targeted deletion | |
| NC ablation | PS, AA | Diphtheria toxin 24 | Genetic ablation of Wnt1-Cre positive cells | |
| Endothelium | Disorganized yolk sac vascularization, AA | Nrp1 43 | Global deletion | |
| OFT myocardium proliferation | Right-sided heart hypoplasia | Pitx2 44 | Mef2c-Cre or Isl1Cre targeted deletion | |
| NC differentiation | OA, valve defects, AA | Ptk2 45 | Wnt1-Cre targeted deletion | |
| Post-migratory NC-derived mesenchyme | SMC differentiation, AA | Rac1 46 | Wnt1-Cre targeted deletion | |
| NC deficiency | Arch artery | Rara, Rarb, Rarg 47 | Global deletion | |
| NC migration | DORV, AA | Sema3c 20 | Global deletion | |
| NC apoptosis/colonization | EC and PA hypoplasia, AA | Smad4 48,49 | Wnt1-Cre targeted deletion | |
| NC differentiation | PA hypoplasia | Smad7 50 | Inducible Smad7 overexpression in Wnt1-Cre expressing cells | |
| Endocardium | DORV, AA | Sox4 51 | Global deletion | |
| SHF morphogenesis | Arch artery | Tbx1 52–54 | Global deletion | |
| SHF morphogenesis | Right-sided heart hypoplasia | Tbx20 55 | Hypomorphic deletion | |
| NC morphogenesis | Arch artery | Tgfbr1 56 | Wnt1-Cre targeted deletion | |
| NC morphogenesis | Arch artery | Tgfbr2 57 | Wnt1-Cre targeted deletion | |
| Wnt5a signaling to NC from pharyngeal mesoderm | DORV, TGA, AA | Wnt5a 58 | Global deletion | |
| DORV | Endocardial EMT | EC hypoplasia, vascular hemorrhage | Birc5 59 | Tie1-Cre targeted deletion |
| Cardiomyocytes | OA | Bmp2 and Bmp4 60 | Global double heterozygous | |
| Mesenchymal BMP signaling | AV cushion, thickened valve leaflets | Bmpr2 61 | Tie2-Cre or Wnt1-Cre targeted deletion | |
| CNC apoptosis | TGA, PTA, AA | Fgf8 62,63 | Global hypomorph | |
| OFT myocardium defects | PTA, valve dysmorphism | Fgfr1c/2c isoforms 64 | Isl1Cre targeted deletion | |
| CNC proliferation and migration | Hand2 65 | Wnt1-Cre targeted deletion | ||
| CNC migration/apoptosis | PTA, TOF, valve hypoplasia | Msx1 and 2 66 | Double global deletion | |
| Endothelial cell proliferation | EC hyperplasia, myocardial hypoplasia | Nf1 67 | Tie2-Cre targeted deletion | |
| CNC specification | VSD | Pax3 24 | Tfap2aIRESCre targeted deletion | |
| Myocardial function during OFT alignment | Myocardial hypoplasia | Smad4 68 | Mu-Cre | |
| CNC specification | PTA, TOF, PS, AA | Tfap2a 69 | Global deletion | |
| Endocardial maturation | OA | Tgfb2 70,71 | Global deletion | |
| Endothelial cell-derived OFT endocardial cushion | Thinned myocardium | Zfpm1 72 | Global, Wnt1-Cre or Tie2-Cre targeted deletion | |
| TGA | Left-right patterning | DORV, heterotaxy, AA | Acvr2b 73 | Global deletion |
| Left-right patterning | Heterotaxy | Cfc 74 | Global deletion | |
| Left-right patterning | Heterotaxy | Gdf1 75 | Global deletion | |
| Conotruncal rotation | Coronary artery defects, EC hyperplasia | Hspg2 76–78 | Global deletion | |
| OA | CNC | PTA, DORV, AA | Cited2 79,80 | Global deletion |
| Decreased function within post-migratory NC | PS | DNMAML 81 | Wnt1-Cre and Pax3Cre inhibition of Notch | |
| CNC | PTA, DORV, AA | ECE1 82,83 | Global deletion | |
| CNC | PTA, DORV, AA | ECE1 and ECE2 83 | Global deletion | |
| Decreased SHF proliferation and CNC numbers | Arch artery | Hes1 84,85 | Global deletion | |
| CNC | PTA, DORV, AA | Spp1 82,86,87 | Global deletion |
AA, arch artery; CNC, cardiac neural crest; DORV, double outlet right ventricle; EC, endocardial cushions; NC, neural crest; OA, overriding aorta; OFT, outflow tract; PA, pharyngeal arch; PS, pulmonary stenosis; PTA, persistent truncus arteriosus; SHF, secondary heart field; SMC, smooth muscle cells; TGA, transposition of the great arteries; TOF, Tetralogy of Fallot.
GENESIS OF THE OFT
The mammalian OFT conduit is composed of migratory progenitor cells (i.e., CNC, SHF, and resident primary heart field cells) that give rise to endocardial and myocardial cells as well as cells derived from EMT within the OFT itself (i.e., endocardial cushion cells; Figure 2). Not only are multiple lineages and extra-cardiac colonization of the heart/OFT from distinct embryonic regions required for dynamic OFT development, migration of these extra-cardiac lineages into the heart requires precise spatiotemporal regulation and lineage–lineage signaling/feedback for normal morphogenesis to occur. Compounding this complexity, the beating heart is becoming coupled to the metabolic needs of the developing embryo even as the OFT is being formed from several disparate cell types.88,89
FIGURE 2.
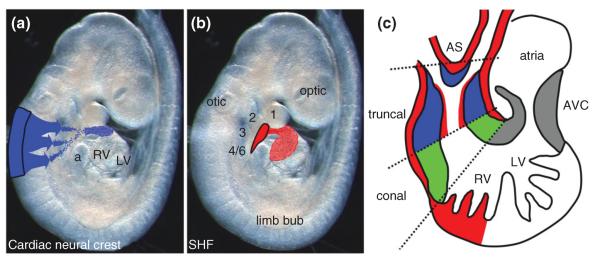
Genesis of the OFT. (a,b) E9.5 mouse embryo illustrating origin (black outline) and migration pathways of CNC (blue in a) to OFT truncal cushions and SHF (red in b) to OFT myocardial cuff and overlying endocardial cells within truncal region. (c) Schematic shows the locations of OFT colonization by the extra-cardiac CNC (blue), SHF (red) and the location of the EMT-derived conal endocardial cushions (green). Additionally, the position of the aorticopulmonary septum is indicated by * symbol. a, atria; AS, aortic sac; AVC, aorto-ventricular cushions; otic, otic placode; optic, optic placode, 1–6, first to sixth aortic arches; LV left ventricle; RV, right ventricle.
PHF and SHF Lineages
The primitive embryonic heart tube begins beating within hours of formation, around embryonic (E) day 8–8.5 in mice,25,90 and days 21–2391 in human embryos. As briefly revealed above, different segments of the OFT arise from distinct progenitor pools both from within the looped heart itself, as well as distinct populations of invading extra-cardiac cells. The initial linear heart tube is derived from the paired lateral precardiac mesoderm of the primary heart field (PHF), and contributes primarily to the eventual LV and parts of the atria.92 Subsequently, additional cardiomyocytes migrate into the heart from SHF pharyngeal mesoderm (E8 in mice93–95 and end of the third week in humans); and these later additions contribute to the RV, ventricular septum (or a part of ventricular septum), and OFT, while other cells contribute to areas of the atria.27,96–98 The SHF grows out from the cardiac crescent to the arterial pole of the primitive LV and forms a primitive outlet, the OFT precursor. At this stage, the PHF cells occupy the greater part of the single straight heart tube, and despite the use of several transgenic marking systems the early contribution of SHF to the heart tube is still imprecise.99–101 Moreover, the mechanisms governing SHF migration into the heart are unclear: for instance, it remains to be determined whether SHF cells migrate individually or as a sheet, how they communicate with each other and the PHF derivatives, and if the SHF cells respond to stimuli from the PHF. Additionally, the SHF is heterogeneous and some SHF cells may populate the endocardial cushions,26,94 although whether they migrate into the endocardial cushion or enter as a result of EMT from the endocardium remains ambiguous.102 Simultaneous with rightward looping of the heart tube, a rapid increase in the length of the primitive OFT occurs with the addition of SHF-derived myocardial precursors from the pharyngeal mesoderm.96,101,103 As SHF derivatives colonize the primitive OFT there is a co-incident downregulation of SHF progenitor proliferation markers (including CTNNB1, SHH, BMPs) and activation of early markers of the cardiomyocyte lineage (including Nkx2.5, Tbx5, Tbx20, Gata4, Gata5, and Gata6; reviewed in Ref 100). The OFT is often described as two regions, the `truncal' portion being adjacent to the RV and the truncal portion being further away from the heart and adjacent to the pharyngeal arches. Conal myocardium is added before truncal myocardium and hence is proximal to the distal truncal myocardium (Figure 2). Although adjacent to each other in the elongated OFT, the conal and truncal OFT have distinct characteristics and the truncal/conal boundary is thought to play an important role in remodeling of the heart. The interface of the conal (EMT-derived) and truncal (CNC-derived) endocardial cushions is believed to be important for the remodeling process of the systemic and pulmonary circulations and to help establish positioning of the emergent semi-lunar valves.30,104,105 Directional signaling from the myocardial sheath surrounding the OFT is important for normal endocardial cushion morphogenesis, and there is evidence that the conal and truncal myocardium have distinct characteristics from chick studies.104 It seems reasonable to consider that conal and truncal myocardium exert distinct influences on the EMT and CNC-derived cushions in mice; however, it remains unclear whether the conal and truncal myocardium in higher vertebrates maintain the unique properties that have been observed in avian species. Preceding differentiation into mature cardiomyocytes, SHF-derived myocar-dial precursors may also directly influence semi-lunar valve formation.34 Moreover, the conal myocardium is non-trabeculated and is able to induce endocardial EMT to seed the proximal OFT endocardial cushions, which is central during closure of the ventricular septum and semi-lunar valve development.106In vitro approaches indicate that chick myocardial differentiation of SHF progenitors requires BMP signaling combined with downregulation of the FGF/Ras/Erk pathway, and that the FGF signaling cascade may maintain proliferation of SHF progenitors until OFT colonization.107 After addition of the myocardium to the truncal myocardial cuff around the OFT, SHF cells differentiate into smooth muscle cells near the base of the remodeling aorta and pulmonary artery and populate regions adjacent to the more distal walls of the great arteries that are CNC-derived.108 Indeed, impaired SHF proliferation and function can lead to severe conotruncal hypoplasia and concomitant CHDs.94,109–111 Early conotruncal hypoplasia is primarily a defect in SHF colonization of the arterial pole of the primitive heart tube, and the importance of SHF in heart tube and OFT elongation has been demonstrated using several distinct approaches. Moreover, surgical removal of chick SHF progenitors leads to OA and pulmonary stenosis or atresia, which are key components of TOF.27 Ablation of genes that are highly expressed in mouse SHF (such as ISL1112 and MEF2c113), conditional inhibition of Notch signaling in SHF derivatives,114 and regional application of a cell cycle inhibitor to the anterior pharyngeal mesoderm prior to migration of SHF derivatives95 all resulted in failed heart tube elongation, looping, and hypoplastic right-sided heart, atria, and OFT, underscoring the importance of this specialized cell lineage in OFT development.
Endocardial Cell Lineage
The endothelial cells closely juxtaposed to the myocardium in the heart, termed the endocardium, are derived from early cardiac mesoderm.115 However the origin of the cardiac endocardial lineage remains uncertain as different experimental systems have provided divergent models. From data obtained mainly from mouse models and cell lineage-mapping approaches, it is thought that mammalian endocardial and myocardial cells are derived from a common multipotent progenitor within the cardiac mesoderm.102,116–119 However, using both chick and zebrafish embryo methodologies, it believed that endocardial cells within the cardiac mesoderm are pre-specified during primitive streak stages, prior to migration to the cardiac crescent, to become endocardium.120,121 Furthermore, cell fate approaches using precise inducible lineage-mapping techniques, clonal analysis and multicolor reporter constructs (i.e. those reviewed in Ref 122), as well as birth dating and live imaging of the progenitor cell populations may help in resolving these inconsistencies. Nevertheless, the endocardium plays two key roles during OFT morphogenesis, principally during endocardial cushion morphogenesis and also as a regulator of OFT myocardial proliferation.
