

Abstract
Insulin binding changes conformation of the insulin receptor kinase (IRK) domain and initiates glucose uptake through the insulin, IGF-1, phosphatidyl inositol 3-kinase (PI3K), and MAPK pathways; human biliverdin reductase (hBVR) is an IRK substrate and pathway effector. This is the first report on hBVR peptide-mediated IRK activation and conformational change. 290KYCCSRK, which increased IRK Vmax without changing Km, stimulated glucose uptake and potentiated insulin and IGF-1 stimulation in 4 cell lines. KYCCSRK in native hBVR was necessary for the hBVR and IRK cross-activation. Peptide treatment also activated PI3K downstream effectors, Akt and ERK, phosphorylation, and Elk transcriptional activity. In cells transfected with CMV-regulated EGFP-VP-peptide plasmid, C292→A mutant did not stimulate glucose uptake; K296→A decreased uptake and kinase activity. KEDQYMKMTV, corresponding to hBVR's SH2-binding domain, was a potent inhibitor of glucose uptake and IRK. The mechanism of action of peptides was examined using cells expressing IRK (aa 988–1263) activated by coexpressed KYCCSRK. Three active cys-mutants of IRK, with fluorophore coupled to cysteines, C1056, C1138, or C1234, were examined for changes in fluorescence emission spectra in the presence of peptides. KYCCSRK and KEDQYMKMTV bound to different sites in IRK. The findings identify novel agents for activating or inhibiting insulin signaling and offer a new approach for treatment of type 2 diabetes and hypoglycemia.—Gibbs, P. E. M., Lerner-Marmarosh, N., Poulin, A., Farah, E., Maines, M. D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor.
Keywords: diabetes, heme metabolism, protein kinases
Binding of insulin and insulin-like growth factor (IGF)-1 and -2 to insulin receptor (IR) stimulates transmembrane tyrosine kinase activity; the initial step in the signaling event consists of conformational change in the extracellular domain of the receptor (1). The receptor is a heterotetramer of 2 extracellular insulin-binding (α) subunits joined covalently by disulfide bonds and 2 β subunits. The 2 β subunits are composed of 3 domains: extracellular, which is disulfide bonded to the C-terminal region of the α subunits (1), transmembrane, and cytosolic. Insulin binding stimulates autophosphorylation of 3 key tyrosines, Y1158,1162,1163, in the activation (A) loop of the cytosolic compartment, thereby inducing conformational change of the A loop. The change is necessary for ATP to access the glycine-rich loop and binding of tyrosine phosphorylation sites of substrates containing YMXM sequences to the catalytic and A loops (2–5). Proteins that contain YMXM, when tyrosine phosphorylated by insulin receptor kinase (IRK), become the recognition sites for the src-homology 2 (SH2) domain containing proteins (6, 7) that include insulin receptor substrate (IRS)-1 and phosphatidyl inositol 3-kinase (PI3K) and allow assembly of multiprotein signaling complexes.
IR has a high degree of sequence and structural identity with the IGF receptor. The kinase domains of the 2 receptors are more closely related at the sequence level than the full-length proteins and have nearly identical structures (8). IGF-1 and IGF-2 may act by binding to either their specific receptor (IGFR) or to IR (9–11). IGF-2, in particular, binds more strongly to the A isoform of IR, which includes the short exon 11 absent from the B form (11). IGF-1 and insulin both are capable of stimulating glucose uptake in nondiabetic skeletal muscle cells (12).
Biliverdin reductase (BVR) is an evolutionarily conserved 296-residue soluble protein that was initially considered solely in context of reduction of biliverdin, the product of the heme oxygenase (HO) isozymes HO-1 and HO-2, to bilirubin, an antioxidant (13). Subsequently, human BVR (hBVR) was found to function in the insulin, IGF-1, PI3K, and MAPK signaling pathways (14). As predicted by the crystal structure of BVR-NADH complex, the N-terminal half of the protein houses the adenine nucleotide binding pocket, and the C-terminal half forms a 6-stranded β sheet; the C-terminal helix links the 2 halves (15). hBVR terminates with a KYCCSRK sequence, which contributes to Zn2+ binding (16). Several functional regulatory motifs and sequences shared by proteins that function in insulin and IGF-1 signaling pathways are present in the C-terminal half of the protein (17–19). Most motifs are accessible for protein-protein interaction (14, 15). The most noteworthy is the specific IRK phosphorylation site, Y198MKM (20), as well as Y228 in the YLSF motif, which meets YΦSΦ criteria (7). In response to insulin- and IGF-1-activated IRK, Y198, Y228, and Y291, in the sequence E286EIQKYCCSRK, are phosphorylated (20). Subsequently, activated hBVR modulates signaling activity of various effectors in the insulin, IGF-1, PI3K, and MAPK signaling pathways, including protein kinase C (PKC)-βII and PKCδ. hBVR is the cytoplasm ↔ cell membrane translocator and transporter of PKCβII in cells treated with phorbol myristate acetate (17); this Ser/Thr kinase is associated with pathophysiological disorders of type 2 diabetes that are linked to disrupted homeostasis of glucose metabolism (21). hBVR is also a scaffold or bridge protein for PKCδ/ERK2- and MEK1/ERK1/2-mediated activation of Elk1, NFκB, and iNOS (19) and is a nuclear ↔ cytoplasm transporter of ERK1/2 (22). MAPK/ERK signaling was shown to regulate insulin sensitivity of cells and in type 2 diabetes (23). Similarly, iNOS, NFκB, and PKCδ are involved in cellular glucose metabolism (24–26).
We have examined whether hBVR motifs that display regulatory activity in intact hBVR retain this activity when contained in short synthetic peptides. After testing several peptides, we concluded that hBVR kinase activating effect was singularly specific to 290KYCCSRK and not to other peptides, based on regulatory motifs in hBVR (19, 27). One notable characteristic of the primary structure of BVR is the evolutionary conservation in species ranging from unicellular Synechocystis to human of a handful of amino acids including the first C in KYCCSRK, which corresponds to C292 in intact protein. Because hBVR activity in the insulin and IGF-1 signaling via PI3K and MAPK signaling and binding to IRK is dependent on retaining a specific structural conformation and kinase competency, we questioned whether, in the absence of hBVR, protein KYCCSRK by itself can interact with IRK and affect glucose uptake. Furthermore, in order to determine the mechanism by which hBVR-based peptides might regulate IRK, we tested whether peptides altered the secondary structure of IRK. For this, we analyzed association of the peptide with the kinase domain by generating a series of mutant IRK proteins (C1138,1234,1245→A, C1056,1234,1245→A, C1056,1158,1245→A) that each contained only 1 cysteine residue for labeling with a fluorophore. Changes in the secondary structure of the kinase domain in the presence of peptide were expected to be reflected by specific changes in the emission spectra of the fluorophores.
The results of this investigation reveal that a 7-residue peptide can, on its own, act as an activator of IRK autophosphorylation, kinase activity, and glucose uptake. The observed structural changes offer the potential for further exploration of the mode of peptide activation or inhibition of IRK, which may be exploited as means of designing specific peptide derivatives. Considering the universality of type 2 diabetes, we propose that this peptide has a place in the limited panel of compounds currently available for increasing glucose uptake.
MATERIALS AND METHODS
Materials
Human insulin, 2-deoxyglucose, and dithiothreitol (DTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA); human skeletal muscle cells were from Invitogen (Carlsbad, CA, USA); IR β subunit (IRK), IGFR β subunit [IGFR kinase (IGFRK)], IRS, and IRS Y608 peptide substrate were obtained from Enzo Life Sciences (Farmindale, NY, USA); [γ-32P]-ATP was obtained from PerkinElmer (Wellesley, MA, USA); and [1-3H]-2-deoxyglucose was obtained from Amersham (Arlington Heights, IL, USA. BVR peptides were synthesized in both myristoylated and native forms by EZBiolab (Westfield, IN, USA). Cy3 maleimide derivative (Cy3-MAL) was obtained from Combinix (Los Altos, CA, USA), and all antibodies were obtained from Cell Signaling (Beverly, MA, USA).