In response to conal OFT myocardial signals to the adjacent endocardium, the endocardium undergoes EMT to give rise to a migratory population of mesenchymal cells that seed the acellular endocardial cushion prior to normal semilunar cardiac valve development. Multiple signaling pathways and transcription factors, including TGFB, BMP, FGF, and WNT signaling,33,36,123–125 regulate this EMT process and subsequent cardiac valve development. The early cardiac cushions are required to prevent retrograde blood flow. Significantly, abnormal regurgitant flow in the aorta evident as early as E12.5 within Nfatc1 null embryos has been shown to result in altered physiological parameters that may culminate in the mid-gestational lethality observed.29 Indeed, recent mouse lineage-mapping experiments using the Nfatc1 promoter have revealed that NFATC1 transcription factor plays a key role in coordinating the timing and location of EMT within the OFT via suppression of Snai1 and Snai2, two key transcriptional factors for initiation of EMT.30 After fusion of the OFT cushions and septation, the truncal OFT endocardial cushions further remodel into the mature aortic and pulmonary valves.39,126 Additionally, the endocardium is known to play a critical role during heart development via the regulation of cardiomyocyte proliferation. Multiple pathways, including the NRG1/ERBB2/B4, Notch, VEGF, ANG, and FGF signaling pathways, are important for endocardial myocardial signaling and many of these act as mitogens and promote cardiomyocyte differentiation.125,127,128
CNC Lineage
Disruption of neural crest cell (NCC) development, either cell-autonomously or non-cell autonomously, results in many forms of human birth defects, including Waardenburg, DiGeorge, Neurofibromatosis, Hirschsprung, Treacher-Collins, Leopard, Axenfeld-Rieger, and CHARGE syndromes; many of which include defects in pharyngeal arch arteries (AAs) and/or cardiac OFT, such as congenital vascular rings, PTA, or pulmonary atresia. The neural crest (NC) related congenital defects are often called neurocristopathies.129
NCC derivatives are absolutely essential for OFT remodeling,130 specifically during the OFT septation process and for patterning of the great vessels. NCCs compose a multipotent migratory embryonic lineage that is derived from the border zone of the neural plate/surface ectoderm.131 NCC specification occurs embryonic (E) days 7.75–8 (corresponding to day 16/17 in human embryos) within mouse neural folds in the dorsal domain of the developing NT/spinal cord. The CNC is a subset of the NC that originate from the lower hindbrain between the otic placode and fourth somite and is absolutely required for normal heart development.22,23,132–134 Following specification, EMT confers morphological changes that allow E8–E8.5 CNC subpopulation to migrate over and through a meshwork of loose ECM or along dense basement membranes and where they encounter a great diversity of substrates135 on their way to the aortic sac and further on into the OFT truncal cushions. Several key signaling molecules (BMP, FGF, Notch, RA, WNT) are thought to participate in both induction and later steps of NCC development. The induction of NCC emigration from within the neural tube triggers the expression of a specific set of transcription factors, collectively known as border specification genes (including Msx1, Msx2, Pax3, and Zic1) that, along with signaling molecules, are thought to direct the expression of NCC transcriptional specifiers (including Tfap2a, Foxd3, Crabp1, Snai2, and Sox10). These specifiers in turn regulate the appearance of later NCC effector genes responsible for NCC migration and differentiation (including Sox9, Sox10, Cdh7, Col2a1, Neurog1, and Plxna2). The expression of these genes appears in a temporal fashion during embryogenesis, and marks the progression of NCC morphogenesis (reviewed in Ref 136).Cell-cell interactions [via Connexins and Cadherin 2 (CDH2), formerly known as N-cadherin] between NCC within the same migratory stream are thought to contribute to the generation of cell polarity and guide overall direction.137 In addition, interactions with neighboring non-NCC lineages are thought to be essential for the interpretation of external cues.138–142 NCC migration is followed by terminal differentiation at sites of colonization,143 but the specifiers and signals responsible for cessation of CNC migration and differentiation during mesenchymal condensation and formation of the muscular OFT septum are yet to be identified.
Specifically, the CNC subpopulation is thought to migrate via the aortic sac and begin to colonize the truncal OFT cushions from E9 onwards in mouse embryos (corresponding to days 20–22 in human) and continue to populate the truncal cushions until approximately E11 and give rise to the aorticopulmonary septa.5,7,22,24 The aortic sac is the non-muscularized connection between the primitive OFT and the arch arteries (AAs; Figure 2). The OFT septum, which physically divides the systemic circulation from the pulmonary circulation, begins as a shelf-like structure distal to the OFT.39 The shelf forms between the fourth aortic AA (eventual aorta) and sixth aortic AA (eventual pulmonary artery) and it grows into the distal OFT toward the proximal OFT.7,22,24,144 Subsequently, as the shelf grows proximally, it comes into continuity with the bulging spiraling OFT cushions.39 Rotation of the OFT is a seminal event in proper OFT alignment,145 and reduced rotation has been correlated to DORV, OA, and other OFT remodeling defects in humans.146 The OFT shortens at the same time it undergoes rotation and shifts to a medial position above the presumptive ventricular septum. Thus, OFT septation of the elongated spiraling OFT occurs with fusion of the single aorticopulmonary septum and enlarged endocardial cushions following significant CNC colonization of the truncal cushions. Subsequently, the arrest of NCC migration is sometimes coupled with a reaggregation process147; however, the environmental signals that directly influence when, where, and how the CNC subpopulation migrates and begins to differentiate are yet to be identified.
In addition to controlling OFT remodeling, CNC-derivatives are essential for E10 pharyngeal AAs remodeling that gives rise to the great vessels exiting the heart. Specifically, CNC contribute to vascular smooth muscle cells of the aortic arch and eventually differentiate to form some of the smooth muscle cells within the adult arterial wall. Mammals have five branchial arches, each of which contains an AA (numbered 1–4 and 6). Blood flows out of the heart via branchial AAs to circulate throughout the embryo. Although the arterial system initially forms symmetrically, in the mature organism, the left fourth and sixth AAs persist and give rise to the aortic arch and pulmonary trunk, whereas the right fourth and sixth AAs regress.22 Although CNC colonize the fourth and sixth AAs equivalently,130 and reduced CNC function invariably results in a wide variety of AA defects,22,24,50,148,149 CNC are thought to be essential for the persistence of an AA but not its formation.150 CNC also colonize the dorsal root ganglia (DRG), thyroid, and parathyroid and are the precursors of the Schwann cell lineage.132,133,151,152 Recent lineage-mapping data suggest that CNC may also orchestrate E11–E13 valve remodeling35,126 and give rise to glia cells around the conduction system153,154; however, their precise role in these unexpected locations remains to be fully elucidated and these lineage-mapping results are yet to be tested functionally.
Aberrant CNC morphogenesis is associated with an astonishing array of structural CHDs including severe conotruncal defects (e.g., PTA and concomitant membranous VSDs) and less severe cardiac phenotypes (e.g., DORV). The range of CNC-associated phenotypes suggests a continuum where the relative severity of NC deficiency dictates the severity of the CHD. Identification of the molecular mechanisms underlying CNC specification has remained elusive. Similarly, there is almost nothing known about the role of CNC within the OFT itself, nor the reason why so many mouse KO and chick NC ablations almost uniformly result in early embryonic lethality,19,132,155 as the presence of ductus arteriosus and placenta preclude the requirement of septation of the OFT and division of pulmonary and systemic circulations until birth. Moreover, our understanding of NCC development in human embryos remains largely uncharacterized, despite the role the NCC plays in several human pathologies. However, examination of several molecular markers identified in mouse studies has revealed conservation in human embryos from Carnegie Stages 12 to 18.156 Significantly, expression of Sox9, Sox10, and Pax3 transcription factors was demonstrated within premigratory NCC, while actively migrating NCC display the additional transcription factors Pax7 and Tfap2a. Combined, this has meant elucidation of a CNC-specific role causing actual functional in utero lethality during heart development remain vague, although their requirement during structural remodeling is unquestioned.
What Developmental Biology Can Tell Us About the Mechanism/s that Cause Persistent Truncus Arteriosus
PTA, also known as common arterial trunk, is defined by a single undivided arterial trunk exiting the heart and is principally thought to be a result of OFT septation failure. The lack of OFT septation prevents separation of pulmonary and systemic circulations at birth, usually requiring neonatal surgical repair to prevent cyanosis and pulmonary failure due to the presence of a concomitant VSD. In humans, the common trunk may arise from the RV or LV, or more commonly, the PTA may override the VSD.157 Intriguingly, PTA typically arises from the RV in most mouse mutants, particularly when NCC and SHF restricted Cre recombinase drivers are used. However, recent conditional inactivation of the histone trimethyltransferase Ezh2 using Nkx2.5-Cre resulted in overriding PTA.158 It is unclear why there appears to be a species-specific difference between mouse and human in the location of PTA. Further studies are required to determine whether mutations within distinct progenitor cell populations, developmental timing of genetic insults, unique physiological demands, or hemodynamic stresses primarily mediate the final position of PTA.
Clinically, PTA is classified into type-I (reflecting partial septation of the OFT once it has exited the heart, as the OFT partially divides into separate pulmonary and aortic arch elements); type-II (in which only a single OFT exits the heart), and type-III (in which there are two lateral pulmonary arteries). However, from a developmental perspective these types more likely reflect a continuum of severity and the degree to which CNC morphogenesis is compromised. Multiple underlying cellular mechanisms can give rise to the same phenotypic structural PTA defect.5 This complicates the analysis of the contributory primary defects and lineages involved in OFT pathogenesis. Nevertheless, the CNC lineage is known to play a fundamental and unquestioned role during OFT septation, initially demonstrated via landmark surgical neural fold ablation studies performed in chick embryos.18 These microablation studies confirmed the importance of CNC cells to normal cardiovascular development, as PTA invariably resulted following their removal at the neural plate stage. These ablations often led to failed OFT elongation,108,159,160 supporting a cell-autonomous role of CNC and further suggesting that the CNC may directly contribute to OFT elongation. However, using a transgenic model employing Cre/loxP technology and spatiotemporally restricted expression of a diphtherotoxin (DTA) subunit with the NCC-specific Wnt1-Cre driver, genetic ablation of the mouse CNC results in fully penetrant PTA, but a normal length OFT, despite absence of any CNC migration from the neural tube (previously demonstrated by Olaopa et al.24 and illustrated in Figure 3). Combined, these NCC ablation approaches confirm that the absence of CNC subpopulation causes PTA due to a lack of mesenchymal contribution to the aorticopulmonary septum and failed septation. However, the role of the CNC within OFT elongation remains unclear, and this ambiguity could be due to species-specific effects, the extensiveness of the CNC ablation or an indirect non-cell-autonomous effect upon adjacent lineages.
FIGURE 3.

Genetic neural crest cells (NCC) ablation in Wnt1-Cre;R26-EGFP-DTA embryos results in gross morphological defects including persistent truncus arteriosus (PTA). Gross examination under UV light demonstrates lack of craniofacial structures (a, open arrowhead) and internalized eyes following genetic NCC ablation in Wnt1-Cre;R26-EGFP-DTA embryos (a) compared to R26-EGFP-DTA alone (b) and littermates lacking the transgene (c) at E15 (with brightfield insets). Subsequent transverse sectioning and counterstaining with αSMA immunohistochemistry (brown DAB staining) demonstrates reduced outflow tract (OFT) smooth muscle cells surrounding a grossly abnormal heart and OFT with PTA in Wnt1-Cre;R26-EGFP-DTA embryos (d) compared to normal OFT morphogenesis in R26-EGFP-DTA controls (e). Note that PTA OFT has four valve leaflets (indicated by * in d) compared to two normal pulmonary leaflets in normal hearts (indicated by * in e). Panels (f) and (g) illustrate the associated interventricular septal defects Wnt1-Cre;R26-EGFP-DTA embryos with OFT defects (f) compared to closed septum between left and right ventricles in control (g) embryos. (Reprinted with permission from Ref 24. Copyright 2011 Elsevier Ltd)
Congenital malformations are due primarily to aberrant gene expression, which ultimately leads to poor cardiac output both in utero and after birth.161 Moreover, the identification of genes that mark specific cardiac lineages and are mis-expressed during CHD pathogenesis12,13,16,22,24,132–134 has enabled investigators to molecularly monitor and analyze the fate of the CNC lineage. The advent of transgenic Cre recombinase mouse lines, which allow Cre/loxP-mediated lineage traces to be performed of the marked CNC and their progeny without changing the properties of the marked cells, has provided a number of important insights. Several transgenic mouse Cre lines are now used to study CNC cell migration (listed in Table 3), providing a comprehensive atlas of the structural contribution of CNC cells and their derivatives to the OFT. The most extensively employed NCC-restricted Cre line within heart development is Wnt1-Cre.130 The Wnt family of transcription factors has a proven and critical role within early CNC specification,162 and Wnt1-Cre marks NCCs within the cranial neural tube beginning E8 and within the emigrating CNC approximately E8.25.24,163 Similarly, transgenic 1.6 kb Pax3-Cre line164 also drives dorsal neural tube and NCC-restricted reporter expression from E8 to E8.25 onwards;163 while P0-Cre drives NCC-specific reporter expression, including the CNC subpopulation, only after they have emigrated from the neural tube.165 Several other Cre lines are also used for OFT analysis (Table 3), including the Tfap2a-Cre knock-in line166 that is expressed earlier than Wnt1-Cre within the CNC lineage at E8,24 but is additionally expressed within the surface ectoderm. Interestingly, global Wnt1 knockout embryos do not exhibit any NCC abnormalities nor CHDs, but double Wnt1/Wnt3a knockouts are lethal by E10 and the NCC are stuck within the dysmorphic neural tube.167 However, both global Pax3 and Tfap2a mutants exhibit OFT defects.19,168 Although we are fortunate to have several CNC Cre lines at our disposal, it should be noted that it has not yet been established whether any of these important mouse tools comprehensively mark all CNC and whether any of them are expressed early or extensively enough to mark the different waves of emigrating NCC subpopulations.
TABLE 3.