Plasmids
Cloning of wild-type (WT) hBVR has been described elsewhere (20). C-terminally truncated protein (BVR1-285), intended to yield a protein lacking the KYCCSRK sequence while retaining enough of the C-terminal helix to stabilize the secondary structure, was amplified using a 3′ primer designed to introduce a termination after A285. Plasmids for prolonged expression of peptides in mammalian cultures were constructed in pEGFP-C1 (Clontech, Mountain View, CA, USA). A DNA duplex to express the retroviral P2A peptide that is spontaneously cleaved during translation (28) was cloned downstream of the EGFP open reading frame between the BglII and XmaI sites to give pEGFP-P2A; this introduced novel SalI and PspOM I sites to enable insertion of duplex DNA oligomers encoding the peptides.
Throughout this study, we have used the sequence coordinates of mature IR, as used in crystallographic studies of the kinase domain (4, 29). Sequence encoding IR residues 988–1263, containing the active site of IRK, was cloned between the NheI and XhoI sites of pET-28a. Expression of the sequence in Escherichia. coli yielded a protein with an N-terminal his6 tag. Selected residues in this IRK fragment were mutated (to alanine, unless otherwise indicated) by site-directed mutagenesis; for proteins with multiple mutations, site-directed mutagenesis was repeated as necessary. The DNA encoding IRK residues 988–1263, including the his6 tag, was amplified from the bacterial expression vector and cloned into pcDNA3 for expression in mammalian cells. All constructs were verified by sequencing.
IR tyrosine kinase activity
IRK was assayed as previously reported (20), using 300 μM IRS-1 Y608 peptide substrate. Here, 20 μM hBVR-based peptides were added as activator or inhibitor. In some experiments, hBVR (0.1 μg/μl) or synthetic hBVR-based peptides (20 μM) were themselves used as substrates. Reaction products were detected by liquid scintillation counting (20) or by gel electrophoresis and autoradiography, as appropriate. Kinetic analyses of IRK activity were carried out in experiments using varying concentrations of substrates, activator, or inhibitors. IGFRK domain autophosphorylation in the presence of 20 μM peptide was determined under reaction conditions recommended by the manufacturer (Enzo Life Sciences); reactions were started by addition of [32P]-ATP to a final concentration of 25 μM.
Cell culture and transfection
Cultures of human cell lines were maintained in DMEM with 10% FBS. Human embryonic kidney 293 (HEK293) cells were transfected with plasmids using Transfectin lipid reagent (Bio-Rad, Richmond, CA, USA). Transfected cells were serum starved in DMEM containing 0.1% FBS for up to 24 h prior to treatment with myristoylated peptide, insulin, or IGF-1 (22). Human skeletal muscle cells were maintained in 1:1 DMEM:F12 containing 10% FBS. At 1 d prior to glucose uptake analysis, the medium was changed to low-glucose (5 mM) DMEM with 10% FBS.
Protein expression
Protein expression from plasmids in E. coli strain BL-21 was induced by 60 μM IPTG. His6-tagged IRK in inclusion bodies was solubilized in buffers containing 1 M KCl, 6 M urea and affinity purified with Ni-NTA-agarose, followed by chromatography on Q-Sepharose; IRK was eluted from the gel bed with a gradient of 0–200 mM KCl in Tris-urea buffer. Fractions containing IRK were concentrated and dialyzed sequentially against 4, 2, and 0 M urea in 50 mM Tris (pH 7.5).
HEK cells were cotransfected with pcDNA-IRK construct and either the KYCCSRK-expression plasmid or empty vector. Cells were washed with PBS, harvested, and lysed in 0.1 M sodium phosphate buffer containing protease and phosphatase inhibitors and 0.1% Nonidet P-40. Lysate protein (400 μg) was incubated with anti-His-tag antibody (Cell Signaling, Danvers, MA, USA), followed by addition of protein-A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The bound immunoprecipitate was collected by centrifugation and washed, and the IRK activity was measured as described above.
BVR autophosphorylation and kinase activity
BVR kinase activity was measured at pH 8.4 in the presence of MnCl2, as described previously (20). Reaction products were detected as above.
Glucose uptake
Glucose uptake was assessed using 2-deoxy 1-[3H] glucose as before (20). Cells in 24-well plates were grown to near confluence and starved in calcium- and magnesium-supplemented PBS (Invitrogen) containing 1 mg/ml BSA for 2 h. Myristoylated peptides (20 μM) were added, and incubation continued for a further 2 h, followed by treatment with 100 nM insulin or 40 ng/ml IGF-1 for 15 min. Deoxyglucose (0.2 mM, containing 1 μCi/ml [3H]-deoxyglucose) was added for the times indicated in the figure captions. Cells were washed 3 times with cold PBS and solubilized in 150 μl 50 mM NaOH. Radioactivity was measured and normalized to protein concentration. Assessments were made in triplicate, and experiments were repeated 3 times. Glucose uptake in cells transfected with the peptide expression plasmids was measured 24–48 h after DNA addition.
Akt, ERK, and Elk1 activation
HEK cells were incubated in low-glucose DMEM with 0.1% FBS overnight. Depending on the experiment, cells were treated with 20 μM myristoylated peptide for up to 2 h, and insulin or IGF for 15 min, as noted in figure captions. Cell lysates were probed consecutively by Western blotting for phosphorylated Akt and Akt1 plus Akt2, or phosphorylated ERK and ERK2. Activation of Elk1 transcription factor in response to KYCCSRK was determined by a luciferase assay (19, 22). Cells were transfected with an Elk1-dependent luciferase reporter system and treated with 20 μM peptide for 12 h (additional peptide was added at 3 h interval), and the luciferase activity was measured.
Fluorometry
A series of IRK triple-cysteine mutants was made in the pET-IRK construct by serial site-directed mutagenesis leaving only Cys1056, Cys1138, or Cys1234 intact. In addition, the R1164→Q and E1159TD→KTK mutations were introduced into the mutant retaining Cys1234. Purified proteins, while still in 6 M urea, were reduced with Tris-(2-carboxyethyl)-phosphine hydrochloride, followed by incubation with Cy3-MAL (30). Labeled proteins were purified by Ni-NTA chromatography in the presence of 6 M urea, followed by dialysis, as above. Fluorescence spectra were determined for 0.1–0.25 nmol protein in the presence and absence of 10 μM myristoylated peptide using a Fluoromax 4 fluorometer (Horiba Jobin Yvon, Palaiseau, France). Excitation was at 540 nm, and emission spectra were recorded from 555 to 750 nm.
Statistics
GraphPad Prism software (GraphPad, San Diego, CA, USA) was used for analyses of glucose uptake and kinase assay data by 1-way ANOVA, and for nonlinear regression analyses of enzyme kinetic data.