Cre Recombinase Mice Commonly Used Within OFT Development Studies
| Lineage | Cre | Spatiotemporal Expression (by stage of onset) | Genetic System | References |
|---|---|---|---|---|
| Neural crest | Tfap2aIRESCre | E7.5 neural folds; E8.0 CNC and ectoderm | 3′ UTR Knock-in | 166 |
| Wnt1-Cre | E8.0 cranial-caudal onset within neural plate; E8 cranial NC, E8.25 cardiac NC | Transgenic | 130 | |
| Wnt1-Cre-ER™ | Temporal regulation and as above | Tamoxifen-inducible transgenic | 169 | |
| Pax3 Cre | E8.0 dorsal neural tube and emigrating cranial and caudal NC; as well as somites and muscle derivatives | Knock-in | 170 | |
| 1.6kb Pax3-Cre | E8.0 neural tube and NC restricted; some ectopic expression in lumbosacral and myocardial cells and not expressed in all sections of dorsal neural tube that normally express endogenous Pax3 | Transgenic | 164 | |
| Ht-PA-Cre | E8.5 NC within cranial-caudal onset | Transgenic | 171 | |
| P0-Cre | E9.0+ migratory NC restricted | Transgenic | 165 | |
| Isl1Cre * | Some CNC* as they migrate through SHF | Cre knock-in | 172 | |
| Myocardial | Mesp1Cre | E7.5 extra-embryonic and lateral mesoderm; E9.5+ heart tube myocardium, amnion contiguous with closing foregut | Knock-in | 173 |
| cTnT-Cre | E7.5+ myocardium | Transgenic | 174 | |
| Nkx2.5Cre | E7.5 cardiac crescent; E8.0 myocardium; E9.5+ myocardium and first pharyngeal arch ectoderm | Knock-in | 175 | |
| Nkx2.5-Cre | E7.5 anterior splanchnic mesoderm, pharyngeal endoderm adjacent to cardiac mesoderm; E8.0+ atrial and ventricular myocardium | Transgenic | 176 | |
| Myl2Cre | E7.5 cardiac precursors of cardiac crescent; E8.0+ ventricular precursors, definitive ventricular tissue, and slow-twitch skeletal muscle | Knock-in | 177 | |
| Gata6-Cre | E7.5 restricted posterior cardiac crescent; E8.0–E8.5 sparse expression restricted within lateral heart tube; E9.5 myocardium of posterior right AV canal, sparse expression in myocardium of posterior left and anterior AV canal. | Transgenic | 178 | |
| Gata5-Cre | E9.5–E10 caudal surface of the common ventricular chamber myocardium; E12 body wall, hepatic capsule/kidneys | Transgenic | 179 | |
| αMHC-Cre | E9.5+ myocardium | Transgenic | 180 | |
| MerCreMer | Temporally controllable regulation within myocytes | Tamoxifen-inducible transgenic | 181 | |
| Mu-Cre | E11.5+ myocardium and background dependent expression in skeletal muscle | Transgenic | 68 | |
| β-MHC-Cre | E12.5 myocardium; E14.5 myocardium; E17.5 myocardium | Transgenic | 182 | |
| Secondary heart field | Isl1 Cre * | Pharyngeal endoderm and crescent mesoderm; first pharyngeal pouch and endoderm, LV precursors in ventral heart tube, splanchnic mesoderm, SHF | Cre knock-in | 172 |
| Mef2c-Cre | E7.75 medial cardiac crescent; E8.25 pharyngeal mesoderm; E9.0–E9.5 right ventricular myocardium, pharyngeal mesoderm, branchial arches | Transgenic | 26 | |
| Nkx2-5Cre | E7.5 cardiac crescent; E8.0 myocardium; E9.5 myocardium and first pharyngeal arch ectoderm | Knock-in | 175 | |
| Nkx2-5-Cre | E7.5 anterior splanchnic mesoderm, pharyngeal endoderm adjacent to cardiac mesoderm; ≥E8.0 atrial and ventricular myocardium; E11.5–E12.5 distal thyroglossal duct, pharyngeal endoderm, tongue, spleen, and distal stomach | Transgenic | 176 | |
| Pitx2cre | E9.5 left splanchnic mesoderm; E10.5 left atrium, primary interatrial septum, left dorsal mesocardium, and right ventricular and interventricular myocardium | Knock-in | 183 | |
| Endocardial | Tie1-Cre | E8+ endothelial specific, extensively expressed in endocardial cushions | Transgenic | 184 |
| Tie2-Cre | E7 extra-embryonic mesodermal component of visceral yolk sac; E8+ endothelial cells and endocardial cushions; E11.5 developing valve leaflets, mesenchymal cells of proximal OFT septum | Transgenic | 185 | |
| KdrCre | E8.5 vascular endothelial cells and hematopoietic cells; E10.5 small percentage of myocytes; E11-Adult progressively increasing percentage of cardiac and skeletal muscle cells | Knock-in | 118 | |
| VE-cad-Cre | E9.5 basilar arteries, intersomitic vessels, and endocardium; E10.5 dorsal aorta; P0 liver | Transgenic | 186 | |
| Nfatc1enCre | E10.5 atrioventricular canal; E10.5–E11.5 expanded atrioventricular canal and into OFT | Knock-in | 30 |
OFT, outflow tract.
Please see Ref 187.
Although it is well established that a combination of BMP, WNT, FGF, retinoic acid (RA), and Notch/Delta signaling are important for the specification of NCC lineage from dorsal neural progenitors162,188,189; surprisingly, the mechanism responsible for specification and induction of the CNC subpopulation is less well understood. However, BMP signaling is thought to be important in regulating a subset of CNC cells as they undergo migration to the OFT, as BMP-dependent NCC have been shown to be important during OFT remodeling. In mouse embryos where combined TGFB and BMP signaling were significantly downregulated via Wnt1-Cre-mediated Smad7 antagonist overexpression, CNC migration was impaired due to elevated CNC-specific apoptosis leading to PTA.50 Similarly, Wnt1-Cre conditional deletion of the downstream TGFB and BMP signaling co-effector Smad4, results in elevated CNC apoptosis, hypocellular OFT cushions, and PTA.48,49 In chick embryos, retrovirally induced Noggin overexpression in developing hearts leads to decreased CNC migration into the OFT and a variable array of OFT defects (possibly dependent on intra-experimental variance in viral efficacy) including DORV and PTA.190 These results are consistent with additional studies in chick that exogenously applied Noggin191,192 and surgical implantation of Noggin-expressing notochords into the open neural folds192 (notochord is known to inhibit NCC migration193) all of which resulted in failed NCC migration from the neural tube. While excessive Noggin results in reduced CNC migration to the OFT, global loss of Noggin in mouse results in increased apoptosis within the NCC-containing neural tube at E9.0.194 Given the extensive efforts to delineate the importance of BMP signaling in NCC function and cardiac development, future studies should focus on temporal or spatially restricted modulation of Noggin expression to determine what role it may play in OFT morphogenesis itself.
The role of directional BMP signaling directly to the CNC during OFT morphogenesis is further supported by failed CNC morphogenesis resulting in PTA following cardiac-specific deletion of Bmp4.174,195Bmp4 expression within SHF derivatives, including the OFT myocardial cuff, is critical for OFT septation and conditional ablation of Bmp4 via Mef2c-Cre results in PTA, as previously demonstrated by McCulley et al.196 and illustrated in Figure 4. Furthermore, NCC-restricted deletion of Acvr1 (formerly known as Alk2) BMP receptor signaling also resulted in PTA and abnormal maturation of the aortic arch, reminiscent of common forms of human CHD.40 Type-II Bmp receptors are also critical for OFT morphogenesis and while global Bmpr2 deletion is not instructive in cardiac development (mutants die during gastrulation197) global Bmpr2 hypomorphism results in PTA.198 Similarly, migration of mutant Acvr1 CNC to the OFT was impaired, and differentiation of CNC into smooth muscle cells surrounding the aortic AAs was deficient. However, Wnt1-Cre targeted deletion of Bmpr1a (formerly known as Alk3) resulted in a shortened OFT with defective septation leading to PTA despite normal CNC migration and colonization of the OFT.199 Although marker gene analysis or organization of the CNC within the Bmpr1a mutant OFT septum itself was not analyzed, these data were interpreted as Bmpr1a may not be necessary for early CNC biology. An intriguing alternative is that the hyperplastic OFT cushions within these Bmpr1a;Wnt1-Cre mutant mice precluded any functional need for the CNC to mediate OFT septation. The apparent discrepancy regarding the importance of BMP signaling for CNC morphogenesis may also be due to the respective cell-autonomous requirements and redundancy of ubiquitous Acvr1 versus ubiquitous Bmpr1a or it could be that the timing of NCC-restricted deletion may explain differential effects upon CNC morphogenesis. Specifically, global Bmpr1a nulls fail to gastrulate200 while global Acvr1 nulls arrest at early gastrulation. Alternatively, the finding that Nkx2.5-Cre deletion of Bmp4 (an Bmpr1a ligand) results in defective OFT septation, aortic arch interruptions, abnormal AA remodeling with decreased smooth muscle recruitment, and hypoplastic conotruncal cushions.195 As Nkx2.5-Cre is absent from the CNC lineage and a known SHF marker, this suggests that indirect reciprocal BMP signaling from the SHF to CNC is also key during normal OFT septation.
FIGURE 4.

Bmp4 expression in Mef2cCre expressing cells is necessary for outflow tract (OFT) septation. Transgenic Mef2c-Cre is expressed in second heart field (SHF)-derived structures of the right heart and OFT (b and c). Bmp4 is expressed in the pharengeal ectoderm (d) and OFT myocardium (e). Conditional ablation of Bmp4 in Mef2c-Cre expressing cells reduces Bmp4 expression in OFT myocardium (g) and results in persistent truncus arteriosus (PTA). (Reprinted with permission from Ref 196. Copyright 2008 John Wiley and Sons).
Normal SHF morphogenesis is critical for OFT septation; as deletion of Tbx1,52,53,201Isl1,94Mef2c,109Tbx20,55 and Aldh1a2202 all result in SHF dysmorphism and cardiac defects (including failed OFT formation, rotation, and septation). Due to complex genetic interactions between genes critical for SHF morphogenesis, it has remained difficult to define the precise signaling and transcriptional cascades within the SHF that are important for OFT formation and remodeling. However, exciting work has begun to delineate a few of the key upstream factors involved in SHF signaling. As a prime example, TBX1 provides critical transcriptional regulation of SHF morphogenesis. TBX1 has been tightly associated with DiGeorge syndrome (22q11DS) through several important studies.52–54,203,204 While Tbx1 null mice develop fully penetrant PTA (including other main aspects of the DiGeorge syndrome), Tbx1 heterozygotes develop normal, fully septated OFTs and do not develop the full spectrum of 22q11DS-associated defects, although they do infrequently present AA remodeling defects.53,205 This is in contrast to humans who develop malformations associated with 22q11DS in a haplo-insufficient manner, suggesting either a species difference in DiGeorge syndrome pathogenesis between mouse and human or the possibility that additional gene(s) and/or genetic deletions may contribute to DiGeorge syndrome in humans.206 Global and mesodermal specific (T-Cre transgene) deletion of Tbx1 results in structural heart defects including PTA, as well as increased expression of Aldh1a2, Tbx5, and Gata4, and decreased expression of Isl1 and Nkx2–6.110 Interestingly, overexpression of Tbx1 also results in cardiac and OFT defects despite decreasing the expression of Aldh1a2, Tbx5, and Gata4, and increasing the expression of Nkx2–6 (opposite effect of Tbx1 ablation110). It is important to understand that while TBX1 may transcriptionally regulate Aldh1a2 (responsible for RA synthesis), RA conversely inhibits Tbx1 expression in vivo and in vitro.207 Considering the acknowledged importance of RA for CNC morphogenesis in normal OFT septation (see below), an intriguing possibility arises whereby Tbx1 deficiency may affect not only SHF cells, but also CNC cells that are involved in OFT remodeling. However, whether there is functional overlap or signaling between SHF and CNC in Tbx1 deficiency is not clear, especially considering the apparent inter-lineage signaling that is critical for normal OFT formation.
In attempting to elucidate the early NCC signaling germane to cardiac development, mouse PAX3 has become an interesting avenue of investigation. Mouse PAX3 is a paired and homeobox-containing transcription factor that is one of the earliest expressed NCC genes (approximately E7.5) and maintains dorsally restricted expression throughout neural tube closure, following which Pax3 expression is ventrally expanded.208 Global Pax3 knockout mice exhibit significantly reduced CNC migration resulting in PTA and mid-gestation lethality,19,24,151,209,210 and NCC-restricted deletion reveals Pax3 is essential for early CNC progenitor formation, but is not required for subsequent CNC progeny morphogenesis involving their migration to the heart or during septation of the OFT itself.24 Within a small minority of CNC-related mouse mutants, Pax3 null mice exhibit severe CNC emigration and migration deficiencies (previously demonstrated by Olaopa et al.24 and illustrated in Figure 5; similar to the chick microsurgical and DTA genetic cell ablation models), rather than the more usual post-migratory NC differentiation defects often seen in many Wnt1-Cre-driven conditional knockout models.24 Using a conditional approach, it has been recently demonstrated that Pax3 acts upstream of (or earlier than) Wnt1-Cre and co-incident with Tfap2a-Cre temporal expression within NC cells.24 This demonstrates that PAX3 is likely to be one of the first NCC-restricted transcription factors required for CNC specification and generation of sufficient CNC numbers required for completion of OFT septation. Downstream, Pax3 has been shown to be required to repress Msx2 transcription factor expression.211 Significantly, Msx2 is co-expressed with Pax3 within NCC-containing dorsal neural tube domain. Intercrossing Pax3 mutant allele and Msx2 global nulls (viable and without CHDs), revealed that CNC deficiency in Pax3 mutants is caused by upregulation of Msx2, as Pax3/Msx2 double mutant embryos no longer exhibited the PTA defects seen in Pax3 mutants alone. More recently, the widely expressed PBX1 homeodomain transcription factor has been shown to be able to directly activate Pax3, leading to repression of its target gene Msx2 in NCC lineage.163 PBX1 forms heterodimeric complexes with several neural tube-restricted Hox homeodomain proteins, as well as MEIS/PREP proteins, and global deletion of Pbx1 results in PTA. Moreover, global Pbx1 mutant embryos lose a transient burst of Pax3 expression in premigratory CNC that ultimately specifies CNC function for OFT development, but does not regulate subsequent CNC migration to the heart. Thus, Pbx1 is thought to act upstream and directly activate the Pax3 transcriptional promoter to drive Pax3 expression within the neural tube.163 Combined, these data suggest that PBX1-induced Pax3 expression is required to repress Msx2 within the neural tube. Furthermore, these data suggest that partial rescue of OFT defects observed in Msx2/Pbx1 double knockouts may further indicate that PBX1 regulates expression of other, unidentified transcription factors in the CNC lineage independent of the Pax3-Msx2 regulatory pathway.
FIGURE 5.