RESULTS
hBVR C-terminal 11 residues, a sequence that includes 290KYCCSRK, which is essential for stimulation of IRK autophosphorylation by hBVR, phosphorylation of hBVR by IRK, and hBVR autophosphorylation
Earlier studies had indicated that hBVR was both a substrate for, and an activator of, IRK (20). Examination of the hBVR primary sequence suggested that the extreme C-terminal of the protein, at the end of a 29-aa α helix that links BVR's 2 major domains (15), might be a binding site for IRK. We tested whether this region of hBVR is involved in activation of IRK. Two hBVR mutants were examined for their effects on IRK activation and on hBVR kinase activity. Mutation of the C-terminal lysine yielded a protein that was as effective as WT hBVR in stimulating IRK autophosphorylation and, like WT hBVR, was a substrate for IRK (Fig. 1A). Moreover, the K296A mutant retained full hBVR kinase activity (Fig. 1B). Deletion of the entire helix was expected to yield a severely damaged protein; instead, a deletion mutant was constructed that lacked only the terminal 11 residues. The hBVR truncation mutant (hBVR 1–285) only weakly stimulated IRK's autophosphorylation and was not a substrate for the kinase activity. The truncated hBVR was also a very poor kinase compared to WT hBVR.
Figure 1.
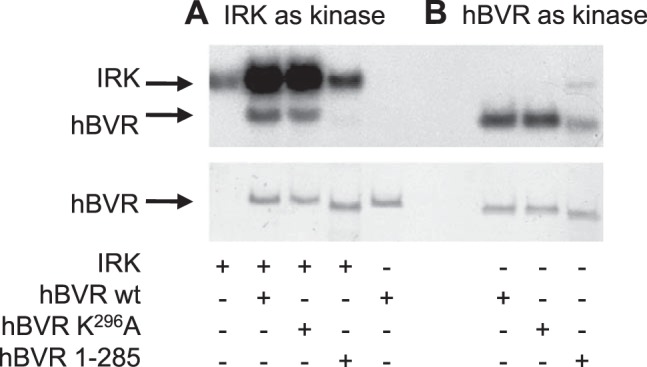
Disruption of the KYCCSRK296 sequence prevents stimulation of IRK autophosphorylation by hBVR, phosphorylation of hBVR by IRK, and hBVR autophosphorylation. A) Kinase reactions were performed as described in Materials and Methods, in the presence or absence of WT or mutant hBVR. Top panel: autoradiograph of [32P]-labeled protein. Bottom panel: Ponceau S-stained blot, used as a BVR loading control. B) WT and K296A proteins are both kinase active under BVR kinase conditions. The kinase reactions were prepared as in A, except that buffer conditions were optimized for BVR kinase activity. IRK is inactive under these conditions.
Kinetic analysis of effect of hBVR-based activator peptide on IRK activity
Since the peptide 290KYCCSRK is an activator of hBVR kinase and reductase activities (27), we tested whether it might activate IRK. Addition of KYCCSRK to IRK resulted in a linear increase in IRK phosphorylation of an IRS-1 peptide substrate, up to 50 μM added KYCCSRK (Fig. 2A), which resulted in an ∼14-fold increase in activity. Because activation could occur by altering either the Km of the enzyme or its Vmax, or possibly both, IRK was assayed with increasing concentrations of substrate peptide in the presence or absence of a fixed (20 μM) concentration of KYCCSRK. The data showed no significant difference in Km (140 μM vs. 127 μM in the absence or presence, respectively, of KYCCSRK) but a 1.6-fold higher Vmax when peptide was present (Fig. 2B).
Figure 2.

KYCCSRK stimulates IRK activity. A) Concentration-dependent activation of IRK by KYCCSRK. IRK was incubated with IRS-1 peptide substrate, together with increasing amounts of KYCCSK. Incorporation of [32P] was normalized to that observed in the absence of KYCCSRK. B) Kinetic analysis of KYCCSRK activation of IRK. IRK was incubated with or without 20 μM KYCCSRK and with varying concentrations of the IRS Y608 substrate peptide. Data were fitted to the Michaelis-Menten equation for determination of Km and Vmax.