Reduced neural crest cells (NCC) emigration and migration in mice embryos lacking Pax3 expression. Dorsal (a) and left lateral (b) views of E9.0 Pax3Δ5 null embryos (a and b, right) demonstrate decreased NCC emigration and migration toward the second PA (indicated by *), when compared to wild-type littermates (a and b, left). First pharyngeal arch and cranial NCC migration are unaffected in Pax3Δ5 nulls. (c) Right lateral views of E9.5 demonstrate that fewer CNC populate the heterozygous third, fourth, and sixth pulmonary artery (PAs) (c, middle embryo), and still even less cardiac neural crest (CNC) colonize the Pax3 null 3/4/6th arches (c, right embryo indicated by white *) compared to wild-type littermates (c, left). Cranial NCC migration is unaffected by loss of Pax3. (d,e) Histology of Pax3Δ5 null embryos demonstrated a lack of Wnt1Cre-marked CNC within the fourth PA (d), AP septum (indicated by * in wild-type in e), and OFT cushions compared to wild-type controls. (Reprinted with permission from Ref 24. Copyright 2011 Elsevier Ltd)
Although there is continued uncertainty regarding CNC lineage pre-specification within the dorsal neural tube prior to emigration versus post-migratory specification of CNC progenitors within the aortic arches and OFT, the extra-cardiac CNC follow a well-described migratory pathway to the heart, prompting investigators to search for chemotactic guidance effectors. Targeted inactivation of Nrp1, a membranous protein that functions as a receptor for class 3 inhibitory axon guidance semaphorin signals, resulted in various types of vascular defects, including impairment in neural vascularization, agenesis and TGA, PTA, and insufficient development of vascular networks within the yolk sac.43 Lack of semaphorin signaling in Nrp1 null mice results in an embryonic lethal phenotype; embryos die at E13 and fail to form the left sixth AA causing the distal end of the pulmonary channel to merge with the aortic arch, and also have an absent left fourth AA that results in a right-sided aortic arch.43 Semaphorins are secreted, transmembrane, and GPI-linked proteins, defined by cysteine-rich semaphorin protein domains, which have been demonstrated to play an important role during CNC OFT development.21 Unlike the Pax3 and NCC ablation models in which initial NCC migration is deficient, Semaphorin3c (Sema3c) mutant CNC cells migrate normally but then fail to invade the OFT cushions. These mutant mice have PTA and interrupted aortic arch, but do not appear to have any complications with other NCC subpopulations.20Sema3c is expressed in the cardiac OFT myocardial cuff as the CNC colonize it, and it is believed that signaling to the CNC, which reciprocally expresses its receptor PLXNA2,21 is important for directing CNC migration and colonization of the OFT. Indeed, Plxna2 is expressed by both migrating and post-migratory CNC and is absent in Pax3 null OFT cushions.21 Significantly, it has been found that the Gata6 transcription factor can directly regulate Sema3c and Plxna2, and that GATA6 mutations can cause PTA in patients,212 strengthening the likelihood that animal studies envisaging the underlying genetic and molecular basis of OFT defects are clinically applicable. Indeed, there are several other examples of complementary expression of proteins and their receptors in CNC cells and their surrounding environment (e.g., BMP4 and ACVR1/BMPR1A, VEGF and NRP1). In fact, a particularly intriguing case is that of RA and its role in CNC migration and differentiation. RA is the active form of vitamin A, and either an excess or reduction of RA levels affects cardiac development. RA functions via its ligand-activated receptors that work as transcription factors. These receptors include the RA receptors (RARs) and Retinoic X receptors (RXRs), which function as both heterodimers and homodimers to transduce RA signaling. Mice deficient for different combinations of these receptors display OFT defects including PTA.213 Interestingly, Wnt1-Cre-mediated lineage tracing revealed that these mutants exhibit normal CNC migration and OFT colonization and suggested that normal aorticopulmonary septation requires not only a sufficient threshold of CNC, but also the normal function of these cells either during their migration or upon their arrival at the OFT.148 This result is different from the reduced CNC colonization of the OFT observed in Pax3 and Sema3c models, which require normal complementary expression of ligand and receptors in both migratory CNC and their surroundings.
While a plethora of signals are required in a delicate spatiotemporal balance just to facilitate SHF and CNC cell colonization of the OFT, the story of OFT development does not end there. In response to CNC infiltration and endocardial cushion EMT and proliferation, endocardial ridges swell until they meet in the middle of the OFT. At the midline of the OFT the endocardial ridges fuse to form a spiral septum. Initial fusion of the spiral septum is dependent on a bulging compact mesenchymal shelf derived from CNC that forms between the fourth and sixth AAs. This spiral septum extends along the axis of the OFT, thus beginning a process that segregates the aortic and pulmonary circulatory systems. Effectively, conditional deletion of focal adhesion kinase (Ptk2) in NCCs using Wnt1-Cre results in normal SHF colonization of the OFT, CNC migration and colonization of the endocardial cushions, and myocardialization of the OFT sheath, but an inability of the OFT to rotate following remodeling. This results in incomplete OFT septation, and a high incidence of PTA.45 PTK2 is a cytoplasmic protein that is activated by integrin and growth factor signaling and is typically associated with migration, proliferation, and differentiation.214 Intriguingly, while CNC differentiation into smooth muscle cells was normal within the OFT wall of Wnt1-Cre mediated conditional Ptk2 mutants, there was reduced smooth muscle differentiation within the AAs, which may have contributed to concomitant AA remodeling defects. Significantly, Ptk2 null CNC failed to form condensed mesenchyme during endocardial ridge fusion as the OFT remodeled, primarily mediating failed OFT septation and resultant PTA. Furthermore, microarray analysis revealed disrupted expression of several genes important in ECM composition; both Heparan sulfate proteoglycan 2 (Hspg2) and SEMA3C were greatly reduced, suggesting CNC are required for normal ECM deposition within the condensed mesenchyme of the OFT endocardial cushions. Indeed a striking example of the importance of the OFT endocardial cushions in OFT septation is provided by the SOX4 transcriptional activator.51Sox4 null embryos exhibit PTA, yet Sox4 is expressed neither in the CNC nor in SHF derivatives, and no abnormalities are observed in either lineage. However, Sox4 is expressed within the endocardial cushion and Sox4 nulls exhibit totally absent conal OFT cushions which explains the presence of PTA in this model, reinforcing the importance of endocardial cushions to OFT development.
Following CNC colonization of the OFT endocardial cushions, extensive remodeling-associated apoptosis is detectable within the endocardial cushions and during ridge fusion and OFT septation.215–217 However, neither the extent of apoptosis within the CNC nor the exact function of the observed apoptosis within endocardial ridge fusion is understood, but as CNC are the most abundant cell type in the OFT endocardial cushions50 and the `hot spot' of apoptosis is at the base of the OFT, it seems reasonable that CNC themselves may undergo apoptosis or regulate adjacent conal cushion apoptosis during normal OFT septation. Unfortunately, it is difficult to directly assess this hypothesis as there are a lack of cellular markers and lineage-mapping tools for post-migratory CNC following colonization of the OFT.22 In other words, there are no tools available to examine the CNC subpopulation that colonizes the OFT in isolation; as any Wnt1-Cre or Pax3Cre-mediated recombination event will affect the NCC from specification onwards. Given the intricate interactions of CNC with the pharyngeal arch mesoderm during migration and their necessity for OFT morphogenesis, understanding the role of a gene in the CNC post-colonization is difficult with many potentially confounding influences of OFT, SHF, and CNC morphogenesis. This is a major obstacle in understanding the role that CNC and SHF cells fulfill following initial colonization of the OFT as all current tools are active prior to colonization. In this respect, temporal Cre (Wnt1-CreERT2169 and Pax3CreERT2) and/or an inducible system (i.e., Dox induction in Tang et al.50) would be useful. Unfortunately there is a current lack of inducible Cre recombinases available for selective use in SHF and NCC lineages. This is unlike the case of cardiomyocytes where MerCreMer is a highly efficient and inducible Cre recombinase specific to cardiomyocytes.181 Future research would benefit from considering temporally restricted genetic manipulation in addition to spatial, cell lineage restricted genetic manipulation.
What Developmental Biology Can Tell Us About the Mechanism/s that Cause DORV
DORV, which accounts for 1–3% of all CHDs,218 occurs when both the aorta and pulmonary trunk arise from the RV.219 A key feature that distinguishes DORV from other OFT defects is that the aorta and pulmonary trunk are recognizable as distinct vessels, but the aorta is improperly aligned over the RV. Thus, DORV is not usually considered an OFT septation defect, but rather a defect in OFT alignment. There are no animal models of isolated DORV, as DORV typically occurs in concert with additional aortic AA, myocardial, and ventricular septation defects. DORV often occurs with partial penetrance in genetic models typically utilized to study other developmental cardiac defects. Given these facts, our ability to understand the underlying etiologies of DORV remains limited.
Elongation is one of the first events during OFT remodeling that is critical for OFT alignment and is thought to be dependent on colonization via the SHF lineage of the arterial pole of the primitive heart tube. Deletion of genes responsible for SHF morphogenesis results in failure of right ventricular formation and abnormal OFT formation, including Isl1,94Mef2c,113,220–222Foxh1,223Foxc1, and Foxc2.224,225 However, there remains considerable controversy as to how cells derived from the SHF are specified to become either myocardial or endocardial cells.102 Using Isl1Cre, Mef2c-Cre, and Nkx2.5-Cre lineage mapping it has been revealed that the SHF may contribute to multiple parts of the heart including RV, ventricular septum, OFT myocardial cuff, coronary arteries, endocardial cushions (partial), and even LV (occasionally).26,94,226 Yet, the question remains as to whether the SHF contributes as broadly to the developing heart as described in the lineage-mapping studies or whether it primarily contributes cellular mass to the myocardium and endocardium of the right heart and the transgenic lineage-mapping tools available are simply more broadly expressed to include other cell lineages inhabiting the Isl1/Mef2c/Nkx2.5 gene expression domain. A recent study that used both experimental chick and transgenic mouse embryo approaches provides compelling evidence that the endocardium of the developing heart may be derived from endothelial cells (Mesp1Cre or Tie2-Cre positive cells) that inhabit the SHF prior to delamination and migration and not from the Isl1Cre expressing cells of the SHF.227 In this carefully designed study, Milgrom-Hoffman et al. utilized several models and experimental approaches, including DiI fate mapping in chick, quail-chick chimeras, time-lapse video tracing of cell migration, Cre/loxP transgenic reporter fate mapping, and transgenic ablation of Kdr in the SHF lineage (via Mesp1Cre and Isl1Cre) and endothelial cells (via Tie2-Cre). Although these studies did not address the role of the Mef2c-Cre expressing cells of the SHF, they do suggest that the OFT endocardium is derived, at least in part, from vascular endothelial cells that have diverged from myocardial lineages within SHF progenitors.
In contributing to the developing embryonic heart, the SHF relies on several signaling pathways to mediate its effects. Specifically, FGF signaling critically intersects with SHF function during OFT development. Ablation of Fgf15 results in OFT defects, including DORV and PTA,228 while global deletion of Fgf8 results in early embryonic death229 and Fgf10 results in normal heart development230 (not informative for OFT morphogenesis). Milder assault in Fgf8 hypomorphs demonstrate dysfunctional OFT remodeling with a range of defects including high penetrance of DORV and a relatively low penetrance of TGA and PTA.62,63 Studies utilizing a myriad of SHF- and NCC-specific Cre's to conditionally ablate Fgf8 have demonstrated an autocrine loop of FGF8 signaling from within the SHF that in some yet-undefined way affects CNC morphogenesis in a paracrine manner. Both autocrine and paracrine FGF8 signaling are critical for OFT morphogenesis as conditional ablation of Fgf8 utilizing Isl1Cre results in fully penetrant PTA at E18-P0.172 Remarkably, Fgf8 heterozygotes developed normally, indicating functional allelic reserve. Additionally, genetic deletion of the sequences that code for the `c' isoforms of FGFR1 and FGFR2 in Isl1Creexpressing cells results in failed SHF morphogenesis and defective cardiac formation, including DORV and PTA (demonstrated previously by Park et al.64 and illustrated in Figure 6). Caveats exist as there is interaction between Fgf8 and genes critical for SHF morphogenesis. For example, conditional ablation of Fgf8 in Isl1Creexpressing cells results in diminished expression of early embryonic Bmp4, Mef2c, Isl1, Wnt11 (disruption of each is independently associated with OFT defects including TGA and PTA73,195,196,231). Further supporting genetic interaction between Isl1 and Fgf8, global deletion of Isl1 results in downregulation of Fgf8 within the SHF.94 It is important to note that the Isl1Cre used in the Fgf8 conditional knockout studies is a loss-of-function allele, and therefore may cause off-target defects due to genetic interaction with genes that are targeted for ablation; however, no apparent morphological or molecular disturbances were observed in isolation with mice containing Isl1Cre.172 Additionally, several fate-mapping studies have demonstrated that Isl1 transgene expression is broad (including cells that form the right as well as LV, OFT, and atria), and recent intersectional fate mapping utilizing both Flp and Cre recombinases demonstrate that some CNC (but certainly not all) can also transiently express the Isl1Cre transgene during migration through the SHF (demonstrated by Engleka et al.187 and shown in Figure 7). In light of these data, the promiscuous expression of Isl1Cre must be taken into consideration when interpreting the functional role of genes critical for OFT morphogenesis. These data also underscore the importance of discovering and designing specific tools to demarcate and conditionally manipulate specific lineages involved in OFT development and remodeling. For example, the Isl1 F2 enhancer transgene is more limited in expression pattern than the full length Isl1Crepromoter,232 and, as such, may prove to be a viable tool for segregating SHF and NCC contribution to OFT morphogenesis. Mef2c demonstrates restricted expression within the SHF lineage of the developing heart, with strong expression within the RV and OFT myocardium but is excluded from the LV and atria.26Mef2c expression may also occur in the endocardium and endocardial cushions of the OFT conus and truncus; however, recent data have demonstrated that Mef2c-Cre fails to ablate calcineurin B expression within endocardial cells and endocardially derived mesenchymal cells.34 Combined, these data suggest that either the Mef2c-Cre may demonstrate differential functional expression patterns based on background genetics or that Mef2c-Cre expressing SHF lineage may not contribute to the endocardium or endocardial cushions as previously suggested. Regardless, it is clear that new and better defined (or more restricted) recombinase and lineage-mapping tools are required to fully elucidate mechanisms critical for OFT morphogenesis. Additional resources on cutting edge techniques and future directions of recombinase technologies for cardiac development are reviewed elsewhere.233
FIGURE 6.

FGF signaling within the Isl1Cre lineage critical for outflow tract (OFT) morphogenesis. Deletion of the sequences that code for the `c' isoforms of FGFR1 and FGFR2 in double heterozygote mice demonstrate grossly (a,b) normal OFT rotation, septation, and orientation. However, Isl1Cre-conditional Fgfr1c and 2c isoform mutants demonstrate double outlet right ventricle (DORV) (c,d) and PTA (e,f). Histological analysis confirmed that OFT develop normally in Fgfr1c/Fgfr2c double heterozygote mice (b), but that combined conditional ablation of Fgfr1c and Fgfr2c isoforms results in OFT defects including DORV (d) and type-I persistent truncus arteriosus (PTA) (f). Ao, aorta; co, conus; PA, pulmonary artery; RV, right ventricle; TA, truncus arteriosus. (Reprinted with permission from Ref 181. Copyright 2008 The Company of Biologists Ltd)
FIGURE 7.

Isl1Cre is functionally expressed in migrating neural crest cells (NCC). Using intersectional fate mapping via Isl1Cre and Wnt1-Flpe along with the RC::FrePe indicator mouse line (which reports dual Flpe and Cre recombination187), it has been recently demonstrated that some Isl1 derivatives in the cardiac outflow tract (OFT) can derive from Wnt1-expressing NCC. (a) RC::FrePe mouse embryos positive for Wnt1-Flpe develop normally, and when Isl1Cre lineage is fate mapped using mCherry red fluorescence, NCC migrating into the craniofacial region maintain mCherry fluorescence in the absence of Isl1Cre expression (b, arrowhead). Additionally, Wnt1-Flpe mapped NCC-derived dorsal root ganglia (DRGs) demonstrate eGFP fluorescence (c, arrows). However, cardiac neural crest (CNC) populating the OFT cushions demonstrate both mCherry (d) and eGFP (e) fluorescence suggesting that some (but certainly not all) of Wnt1-Flpe marked CNC populating the OFT endocardial cushions have expressed Isl1Cre at some time earlier. (Reprinted with permission from Ref 187. Copyright 2012 Wolters Kluwer Health)
In addition to SHF contribution to the OFT endocardium, EMT is also critical for conal OFT remodeling. Conditional deletion of the EMT regulator Nf1 via Tie2-Cre within the endothelial lineage results in enlarged hyperproliferative OFT cushions resulting in failed re-alignment and DORV in mice.67 Tie2-cre is initially expressed within the overlying endocardial cells and later in cells having undergone EMT that are detected as early as E9.25 in the OFT conal endocardial cushions (reviewed in Ref 234; Table 3). Additionally, CNC colonization of the OFT truncus is not altered following Tie2-Cre-mediated Nf1 deletion and septation also occurs normally following Wnt1-Cre- and 1.6 kb Pax3-Cre-mediated Nf1 deletion.67 Similarly, TGFB signaling within the OFT is vital for normal OFT remodeling.57,70,235,236 Although TGFB1, 2, and 3 ligands each exhibit restricted spatiotemporal expression patterns within the developing OFT, only global Tgfb2 knockout embryos exhibit enlarged OFT cushions and DORV.70 While conditional deletion of Smad4, a downstream regulator of TGFB signaling, within CNC via Wnt1-Cre results in PTA (see PTA section above), conditional deletion of Smad4 within the myocardium utilizing α skeletal muscle actin Cre (Mu-Cre), results in DORV with concomitant VSD, thinned myocardium, reduced trabecularization, and mid-gestational lethality.68 These results demonstrate a multi-lineage TGFB signaling requirement during OFT septation and suggest that TGFB signaling plays a cell-autonomous role in both CNC morphogenesis as well as myocardial reorganization that affects OFT alignment. Perhaps more intriguing is the role of Smad4 within the myocardium (unveiled using Mu-Cre mediated recombination) that has a non-cell-autonomous effect on endocardial cushions that mediate OFT alignment. Both conus (endothelial derived) and truncus (CNC derived) endocardial cushions have been implicated in controlling OFT alignment and are both spatiotemporally adjacent to the SHF-derived myocardial cuff during OFT alignment. These results may also indicate that, while CNC primarily mediate OFT septation, failed OFT alignment is a convergent phenotype for multiple mechanisms operating within several cellular lineages of the developing heart. These data also support a separate contribution of CNC and endothelial cells to the truncus and conus endocardial cushions, respectively.
Although BMP signaling is critical for conus cushion formation and truncal cushion septation, BMP signaling has also been implicated in OFT alignment as Bmp2/Bmp4 double heterozygous embryos exhibit partially penetrant DORV.60 Further, global Bmpr2l deletion results in fully penetrant DORV with associated VSD, atrioventricular canal defects, and valve leaflet thickening.61 However, both OFT alignment and myocardial development was normal when the Bmpr2 was deleted in either myocardial or conal Tie2-Cre-positive lineage, but the aorta was abnormally positioned when Bmpr2 was deleted within the CNC via Wnt1-Cre. These results further established that truncal endocardial cushions (as well as conal cushions) are required for proper positioning of the aorta. Combined, these results underline the critical necessity of BMP signaling within the endocardial cushions for normal OFT development.
Signaling factors outside the TGFB superfamily have also been shown to play significant roles in cardiac development such as Presenilin1 and Notch signaling. Presenilin1 enzymatically regulates intracellular cleavage of multiple signaling molecules and transcription factors, including Notch.237 Notch is a membranous protein that, upon binding to its ligand Jagged1, recruits the γ -secretase Presenlin-1 (PSEN1) complex to promote cleavage of the Notch intracellular domain that translocates to the nucleus and forms transcriptional complexes with coactivators to mediate transcriptional regulation. Significantly, Notch signaling has been directly implicated in septation and OFT alignment. While dominant-negative inhibition of Notch signaling within the CNC lineage results in OA (see OA section below), global deletion of Psen1 results in fully penetrant DORV with concomitant VSD and pulmonary stenosis.238 Significantly, Notch inhibition within the SHF affected the development of neighboring OFT lineages, including reduced CNC migration and defective EMT within the OFT endocardial cushions resulting in hypoplastic cushions and reduced BMP signaling.114 These results not only demonstrate a clear role for Notch signaling in OFT morphogenesis, but suggest that OFT alignment defects occur on a sliding scale or continuum. Complete inhibition of Notch signaling by deletion of its downstream effector Psen1 results in the severe OFT alignment defect of DORV, but Notch inhibition by a competitive antagonist results in the mild OFT alignment defect of OA.
As expected, regulation of cell survival within the primitive conal endocardial cushions following EMT is critical for normal OFT morphogenesis and prevention of DORV. BIRC5 (formerly known as Survivin) is a key member of the inhibitor of apoptosis family that prevents cell-programmed death.239 Indeed, targeted deletion of Birc5 in endothelial cells utilizing Tie1-Cre184 greatly reduces conal endocardial cushion size and results in failed alignment of the OFT.59 Although normal CNC migration and colonization of the OFT truncus results in correct septation, the hypoplastic Tie1-Cre;Birc5 mutant conal cushions prevent normal OFT remodeling, rotation, and alignment resulting in DORV. These data further emphasize the distinct roles of the truncal and conal endocardial cushions and significance of apoptosis regulation during OFT remodeling. Similarly, studies using retrovirus overexpression of HIF1A in embryonic chick OFT myocardium prevented apoptosis of OFT cardiomyocytes, resulting in malrotation of the aorta, defects in the subpulmonic infundibulum associated with a small RV, and increased OFT mesenchyme with failure of semil-unar valve formation.240 These studies establish that selective disruption of endothelial-derived conus cushion remodeling without affecting CNC-derived truncus cushion septation can result in DORV. Additionally, myocardial cuff apoptosis plays a role in chick OFT rotation, although this mechanism of OFT remodeling has not been formally demonstrated in mammals. Furthermore, CNC-mediated septation of the OFT truncus is not dependent on the function of conus endocardial cushions (endothelial-derived) during OFT alignment.
What Developmental Biology Can Tell Us About the Mechanism/s that Cause TGA
TGA is the most common cyanotic CHD present in neonatal patients. In TGA, the aorta aberrantly exits from the anatomical RV whereas the pulmonary trunk aberrantly exits from the anatomical LV. There are two types of TGA: D-type and L-type. D-type TGA is very common and accounts for 5% of all cases of CHD241 with an incidence of 1 in 3500–5000 live births,242 while the relatively uncommon L-type TGA occurs when the ventricular morphology is interchanged in conjunction with exchanged great arteries. Given its clinical significance, we will focus on D-type TGA (herein referred to as TGA). TGA can occur either in tandem with a VSD or in isolation and is principally a result of failed OFT rotation and is governed by endocardial cushion formation and remodeling.
Organ heterotaxy is commonly associated with TGA. Normal left-right patterning depends on asymmetric TGFB signaling on the left side of the embryo.243,244 Abnormalities in left-right signaling may be evident as soon as the linear heart tube begins to loop and the embryo undergoes ventral folding.245,246 Of particular importance, the activin-like subclass of TGFB receptors have been tightly linked to left-right heterotaxy and TGA in both humans and mouse models. Global deletion of Acvr2b results in laterality defects and nearly full penetrance of TGA in mice.73 CFC1, another activin-like TGFB receptor, is important for normal development of the OFT as both global deletion in mice74 or mutations of the human ortholog CFC1247 result in aberrant left-right patterning and TGA pathogenesis. As left-right patterning is initiated early in development, usually preceding heart tube elongation, these effects suggest that activin-like TGFB receptors play an early role in embryogenesis of OFT morphogenesis. Moreover, activin-like TGFB receptor ligands have also been implicated in TGA, further strengthening the association between left-right heterotaxy, TGFB signaling, and TGA. For instance, GDF1 is an endogenous ligand of activin-like TGFB receptors and global deletion of Gdf1 in mice75 results in TGA. Significantly, TGA can also be found in patients with genetic mutations of GDF1.248 Furthermore, ectopic expression of mouse GDF1 protein within developing Xenopus embryos results in laterality defects,249 suggesting TGFB in left-right signaling is evolutionarily conserved. Finally, a congenital splice site mutation in NODAL (a ligand-agonist of activin-like TGFB receptors) has also been directly associated with TGA and heterotaxy.250
Although both TGFB receptor mutation and agonist mis-expression studies have demonstrated the importance of left-right patterning, transcriptional regulation upstream of NODAL has also been associated with TGA and heterotaxy. Inactivating mutations in the gene that codes for ZIC3, a zinc finger transcription factor that regulates Nodal expression in the early embryo, also result in heterotaxy and TGA.251 Combined, these results demonstrate that TGFβ signaling via ZIC3-NODAL/GDF/CFC1 pathway critically mediates left-right patterning responsible for normal OFT development. Intriguingly, although missense mutations in human ACVR2B can result in heterotaxy, it does not result in TGA.252 Thus, left-right asymmetry in itself is not sufficient to produce TGA, and while strong and divergent evidence support a role of TGFB in TGA, the mechanisms and cell lineages underlying left-right signaling to induce appropriate OFT rotation are not well described.
As discussed previously, both ECM-rich conal and truncal endocardial cushion morphogenesis are critical for normal OFT rotation and septation. Interestingly, global knockout of Hspg2, which is highly expressed in extracellular matrices, results in failed OFT rotation and TGA76–78 associated with abnormal cushion morphogenesis. Unlike many other genetic models that have a relatively low penetrance of TGA, global loss of Hspg2 causes almost full penetrance of TGA with a very low incidence of other cardiac defects.77 In Hspg2-null mice, truncal endocardial cushions are initially hyperplastic early in OFT remodeling, possibly due to increased CNC migration into the OFT.253 Although the endocardial cushions surprisingly return to normal size during later remodeling events, early hyperplasia of the truncal endocardial cushions in Hspg2-null mice appears sufficient to impede OFT rotation, thereby preventing appropriate orientation of the aorta and pulmonary trunk culminating in TGA.253 Intriguingly, the conal endocardial cushions of Hspg2 nulls did not exhibit any hyperplasia, and as these are not populated by CNC derivatives,77 this may suggest that CNC primarily mediate Hspg2-dependent OFT rotation. Combined, these data suggest cellular interactions between CNC-derived truncus and endothelial-derived conus cushions play a crucial role in OFT orientation and prevention of TGA. Additional potential mechanisms include hemodynamic changes due to restricted blood flow through the OFT adversely affecting OFT alignment, although the molecular mechanisms underlying this hypothesis remain undefined.
What Developmental Biology Can Tell Us About the Mechanism/s that Cause OA
OA is defined as abnormal positioning of the aorta directly above the ventricular septum. There are no clear molecular mechanisms that have been directly linked to OA, and OA is a common comorbidity of both Alagille syndrome and TOF. The development of OA in TOF is likely a secondary effect of altered hemodynamics implicating a uniquely strong role for normal blood flow and pressure in OA. Due to the paucity of isolated OA models we will include discussion of mouse models that develop Alagille syndrome and TOF in this section.
Notch signaling has been implicated in several human diseases,254 including Alagille syndrome, which is an autosomal-dominant multisystem disorder with frequent OA. Patients typically present within 3 months of life with intrahepatic cholestasis, but CHDs and OFT defects (including OA) are the most common comorbidities.255 Mutations in either Notch2,256 or its ligand Jagged1,257 have been causatively linked to Alagille syndrome and OFT defects. Further supporting an instrumental role for Notch signaling in OFT remodeling, mice heterozygous for both Notch2 and Jag1 exhibit fully penetrant OA with frequent pulmonary stenosis, VSD, and atrial-septal defects (ASD).258 Global deletion of Hes1, a mediator of Notch signaling expressed within the pharyngeal mesoderm and endoderm, results in decreased SHF proliferation and CNC colonization in the RV and OFT, respectively, with OA and VSD at low penetrance.84 Furthermore, conditional inhibition of Notch signaling, using a dominant-negative inhibitor of Notch (DNMAML259), induced within Wnt1-Cre or Pax3-Cre CNC resulted in OA, pulmonary stenosis, VSD, and ASD.81 CNC migration and cell numbers were not altered by inhibition of Notch signaling within the CNC, although CNC differentiation into smooth muscle cells surrounding the AAs was greatly diminished. These results suggest that Notch signaling is not important for appropriate CNC number, specification, or migration. However, the similar phenotypes in both Notch2/Jag1 heterozygotes and conditional pan-Notch inhibition within the CNC suggest that NOTCH2/JAG1 signaling within CNC is critical for terminal function of the CNC following colonization of the OFT.