hBVR-based KYCCSRK stimulates insulin-independent and -dependent glucose uptake in cells
Because IRK activation is linked to glucose uptake, KYCCSRK was examined for its effect on glucose uptake in HEK cells. Serum-starved cells were treated with 20 μM myristoylated peptide for 2 h prior to addition of insulin (100 nM for 15 min) and assayed for uptake of [3H]-deoxyglucose. Glucose uptake was enhanced by treatment with either KYCCSRK or insulin, and the two together resulted in a further stimulation of uptake (Fig. 3A). At these concentrations stimulation of uptake by insulin or peptide alone was significant (P<0.05) compared to untreated controls. There was no significant difference in uptake by cells treated with peptide compared with those treated with insulin. Peptide and insulin together were more effective than insulin alone (P<0.01). The effect of the peptide is likely to be exerted solely through the intracellular domain of the receptor. Nonmyristoylated KYCCSRK is not expected to cross the plasma membrane and failed to stimulate glucose uptake by cells (data not shown). HEK cells treated with IGF-1 showed only a very modest increase in uptake in response to 40 ng/ml IGF-1 alone, but a significant increase in the presence of peptide (P<0.05) and a further increase in the presence of both stimuli (Fig. 3B). In other experiments, human hepatoma cells (HepG2 line), human pulmonary artery smooth muscle (PASM) cells, or human skeletal muscle cells were used. Again, it was noted that insulin or the peptide stimulated glucose uptake by HepG2, with further enhancement in cells subjected to both treatments (Fig. 3C). PASM cells were treated with KYCCSRK, followed by stimulation with 40 ng/ml IGF-1 (Fig. 3D). IGF-1 treatment in the absence of peptide stimulated glucose uptake by some 50%, whereas the peptide caused a near 2-fold increase (Fig. 3D). Human skeletal muscle cells responded to peptide in much the same way as the other cell types (Fig. 3E), in that KYCCSRK treatment stimulated both insulin-dependent and -independent uptake. In each cell line, the increased glucose uptake observed after treatment with insulin, IGF-1, or peptide compared to untreated cells was statistically significant. The increased uptake in cells treated with both peptide and insulin was significantly greater than that seen in cells treated with insulin (or IGF-1) alone (Fig. 3). In all cell types, the difference in uptake between cells treated with peptide and those treated with insulin or IGF-1 alone was not significant. Accordingly, the enhancement of glucose uptake by KYCCSRK does not appear cell type- or insulin-dependent.
Figure 3.

hBVR-based KYCCSRK stimulates glucose uptake in cells and enhances the effect of insulin or IGF-1. Stimulation of glucose uptake by KYCCSRK is neither cell-type nor insulin specific. All cells were serum starved overnight and, where indicated, treated with 20 μM myristoyl-KYCCSRK for 2 h prior to treatment with insulin or IGF-1. A) HEK cells: insulin was added to 100 nM for 15 min prior to measuring [3H]-deoxyglucose uptake (see Materials and Methods). B) HEK cells, with or without 40 ng/ml IGF-1 for 15 min. C) HepG2 cells, 100 nM insulin, as in A. D) Human pulmonary artery smooth muscle (PASM) cells, 40 ng/ml IGF-1 as in B. E) Human skeletal muscle cells, insulin-treated, as in A. *P < 0.05, **P < 0.01, ***P < 0.001; ANOVA.
Peptide expression system is effective in stimulating glucose uptake in cells
The use of myristoylated peptides enables delivery into the cell (27) but permits only limited treatment times. Therefore, a plasmid expression system designed to release the peptide during translation was constructed. A fusion gene consisting of EGPF, the self-cleaving P2A viral peptide (31), and sequence encoding the peptide of interest was constructed, with its expression regulated by the human CMV promoter (Fig. 4A). We tested the efficiency of the expression and cleavage of the protein in 2 experiments. In one, a construct encoding HA-tagged hBVR in place of peptide downstream of the cleavage site was used to transfect HEK cells. Western blotting of cell lysates prepared 24 h after DNA addition indicated that hBVR was overexpressed and migrated entirely at the expected position for hBVR, with no detectable EGFP-hBVR fusion protein (not shown). The effect of peptide expression was assayed directly by its effect on glucose uptake, and 48 h after transfection cells were assayed for [3H]-deoxyglucose uptake. There was enhanced uptake in the KYCCSRK-expressing cells at all times up to 30 min after deoxyglucose addition (Fig. 4B) compared to control cells transfected with an empty EGFP vector.