Altered SHF morphogenesis has also been shown to result in OA. Ablation of arterial pole progenitors located on the right side of chick embryos leads to OA and pulmonary stenosis or atresia, which are key components of TOF.27 Moreover, similar to the Hes1 mouse knockout data described above, manipulations in chick embryos that interfere with proliferation of SHF progenitors can also cause OA.23,97
OA is rarely reported in mouse models of OFT defects, and there are currently no genetic models of isolated OA. It is interesting to note that the defining characteristics of OA and the much more commonly reported DORV are very similar. In fact, the only structural distinction between DORV and OA is the relative positioning of the aorta with respect to the ventricular septum. OA, DORV, and TGA (also a rotational defect) may represent a common mechanism that occurs along a continuum of disease severity. In other words, mild (OA), moderate (DORV), and severe (TGA) OFT rotation failure may arise from mild to severe presentation of common mechanisms critical for OFT rotation. Data gathered from patients and human embryos suggest that the OFT rotates during remodeling.260 Although some investigators have called for more formal studies to extensively define OFT rotation,101 studies using radioactive tattoos in chick embryos support the necessity of OFT rotation in normal morphogenesis.145 Some have noted that DORV and TGA share failed OFT rotation, but that TGA also demonstrates a leftward shift to allow pulmonary communication with the LV.261 In this sense, it is reasonable to think that OA would be failed rotation of the OFT that does not shift leftward quite as much as TGA, but more than DORV. It will be interesting in the future to determine whether OA, DORV, and TGA defects may all share a common mechanism underlying defective OFT rotation and medial movement.
SUMMARY
The advent of new cell lineage, fate mapping, and gene trapping tools will enable more precise conditional manipulation within the CNC, SHF, myocardium, endocardium, and various cushions during cardiac development. This will be vital to establishing a full understanding of their respective contributions during OFT development. Moreover, the ever-increasing stock of mouse mutants with OFT defects will provide fresh opportunities to define hierarchies and pathways. The convoluted interactions of the multitude of different signaling pathways in different cell lineages at various times may require thoughtful compound genetic mouse models to begin to unravel the spatiotemporal intricacies at work in OFT development. Additionally, the expanding list of animal models with structural OFT defects that exhibit normal CNC and SHF function will have to be addressed, and functional analysis may aid with these exciting investigations. The ability to establish more concretely the timeframe, cell lineages, and genetic interactions critical to the pathomechanism of CHDs will undoubtedly provide insight into the pathogenesis, and, eventually, the prevention or genetic intervention of CHD.
ACKNOWLEDGMENTS
We are grateful to members of the Conway laboratory for their continuing support and insights. While we have tried to comprehensively review as much of the current cardiovascular development literature as possible, we regret that our review cannot include the entire field's many exciting findings. These studies were supported, in part, by Riley Children's Foundation, Indiana University Department of Pediatrics (Neonatal-Perinatal Medicine), and R01 HL60714 (SJC).
REFERENCES
- 1.Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation. 1971;43:323–332. doi: 10.1161/01.cir.43.3.323. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 3.van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 4.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O'Donnell C, Kittner S, et al. Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 5.Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- 6.Sadler TW, Langman J. Langman's Essential Medical Embryology. Lippincott Williams & Wilkins; Philadelphia, PA: 2005. [Google Scholar]
- 7.Schoenwolf GC, Larsen WJ. Larsen's Human Embryology. 4th ed. Churchill Livingstone/Elsevier; Philadelphia, PA: 2009. [Google Scholar]
- 8.Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol. 2006;21:180–184. doi: 10.1097/01.hco.0000221578.18254.70. [DOI] [PubMed] [Google Scholar]
- 9.Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 10.MacGrogan D, Luna-Zurita L, de la Pompa JL. Notch signaling in cardiac valve development and disease. Birth Defects Res A Clin Mol Teratol. 2011;91:449–459. doi: 10.1002/bdra.20815. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava D. Heart disease: an ongoing genetic battle? Nature. 2004;429:819–822. doi: 10.1038/429819a. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Garg V. Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci. 2006;63:1141–1148. doi: 10.1007/s00018-005-5532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conway SJ, Riley PR. Current state of congenital heart research and clues to future directions. Birth Defects Res A Clin Mol Teratol. 2011;91:421–422. doi: 10.1002/bdra.20822. [DOI] [PubMed] [Google Scholar]
- 15.Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. 2010;68:281–285. doi: 10.1203/PDR.0b013e3181ed17e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 17.Williams JM, de Leeuw M, Black MD, Freedom RM, Williams WG, McCrindle BW. Factors associated with outcomes of persistent truncus arteriosus. J Am Coll Cardiol. 1999;34:545–553. doi: 10.1016/s0735-1097(99)00227-2. [DOI] [PubMed] [Google Scholar]
- 18.Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- 19.Conway SJ, Henderson DJ, Kirby ML, Anderson RH, Copp AJ. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc Res. 1997;36:163–173. doi: 10.1016/s0008-6363(97)00172-7. [DOI] [PubMed] [Google Scholar]
- 20.Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- 21.Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–3080. doi: 10.1242/dev.128.16.3071. [DOI] [PubMed] [Google Scholar]
- 22.Snider P, Olaopa M, Firulli AB, Conway SJ. Cardiovascular development and the colonizing cardiac neural crest lineage. ScientificWorldJournal. 2007;7:1090–1113. doi: 10.1100/tsw.2007.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18:101–110. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olaopa M, Zhou HM, Snider P, Wang J, Schwartz RJ, Moon AM, Conway SJ. Pax3 is essential for normal cardiac neural crest morphogenesis but is not required during migration nor outflow tract septation. Dev Biol. 2011;356:308–322. doi: 10.1016/j.ydbio.2011.05.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly RG, Buckingham ME. The anterior heartforming field: voyage to the arterial pole of the heart. Trends Genet. 2002;18:210–216. doi: 10.1016/s0168-9525(02)02642-2. [DOI] [PubMed] [Google Scholar]
- 26.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 27.Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to Tetralogy of Fallot and pulmonary atresia. Dev Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 28.de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 29.Phoon CK, Ji RP, Aristizabal O, Worrad DM, Zhou B, Baldwin HS, Turnbull DH. Embryonic heart failure in NFATc1−/− mice: novel mechanistic insights from in utero ultrasound biomicroscopy. Circ Res. 2004;95:92–99. doi: 10.1161/01.RES.0000133681.99617.28. [DOI] [PubMed] [Google Scholar]
- 30.Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, Baldwin HS, Zhou B. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ya J, Schilham MW, de Boer PA, Moorman AF, Clevers H, Lamers WH. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ Res. 1998;83:986–994. doi: 10.1161/01.res.83.10.986. [DOI] [PubMed] [Google Scholar]
- 32.Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardialmesenchymal transformation in the developing heart. Development. 1998;125:4359–4367. doi: 10.1242/dev.125.22.4359. [DOI] [PubMed] [Google Scholar]
- 33.Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, Pawlowski S, Rajan S, Doetschman T. Ligand-specific function of transforming growth factor β in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238:431–442. doi: 10.1002/dvdy.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin CY, Lin CJ, Chen CH, Chen RM, Zhou B, Chang CP. The secondary heart field is a new site of calcineurin/Nfatc1 signaling for semilunar valve development. J Mol Cell Cardiol. 52:1096–1102. doi: 10.1016/j.yjmcc.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, Epstein JA. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. doi: 10.1172/JCI44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 37.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105:408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markwald RR, Norris RA, Moreno-Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases: valvular heart diseases. Ann N Y Acad Sci. 2010;1188:177–183. doi: 10.1111/j.1749-6632.2009.05098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jain R, Rentschler S, Epstein JA. Notch and cardiac outflow tract development. Ann N Y Acad Sci. 2010;1188:184–190. doi: 10.1111/j.1749-6632.2009.05099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y, High FA, Epstein JA, Radice GL. N-cadherin is required for neural crest remodeling of the cardiac outflow tract. Dev Biol. 2006;299:517–528. doi: 10.1016/j.ydbio.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, et al. Identification of a Wnt/Dvl/β-Catenin–Pitx2 pathway mediating cell-type-specific proliferation during development. Cell. 2002;111:673–685. doi: 10.1016/s0092-8674(02)01084-x. [DOI] [PubMed] [Google Scholar]
- 43.Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 44.Ai D, Liu W, Ma L, Dong F, Lu MF, Wang D, Verzi MP, Cai C, Gage PJ, Evans S, et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev Biol. 2006;296:437–449. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vallejo-Illarramendi A, Zang K, Reichardt LF. Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. J Clin Invest. 2009;119:2218–2230. doi: 10.1172/JCI38194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas PS, Kim J, Nunez S, Glogauer M, Kaartinen V. Neural crest cell-specific deletion of Rac1 results in defective cell-matrix interactions and severe craniofacial and cardiovascular malformations. Dev Biol. 2010;340:613–625. doi: 10.1016/j.ydbio.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- 48.Jia Q, McDill BW, Li SZ, Deng C, Chang CP, Chen F. Smad signaling in the neural crest regulates cardiac outflow tract remodeling through cell autonomous and non-cell autonomous effects. Dev Biol. 2007;311:172–184. doi: 10.1016/j.ydbio.2007.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nie X, Deng CX, Wang Q, Jiao K. Disruption of Smad4 in neural crest cells leads to mid-gestation death with pharyngeal arch, craniofacial and cardiac defects. Dev Biol. 2008;316:417–430. doi: 10.1016/j.ydbio.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang S, Snider P, Firulli AB, Conway SJ. Trigenic neural crest-restricted Smad7 over-expression results in congenital craniofacial and cardiovascular defects. Dev Biol. 2010;344:233–247. doi: 10.1016/j.ydbio.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de Wetering M, Verbeek S, Lamers WH, Kruisbeek AM, Cumano A, et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380:711–714. doi: 10.1038/380711a0. [DOI] [PubMed] [Google Scholar]
- 52.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 53.Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 54.Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 55.Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, Heidt AB, Mori AD, Arruda EP, Gertsenstein M, Georges R, Davidson L, Mo R, et al. Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development. 2005;132:2463–2474. doi: 10.1242/dev.01827. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Nagy A, Larsson J, Dudas M, Sucov HM, Kaartinen V. Defective ALK5 signaling in the neural crest leads to increased postmigratory neural crest cell apoptosis and severe outflow tract defects. BMC Dev Biol. 2006;6:51. doi: 10.1186/1471-213X-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFβ receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–429. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 58.Schleiffarth JR, Person AD, Martinsen BJ, Sukovich DJ, Neumann A, Baker CV, Lohr JL, Cornfield DN, Ekker SC, Petryk A. Wnt5a is required for cardiac outflow tract septation in mice. Pediatr Res. 2007;61:386–391. doi: 10.1203/pdr.0b013e3180323810. [DOI] [PubMed] [Google Scholar]
- 59.Zwerts F, Lupu F, De Vriese A, Pollefeyt S, Moons L, Altura RA, Jiang Y, Maxwell PH, Hill P, Oh H, et al. Lack of endothelial cell survivin causes embryonic defects in angiogenesis, cardiogenesis, and neural tube closure. Blood. 2007;109:4742–4752. doi: 10.1182/blood-2006-06-028068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldman DC, Donley N, Christian JL. Genetic interaction between Bmp2 and Bmp4 reveals shared functions during multiple aspects of mouse organogenesis. Mech Dev. 2009;126:117–127. doi: 10.1016/j.mod.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beppu H, Malhotra R, Beppu Y, Lepore JJ, Parmacek MS, Bloch KD. BMP type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Dev Biol. 2009;331:167–175. doi: 10.1016/j.ydbio.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- 63.Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development. 2008;135:3599–3610. doi: 10.1242/dev.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holler KL, Hendershot TJ, Troy SE, Vincentz JW, Firulli AB, Howard MJ. Targeted deletion of Hand2 in cardiac neural crest-derived cells influences cardiac gene expression and outflow tract development. Dev Biol. 2010;341:291–304. doi: 10.1016/j.ydbio.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishii M, Han J, Yen HY, Sucov HM, Chai Y, Maxson RE., Jr. Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development. 2005;132:4937–4950. doi: 10.1242/dev.02072. [DOI] [PubMed] [Google Scholar]
- 67.Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA. Nf1 has an essential role in endothelial cells. Nat Genet. 2003;33:75–79. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Azhar M, Wang PY, Frugier T, Koishi K, Deng C, Noakes PG, McLennan IS. Myocardial deletion of Smad4 using a novel α skeletal muscle actin Cre recombinase transgenic mouse causes misalignment of the cardiac outflow tract. Int J Biol Sci. 2010;6:546–555. doi: 10.7150/ijbs.6.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brewer S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP-2α in cardiac outflow tract morphogenesis. Mech Dev. 2002;110:139–149. doi: 10.1016/s0925-4773(01)00579-2. [DOI] [PubMed] [Google Scholar]
- 70.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-β(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- 72.Katz SG, Williams A, Yang J, Fujiwara Y, Tsang AP, Epstein JA, Orkin SH. Endothelial lineage-mediated loss of the GATA cofactor Friend of GATA 1 impairs cardiac development. Proc Natl Acad Sci U S A. 2003;100:14030–14035. doi: 10.1073/pnas.1936250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997;11:1812–1826. doi: 10.1101/gad.11.14.1812. [DOI] [PubMed] [Google Scholar]
- 74.Gaio U, Schweickert A, Fischer A, Garratt AN, Muller T, Ozcelik C, Lankes W, Strehle M, Britsch S, Blum M, et al. A role of the cryptic gene in the correct establishment of the left-right axis. Curr Biol. 1999;9:1339–1342. doi: 10.1016/s0960-9822(00)80059-7. [DOI] [PubMed] [Google Scholar]
- 75.Rankin CT, Bunton T, Lawler AM, Lee SJ. Regulation of left-right patterning in mice by growth/differentiation factor-1. Nat Genet. 2000;24:262–265. doi: 10.1038/73472. [DOI] [PubMed] [Google Scholar]
- 76.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gonzalez-Iriarte M, Carmona R, Perez-Pomares JM, Macias D, Costell M, Munoz-Chapuli R. Development of the coronary arteries in a murine model of transposition of great arteries. J Mol Cell Cardiol. 2003;35:795–802. doi: 10.1016/s0022-2828(03)00134-2. [DOI] [PubMed] [Google Scholar]
- 78.Arikawa-Hirasawa E, Watanabe H, Takami H, Has-sell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–358. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- 79.Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- 80.Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, Yuan S, Prabhakar NR, Herrup K, Conlon RA, et al. The essential role of Cited2, a negative regulator for HIF-1α, in heart development and neurulation. Proc Natl Acad Sci USA. 2002;99:10488–10493. doi: 10.1073/pnas.162371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, de Wit D, Emoto N, Hammer RE. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 83.Yanagisawa H, Hammer RE, Richardson JA, Emoto N, Williams SC, Takeda S, Clouthier DE, Yanagisawa M. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J Clin Invest. 2000;105:1373–1382. doi: 10.1172/JCI7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rochais F, Dandonneau M, Mesbah K, Jarry T, Mattei MG, Kelly RG. Hes1 is expressed in the second heart field and is required for outflow tract development. PLoS One. 2009;4:e6267. doi: 10.1371/journal.pone.0006267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Bueren KL, Papangeli I, Rochais F, Pearce K, Roberts C, Calmont A, Szumska D, Kelly RG, Bhattacharya S, Scambler PJ. Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev Biol. 2010;340:369–380. doi: 10.1016/j.ydbio.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- 87.Yanagisawa H, Hammer RE, Richardson JA, Williams SC, Clouthier DE, Yanagisawa M. Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aortic arch patterning in mice. J Clin Invest. 1998;102:22–33. doi: 10.1172/JCI2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hove JR, Koster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421:172–177. doi: 10.1038/nature01282. [DOI] [PubMed] [Google Scholar]
- 89.Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev Cell. 2006;11:845–857. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 90.Kaufman Nunn M. The Atlas of Mouse Development. Academic Press; London: 1995. [Google Scholar]
- 91.Larsen WJ. Human Embryology. Churchill Livingstone; New York: 1993. [Google Scholar]
- 92.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 93.Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- 94.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van den Berg G, Abu-Issa R, de Boer BA, Hutson MR, de Boer PA, Soufan AT, Ruijter JM, Kirby ML, van den Hoff MJ, Moorman AF. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ Res. 2009;104:179–188. doi: 10.1161/CIRCRESAHA.108.185843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buckingham M, Relaix F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol. 2007;23:645–673. doi: 10.1146/annurev.cellbio.23.090506.123438. [DOI] [PubMed] [Google Scholar]
- 97.Dyer LA, Kirby ML. The role of secondary heart field in cardiac development. Dev Biol. 2009;336:137–144. doi: 10.1016/j.ydbio.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vincent SD, Buckingham ME. How to make a heart: the origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41. doi: 10.1016/S0070-2153(10)90001-X. [DOI] [PubMed] [Google Scholar]
- 99.Abu-Issa R, Waldo K, Kirby ML. Heart fields: one, two or more? Dev Biol. 2004;272:281–285. doi: 10.1016/j.ydbio.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 100.Evans SM, Yelon D, Conlon FL, Kirby ML. Myocardial lineage development. Circ Res. 2010;107:1428–1444. doi: 10.1161/CIRCRESAHA.110.227405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Okamoto N, Akimoto N, Hidaka N, Shoji S, Sumida H. Formal genesis of the outflow tracts of the heart revisited: previous works in the light of recent observations. Congenit Anom (Kyoto) 2010;50:141–158. doi: 10.1111/j.1741-4520.2010.00286.x. [DOI] [PubMed] [Google Scholar]
- 102.Harris IS, Black BL. Development of the endocardium. Pediatr Cardiol. 2010;31:391–399. doi: 10.1007/s00246-010-9642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Franco D, Meilhac SM, Christoffels VM, Kispert A, Buckingham M, Kelly RG. Left and right ventricular contributions to the formation of the interventricular septum in the mouse heart. Dev Biol. 2006;294:366–375. doi: 10.1016/j.ydbio.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 104.Brown CB, Baldwin HS. Neural crest contribution to the cardiovascular system. Adv Exp Med Biol. 2006;589:134–154. doi: 10.1007/978-0-387-46954-6_8. [DOI] [PubMed] [Google Scholar]
- 105.Thompson RP, Sumida H, Abercrombie V, Satow Y, Fitzharris TP, Okamoto N. Morphogenesis of human cardiac outflow. Anat Rec. 1985;213:578–586. 538–579. doi: 10.1002/ar.1092130414. [DOI] [PubMed] [Google Scholar]
- 106.van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, Wessels A. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–490. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- 107.Hutson MR, Zeng XL, Kim AJ, Antoon E, Harward S, Kirby ML. Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. Development. 2010;137:3001–3011. doi: 10.1242/dev.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. 2005;281:66–77. doi: 10.1016/j.ydbio.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 109.Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 110.Liao J, Aggarwal VS, Nowotschin S, Bondarev A, Lipner S, Morrow BE. Identification of downstream genetic pathways of Tbx1 in the second heart field. Dev Biol. 2008;316:524–537. doi: 10.1016/j.ydbio.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 112.Lin L, Bu L, Cai CL, Zhang X, Evans S. Isl1 is upstream of sonic hedgehog in a pathway required for cardiac morphogenesis. Dev Biol. 2006;295:756–763. doi: 10.1016/j.ydbio.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 113.Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–4574. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- 114.High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest. 2009;119:1986–1996. doi: 10.1172/JCI38922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ishii Y, Langberg J, Rosborough K, Mikawa T. Endothelial cell lineages of the heart. Cell Tissue Res. 2009;335:67–73. doi: 10.1007/s00441-008-0663-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–117. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- 117.Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 118.Motoike T, Markham DW, Rossant J, Sato TN. Evidence for novel fate of Flk1+ progenitor: contribution to muscle lineage. Genesis. 2003;35:153–159. doi: 10.1002/gene.10175. [DOI] [PubMed] [Google Scholar]
- 119.Masino AM, Gallardo TD, Wilcox CA, Olson EN, Williams RS, Garry DJ. Transcriptional regulation of cardiac progenitor cell populations. Circ Res. 2004;95:389–397. doi: 10.1161/01.RES.0000138302.02691.be. [DOI] [PubMed] [Google Scholar]
- 120.Cohen-Gould L, Mikawa T. The fate diversity of mesodermal cells within the heart field during chicken early embryogenesis. Dev Biol. 1996;177:265–273. doi: 10.1006/dbio.1996.0161. [DOI] [PubMed] [Google Scholar]
- 121.Wei Y, Mikawa T. Fate diversity of primitive streak cells during heart field formation in ovo. Dev Dyn. 2000;219:505–513. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1076>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 122.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 123.Brown CB, Boyer AS, Runyan RB, Barnett JV. Requirement of type III TGF-β receptor for endocardial cell transformation in the heart. Science. 1999;283:2080–2082. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- 124.Delot EC. Control of endocardial cushion and cardiac valve maturation by BMP signaling pathways. Mol Genet Metab. 2003;80:27–35. doi: 10.1016/j.ymgme.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 125.Zhang J, Chang JY, Huang Y, Lin X, Luo Y, Schwartz RJ, Martin JF, Wang F. The FGF-BMP signaling axis regulates outflow tract valve primordium formation by promoting cushion neural crest cell differentiation. Circ Res. 2010;107:1209–1219. doi: 10.1161/CIRCRESAHA.110.225318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Snarr BS, Kern CB, Wessels A. Origin and fate of cardiac mesenchyme. Dev Dyn. 2008;237:2804–2819. doi: 10.1002/dvdy.21725. [DOI] [PubMed] [Google Scholar]
- 127.Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 128.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 129.Bolande RP. The neurocristopathies: A unifying concept of disease arising in neural crest maldevelopment. Human pathology. 1974;5:409. doi: 10.1016/s0046-8177(75)80115-8. [DOI] [PubMed] [Google Scholar]
- 130.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 131.Tucker RP. Neural crest cells: a model for invasive behavior. Int J Biochem Cell Biol. 2004;36:173–177. doi: 10.1016/s1357-2725(03)00243-7. [DOI] [PubMed] [Google Scholar]
- 132.Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- 133.Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet. 2000;97:280–288. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 134.Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 135.Perris R, Perissinotto D. Role of the extracellular matrix during neural crest cell migration. Mech Dev. 2000;95:3–21. doi: 10.1016/s0925-4773(00)00365-8. [DOI] [PubMed] [Google Scholar]
- 136.Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- 137.Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012 doi: 10.1016/j.ydbio.2011.12.041. [DOI] [PubMed] [Google Scholar]
- 138.Le Douarin N, Kalcheim C. The Neural Crest. 2nd ed. Cambridge University Press; Cambridge, UK and New York: 1999. [Google Scholar]
- 139.Trainor P, Krumlauf R. Development. Riding the crest of the Wnt signaling wave. Science. 2002;297:781–783. doi: 10.1126/science.1075454. [DOI] [PubMed] [Google Scholar]
- 140.Hall BK. The neural crest and neural crest cells: discovery and significance for theories of embryonic organization. J Biosci. 2008;33:781–793. doi: 10.1007/s12038-008-0098-4. [DOI] [PubMed] [Google Scholar]
- 141.Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456:957–961. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, Mayor R. Collective chemotaxis requires contact-dependent cell polarity. Dev Cell. 2010;19:39–53. doi: 10.1016/j.devcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002;3:453–461. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- 144.High F, Epstein JA. Signalling pathways regulating cardiac neural crest migration and differentiation. Novartis Found Symp. 2007;283:152–161. doi: 10.1002/9780470319413.ch12. discussion 161–154; 238–141. [DOI] [PubMed] [Google Scholar]
- 145.Bajolle F, Zaffran S, Kelly RG, Hadchouel J, Bonnet D, Brown NA, Buckingham ME. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ Res. 2006;98:421–428. doi: 10.1161/01.RES.0000202800.85341.6e. [DOI] [PubMed] [Google Scholar]
- 146.Bostrom MP, Hutchins GM. Arrested rotation of the outflow tract may explain double-outlet right ventricle. Circulation. 1988;77:1258–1265. doi: 10.1161/01.cir.77.6.1258. [DOI] [PubMed] [Google Scholar]
- 147.Lallier TE, Bronner-Fraser M. A spatial and temporal analysis of dorsal root and sympathetic ganglion formation in the avian embryo. Dev Biol. 1988;127:99–112. doi: 10.1016/0012-1606(88)90192-3. [DOI] [PubMed] [Google Scholar]
- 148.Jiang X, Choudhary B, Merki E, Chien KR, Maxson RE, Sucov HM. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech Dev. 2002;117:115–122. doi: 10.1016/s0925-4773(02)00206-x. [DOI] [PubMed] [Google Scholar]
- 149.Byrd NA, Meyers EN. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev Biol. 2005;284:233–245. doi: 10.1016/j.ydbio.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 150.Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev Dyn. 1996;205:281–292. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 151.Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- 152.Jessen KR, Mirsky R. Signals that determine Schwann cell identity. J Anat. 2002;200:367–376. doi: 10.1046/j.1469-7580.2002.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- 154.Gurjarpadhye A, Hewett KW, Justus C, Wen X, Stadt H, Kirby ML, Sedmera D, Gourdie RG. Cardiac neural crest ablation inhibits compaction and electrical function of conduction system bundles. Am J Physiol Heart Circ Physiol. 2007;292:H1291–H1300. doi: 10.1152/ajpheart.01017.2006. [DOI] [PubMed] [Google Scholar]
- 155.Farrell MJ, Burch JL, Wallis K, Rowley L, Kumiski D, Stadt H, Godt RE, Creazzo TL, Kirby ML. FGF-8 in the ventral pharynx alters development of myocardial calcium transients after neural crest ablation. J Clin Invest. 2001;107:1509–1517. doi: 10.1172/JCI9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Betters E, Liu Y, Kjaeldgaard A, Sundstrom E, Garcia-Castro MI. Analysis of early human neural crest development. Dev Biol. 2010;344:578–592. doi: 10.1016/j.ydbio.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adachi I, Seale A, Uemura H, McCarthy KP, Kimberley P, Ho SY. Morphologic spectrum of truncal valvar origin relative to the ventricular septum: correlation with the size of ventricular septal defect. J Thorac Cardiovasc Surg. 2009;138:1283–1289. doi: 10.1016/j.jtcvs.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 158.Chen L, Ma Y, Kim EY, Yu W, Schwartz RJ, Qian L, Wang J. Conditional ablation of Ezh2 in murine hearts reveals its essential roles in endocardial cushion formation, cardiomyocyte proliferation and survival. PLoS One. 2012;7:e31005. doi: 10.1371/journal.pone.0031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yelbuz TM, Waldo KL, Kumiski DH, Stadt HA, Wolfe RR, Leatherbury L, Kirby ML. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–510. doi: 10.1161/01.cir.0000023044.44974.8a. [DOI] [PubMed] [Google Scholar]
- 160.Yelbuz TM, Waldo KL, Zhang X, Zdanowicz M, Parker J, Creazzo TL, Johnson GA, Kirby ML. Myocardial volume and organization are changed by failure of addition of secondary heart field myocardium to the cardiac outflow tract. Dev Dyn. 2003;228:152–160. doi: 10.1002/dvdy.10364. [DOI] [PubMed] [Google Scholar]
- 161.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Basch ML, Bronner-Fraser M. Neural crest inducing signals. Adv Exp Med Biol. 2006;589:24–31. doi: 10.1007/978-0-387-46954-6_2. [DOI] [PubMed] [Google Scholar]
- 163.Chang CP, Stankunas K, Shang C, Kao SC, Twu KY, Cleary ML. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development. 2008;135:3577–3586. doi: 10.1242/dev.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 165.Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212:191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- 166.Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectodermand endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130:6361–6374. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S. Wnt signalling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- 168.Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- 169.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 170.Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 171.Pietri T, Eder O, Blanche M, Thiery JP, Dufour S. The human tissue plasminogen activator-Cre mouse: a new tool for targeting specifically neural crest cells and their derivatives in vivo. Dev Biol. 2003;259:176–187. doi: 10.1016/s0012-1606(03)00175-1. [DOI] [PubMed] [Google Scholar]