Figure 4.
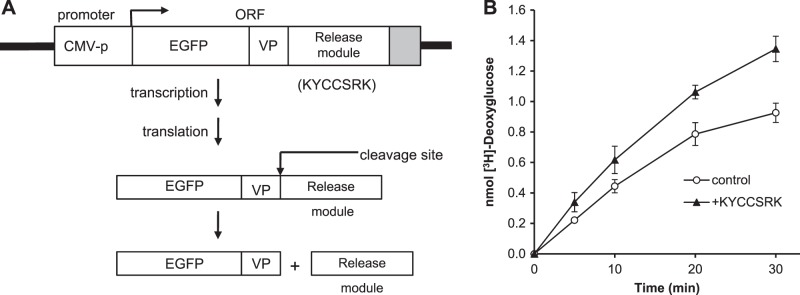
Expression of KYCCSRK in cells stimulates glucose uptake. A) Construct used for peptide expression. Base vector was pEGFP-C1, retaining the CMV promoter (CMV-p) and EGFP open reading frame, and includes a retroviral sequence (VP) that is cleaved during translation (28). Peptide coding sequence was inserted immediately downstream of the cleavage site. B) Expression of KYCCSRK in cells enhances glucose uptake in cells. Cells were transfected with the peptide expression vector encoding KYCCSRK or with empty vector. At 24 h after DNA addition, they were serum starved and subsequently assayed for glucose uptake at the indicated times.
Defining the key KYCCSRK residues for activation of IRK
To determine the specific residues in peptides KYCCSRK that are required for their activation of IRK, a series of modified synthetic versions of KYCCSRK, in which specific residues were replaced by alanine, was examined. Initially, it was found that replacement of Cys293 (peptide position 4) had a modest effect on enhancement of IRK kinase activity, whereas replacement of Cys292 (at position 3) completely abolished activation of IRK (Fig. 5A). Replacement of either lysine by alanine almost completely abolished the activation of the kinase by peptide (Fig. 5B). Conversely, replacement of the tyrosine and arginine residues had little effect on peptide-driven activation of the kinase (data not shown). The effect of modifying essential residues in the KYCCSRK sequence was explored further. As shown in Fig. 5C, inclusion of KYCCSRK in an IRK autophosphorylation reaction resulted in increased incorporation of 32P into IRK. The increase was not seen with a control peptide (KEVVGKD), while changing the tyrosine to phenylalanine and the C-terminal lysine to alanine each led to a modest attenuation of 32P incorporation into IRK. Replacing the cysteine at position 3 with alanine significantly decreased autophosphorylation compared to KYCCSRK. In addition, cells were transfected with the WT peptide expression vector, or with vectors to express KYACSRK or KYCCSRA. After treatment with insulin and [3H]-deoxyglucose, uptake in cells expressing KYCCSRK was higher than that in the empty vector control (Fig. 5D); expression of KYACSRK inhibited insulin-dependent glucose uptake. KYCCSRA expression gave a modest increase of 18% at 30 min, compared to 46% with KYCCSRK. These data indicate that the cysteine residue is of paramount importance in activation of IRK in vitro and on glucose uptake in cells, whereas other residues, notably the C-terminal lysine, may have a less prominent function.
Figure 5.