- 172.Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–2433. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- 174.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–2367. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 176.Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development. 1993;119:419–431. doi: 10.1242/dev.119.2.419. [DOI] [PubMed] [Google Scholar]
- 177.Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRα gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- 178.Davis DL, Edwards AV, Juraszek AL, Phelps A, Wessels A, Burch JB. A GATA-6 gene heart-region-specific enhancer provides a novel means to mark and probe a discrete component of the mouse cardiac conduction system. Mech Dev. 2001;108:105–119. doi: 10.1016/s0925-4773(01)00500-7. [DOI] [PubMed] [Google Scholar]
- 179.Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, et al. Epicardial retinoid X receptor α is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci USA. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Agah R, Frenkel PA, French BA, Michael LH, Over-beek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 182.Parsons SA, Millay DP, Wilkins BJ, Bueno OF, Tsika GL, Neilson JR, Liberatore CM, Yutzey KE, Crab-tree GR, Tsika RW, et al. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. 2004;279:26192–26200. doi: 10.1074/jbc.M313800200. [DOI] [PubMed] [Google Scholar]
- 183.Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–5091. doi: 10.1242/dev.129.21.5081. [DOI] [PubMed] [Google Scholar]
- 184.Gustafsson E, Brakebusch C, Hietanen K, Fassler R. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J Cell Sci. 2001;114:671–676. doi: 10.1242/jcs.114.4.671. [DOI] [PubMed] [Google Scholar]
- 185.Tallquist MD, Soriano P, Klinghoffer RA. Growth factor signaling pathways in vascular development. Oncogene. 1999;18:7917–7932. doi: 10.1038/sj.onc.1203216. [DOI] [PubMed] [Google Scholar]
- 186.Kogata N, Arai Y, Pearson JT, Hashimoto K, Hidaka K, Koyama T, Somekawa S, Nakaoka Y, Ogawa M, Adams RH, et al. Cardiac ischemia activates vascular endothelial cadherin promoter in both preexisting vascular cells and bone marrow cells involved in neovascularization. Circ Res. 2006;98:897–904. doi: 10.1161/01.RES.0000218193.51136.ad. [DOI] [PubMed] [Google Scholar]
- 187.Engleka KA, Manderfield LJ, Brust RD, Li L, Cohen A, Dymecki SM, Epstein JA. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ Res. 110:922–926. doi: 10.1161/CIRCRESAHA.112.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liem KF, Jr, Tremml G, Roelink H, Jessell TM. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell. 1995;82:969–979. doi: 10.1016/0092-8674(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 189.Villanueva S, Glavic A, Ruiz P, Mayor R. Posteriorization by FGF, Wnt, and retinoic acid is required for neural crest induction. Dev Biol. 2002;241:289–301. doi: 10.1006/dbio.2001.0485. [DOI] [PubMed] [Google Scholar]
- 190.Allen SP, Bogardi JP, Barlow AJ, Mir SA, Qayyum SR, Verbeek FJ, Anderson RH, Francis-West PH, Brown NA, Richardson MK. Misexpression of noggin leads to septal defects in the outflow tract of the chick heart. Dev Biol. 2001;235:98–109. doi: 10.1006/dbio.2001.0291. [DOI] [PubMed] [Google Scholar]
- 191.Sela-Donenfeld D, Kalcheim C. Regulation of the onset of neural crest migration by coordinated activity of BMP4 and Noggin in the dorsal neural tube. Development. 1999;126:4749–4762. doi: 10.1242/dev.126.21.4749. [DOI] [PubMed] [Google Scholar]
- 192.Selleck MA, Garcia-Castro MI, Artinger KB, Bronner-Fraser M. Effects of Shh and Noggin on neural crest formation demonstrate that BMP is required in the neural tube but not ectoderm. Development. 1998;125:4919–4930. doi: 10.1242/dev.125.24.4919. [DOI] [PubMed] [Google Scholar]
- 193.Pettway Z, Guillory G, Bronner-Fraser M. Absence of neural crest cells from the region surrounding implanted notochords in situ. Dev Biol. 1990;142:335–345. doi: 10.1016/0012-1606(90)90354-l. [DOI] [PubMed] [Google Scholar]
- 194.McMahon JA, Takada S, Zimmerman LB, Fan CM, Harland RM, McMahon AP. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci USA. 2004;101:4489–4494. doi: 10.1073/pnas.0308466101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.McCulley DJ, Kang JO, Martin JF, Black BL. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev Dyn. 2008;237:3200–3209. doi: 10.1002/dvdy.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol. 2000;221:249–258. doi: 10.1006/dbio.2000.9670. [DOI] [PubMed] [Google Scholar]
- 198.Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–220. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- 199.Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–2218. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- 201.Lindsay EA, Baldini A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Hum Mol Genet. 2001;10:997–1002. doi: 10.1093/hmg/10.9.997. [DOI] [PubMed] [Google Scholar]
- 202.Vermot J, Messaddeq N, Niederreither K, Dierich A, Dolle P. Rescue of morphogenetic defects and of retinoic acid signaling in retinaldehyde dehydrogenase 2 (Raldh2) mouse mutants by chimerism with wild-type cells. Differentiation. 2006;74:661–668. doi: 10.1111/j.1432-0436.2006.00094.x. [DOI] [PubMed] [Google Scholar]
- 203.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 204.Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. 2001;2:858–868. doi: 10.1038/35098574. [DOI] [PubMed] [Google Scholar]
- 205.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 206.Baldini A. Dissecting contiguous gene defects: TBX1. Curr Opin Genet Dev. 2005;15:279–284. doi: 10.1016/j.gde.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 207.Roberts C, Ivins SM, James CT, Scambler PJ. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev Dyn. 2005;232:928–938. doi: 10.1002/dvdy.20268. [DOI] [PubMed] [Google Scholar]
- 208.Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Conway SJ, Godt RE, Hatcher CJ, Leatherbury L, Zolotouchnikov VV, Brotto MA, Copp AJ, Kirby ML, Creazzo TL. Neural crest is involved in development of abnormal myocardial function. J Mol Cell Cardiol. 1997;29:2675–2685. doi: 10.1006/jmcc.1997.0499. [DOI] [PubMed] [Google Scholar]
- 210.Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- 211.Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, et al. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129:527–538. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- 212.Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA. 2009;106:13933–13938. doi: 10.1073/pnas.0904744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lee RY, Luo J, Evans RM, Giguere V, Sucov HM. Compartment-selective sensitivity of cardiovascular morphogenesis to combinations of retinoic acid receptor gene mutations. Circ Res. 1997;80:757–764. doi: 10.1161/01.res.80.6.757. [DOI] [PubMed] [Google Scholar]
- 214.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 215.Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: differentiation and apoptosis. Dev Dyn. 1998;212:373–384. doi: 10.1002/(SICI)1097-0177(199807)212:3<373::AID-AJA5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 216.Keyes WM, Sanders EJ. Regulation of apoptosis in the endocardial cushions of the developing chick heart. Am J Physiol Cell Physiol. 2002;282:C1348–C1360. doi: 10.1152/ajpcell.00509.2001. [DOI] [PubMed] [Google Scholar]
- 217.Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J Clin Invest. 2011;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Obler D, Juraszek AL, Smoot LB, Natowicz MR. Double outlet right ventricle: aetiologies and associations. J Med Genet. 2008;45:481–497. doi: 10.1136/jmg.2008.057984. [DOI] [PubMed] [Google Scholar]
- 219.Walters HL, 3rd, Mavroudis C, Tchervenkov CI, Jacobs JP, Lacour-Gayet F, Jacobs ML. Congenital heart surgery nomenclature and database project: double outlet right ventricle. Ann Thorac Surg. 2000;69:S249–S263. doi: 10.1016/s0003-4975(99)01247-3. [DOI] [PubMed] [Google Scholar]
- 220.Bi W, Drake CJ, Schwarz JJ. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev Biol. 1999;211:255–267. doi: 10.1006/dbio.1999.9307. [DOI] [PubMed] [Google Scholar]
- 221.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vong L, Bi W, O'Connor-Halligan KE, Li C, Cserjesi P, Schwarz JJ. MEF2C is required for the normal allocation of cells between the ventricular and sinoatrial precursors of the primary heart field. Dev Dyn. 2006;235:1809–1821. doi: 10.1002/dvdy.20828. [DOI] [PubMed] [Google Scholar]
- 223.von Both I, Silvestri C, Erdemir T, Lickert H, Walls JR, Henkelman RM, Rossant J, Harvey RP, Attisano L, Wrana JL. Foxh1 is essential for development of the anterior heart field. Dev Cell. 2004;7:331–345. doi: 10.1016/j.devcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 224.Seo S, Kume T. Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev Biol. 2006;296:421–436. doi: 10.1016/j.ydbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 225.Kume T, Jiang H, Topczewska JM, Hogan BL. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001;15:2470–2482. doi: 10.1101/gad.907301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 227.Milgrom-Hoffman M, Harrelson Z, Ferrara N, Zelzer E, Evans SM, Tzahor E. The heart endocardium is derived from vascular endothelial progenitors. Development. 2011;138:4777–4787. doi: 10.1242/dev.061192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
- 229.Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Marguerie A, Bajolle F, Zaffran S, Brown NA, Dickson C, Buckingham ME, Kelly RG. Congenital heart defects in Fgfr2-IIIb and Fgf10 mutant mice. Cardiovasc Res. 2006;71:50–60. doi: 10.1016/j.cardiores.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 231.Zhou W, Lin L, Majumdar A, Li X, Zhang X, Liu W, Etheridge L, Shi Y, Martin J, Van de Ven W, et al. Modulation of morphogenesis by noncanonical Wnt signaling requires ATF/CREB family-mediated transcriptional activation of TGFβ2. Nat Genet. 2007;39:1225–1234. doi: 10.1038/ng2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kang J, Nathan E, Xu SM, Tzahor E, Black BL. Isl1 is a direct transcriptional target of Forkhead transcription factors in second-heart-field-derived mesoderm. Dev Biol. 2009;334:513–522. doi: 10.1016/j.ydbio.2009.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Doetschman T, Azhar M. Cardiac-specific inducible and conditional gene targetingin mice. Circ Res. 2012;110 doi: 10.1161/CIRCRESAHA.112.265066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 235.Gaussin V, Morley GE, Cox L, Zwijsen A, Vance KM, Emile L, Tian Y, Liu J, Hong C, Myers D, et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ Res. 2005;97:219–226. doi: 10.1161/01.RES.0000177862.85474.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19:530–535. doi: 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 238.Nakajima M, Moriizumi E, Koseki H, Shirasawa T. Presenilin 1 is essential for cardiac morphogenesis. Dev Dyn. 2004;230:795–799. doi: 10.1002/dvdy.20098. [DOI] [PubMed] [Google Scholar]
- 239.Cheung CH, Cheng L, Chang KY, Chen HH, Chang JY. Investigations of survivin: the past, present and future. Front Biosci. 2011;16:952–961. doi: 10.2741/3728. [DOI] [PubMed] [Google Scholar]
- 240.Liu H, Fisher SA. Hypoxia-inducible transcription factor-1α triggers an autocrine survival pathway during embryonic cardiac outflow tract remodeling. Circ Res. 2008;102:1331–1339. doi: 10.1161/CIRCRESAHA.107.167858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Samanek M, Voriskova M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999;20:411–417. doi: 10.1007/s002469900502. [DOI] [PubMed] [Google Scholar]
- 242.Martins P, Castela E. Transposition of the great arteries. Orphanet J Rare Dis. 2008;3:27. doi: 10.1186/1750-1172-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Shiratori H, Hamada H. The left-right axis in the mouse: from origin to morphology. Development. 2006;133:2095–2104. doi: 10.1242/dev.02384. [DOI] [PubMed] [Google Scholar]
- 244.Whitman M, Mercola M. TGF-β superfamily signaling and left-right asymmetry. Sci STKE. 2001:re1. doi: 10.1126/stke.2001.64.re1. [DOI] [PubMed] [Google Scholar]
- 245.Harvey RP. Patterning the vertebrate heart. Nat Rev Genet. 2002;3:544–556. doi: 10.1038/nrg843. [DOI] [PubMed] [Google Scholar]
- 246.Ramsdell AF. Left-right asymmetry and congenital cardiac defects: getting to the heart of the matter in vertebrate left-right axis determination. Dev Biol. 2005;288:1–20. doi: 10.1016/j.ydbio.2005.07.038. [DOI] [PubMed] [Google Scholar]
- 247.Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M. CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am J Hum Genet. 2002;70:776–780. doi: 10.1086/339079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Karkera JD, Lee JS, Roessler E, Banerjee-Basu S, Ouspenskaia MV, Mez J, Goldmuntz E, Bowers P, Towbin J, Belmont JW, et al. Loss-of-function mutations in growth differentiation factor-1 (GDF1) are associated with congenital heart defects in humans. Am J Hum Genet. 2007;81:987–994. doi: 10.1086/522890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wall NA, Craig EJ, Labosky PA, Kessler DS. Mesendoderm induction and reversal of left-right pattern by mouse Gdf1, a Vg1-related gene. Dev Biol. 2000;227:495–509. doi: 10.1006/dbio.2000.9926. [DOI] [PubMed] [Google Scholar]
- 250.De Luca A, Sarkozy A, Consoli F, Ferese R, Guida V, Dentici ML, Mingarelli R, Bellacchio E, Tuo G, Limongelli G, et al. Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart. 2009;96:673–677. doi: 10.1136/hrt.2009.181685. [DOI] [PubMed] [Google Scholar]
- 251.Megarbane A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, Kassab R, Loiselet J, Bouvagnet P. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet. 2000;8:704–708. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- 252.Kosaki R, Gebbia M, Kosaki K, Lewin M, Bowers P, Towbin JA, Casey B. Left-right axis malformations associated with mutations in ACVR2B, the gene for human activin receptor type IIB. Am J Med Genet. 1999;82:70–76. doi: 10.1002/(sici)1096-8628(19990101)82:1<70::aid-ajmg14>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 253.Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–164. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- 254.Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signaling in smooth muscle cells during development and disease. Cardiovasc Res. 2012 doi: 10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- 255.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 256.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat. 2001;17:18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 258.McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075–1082. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- 259.Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC, Pear WS. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004;104:1696–1702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- 260.Lomonico MP, Moore GW, Hutchins GM. Rotation of the junction of the outflow tract and great arteries in the embryonic human heart. Anat Rec. 1986;216:544–549. doi: 10.1002/ar.1092160412. [DOI] [PubMed] [Google Scholar]
- 261.Goor DA, Edwards JE. The spectrum of transposition of the great arteries: with specific reference to developmental anatomy of the conus. Circulation. 1973;48:406–415. doi: 10.1161/01.cir.48.2.406. [DOI] [PubMed] [Google Scholar]