Defining the key residues in KYCCSRK for activation of IRK in vitro. A) Cys292 in the KYCCSRK sequence is necessary for enhancing IRK activation. Phosphorylation of the IRS peptide by IRK was assayed in the presence of 20 μM KYCCSRK or the indicated variant peptides; the control had no added peptide. Modified residue is underlined. B) K290 and K296 are both required for activation of IRK by KYCCSRK. IRK was assayed as in A, with KYCCSRK or the indicated mutant peptides. C) C292 is critical for enhancement of IRK autophosphorylation by KYCCSRK. IRK autophosphorylation reactions were incubated without added BVR peptide, or with 20 μM each of the indicated peptides. Modified residue is underlined. Products were detected by autoradiography after SDS gel electrophoresis and transfer to membrane. D) C292 is required for enhancement of glucose uptake by prolonged expression of KYCCSRK in cells. Cells were transfected with peptide expression constructs; 48 h later, they were serum starved, treated with insulin, and assayed for glucose uptake as in Fig. 4C. E) KYCCSRK enhances autophosphorylation of IGFRK. Duplicate reactions with or without 20 μM KYCCSRK were analyzed by gel electrophoresis, as in C.
Because the kinase domains of IR and IGFR are closely related, with 74% amino acid identity, or 82% similarity if conservative replacements are considered, we considered the possibility that the KYCCSRK peptide might also stimulate the kinase activity of IGFRK. To test this, IGFRK domain was incubated in vitro in the presence and absence of KYCCSRK. The presence of the peptide in the reaction mixture resulted in a robust increase in incorporation of 32P from ATP into IGFRK (Fig. 5E).
KYCCSRK activates signaling downstream of IRK and IGFRK
Next, we examined whether the presence of the peptide stimulates activity of kinases downstream of IRK. For this, the effect of KYCCSRK on signaling via the PI3K/Akt pathway was examined in cells treated with myristoylated peptide for 2 h prior to treatment with insulin for 15 min. Pretreatment of insulin-treated cells with KYCCSRK increased phosphorylation of Akt at Ser473 (Fig. 6A). Also, the effect of peptide treatment on activation of ERK by IGF-1 was examined. Cells were treated with peptide and IGF-1 for 15 min or were first treated with KYCCSRK for 15 min, followed by IGF-1 for a further 15 min. In the peptide-treated cells, there was little change in ERK phosphorylation at 15 min, but a clear increase by 30 min (Fig. 6B). As expected, IGF-1 led to increased phosphorylation of ERK, and this was increased by cotreatment with peptide (Fig. 6B). The consequence of ERK activation in the cell was examined. The transcription factor Elk1 is a downstream effector of activated ERK; accordingly, whether ERK/Elk signaling is enhanced by the peptide was investigated There was a significant (P<0.001) 2-fold increase in transcriptional activity in the peptide-treated cells (Fig. 6C). The findings suggest that the peptide is able to elicit enhanced response in the signaling pathway.
Figure 6.

Activation of insulin and IGF-1 signaling by KYCCSRK A) KYCCSRK enhances activation of Akt. Lysates of cells treated with KYCCSRK for 2 h, followed by insulin for 15 min, were analyzed by Western blot, which was probed sequentially with anti-Akt-phosphoS473 and Akt1 + Akt2 antibodies. Blots were further analyzed by densitometry. **P < 0.01. B) Activation of ERK. Cells were treated with KYCCSRK for the indicated periods. Here 40 ng/ml IGF-1 was included for the final 15 min of treatment. Western blot was probed sequentially with anti-phospho-ERK (T202Y204) and anti-ERK2 antibodies. C) Enhanced response of Elk1-dependent transcription by KYCCSR. Cells were transfected with an Elk1-dependent luciferase reporter system (19, 22), incubated overnight in low-glucose medium with 0.1% serum, and then treated with 20 μM KYCCSRK for 12 h (see Materials and Methods). Cell lysates were assayed for luciferase activity, normalized on that of a cotransfected β-galactosidase reporter. ***P < 0.001.
hBVR-based peptides as inhibitors of IRK and of glucose uptake
Building on the observation that hBVR is a substrate for IRK (20), tyrosine-containing hBVR-based peptides with SH2-binding motifs were assessed as substrates for IRK in vitro and compared with the KYCSSRK activator peptide. All 3 were phosphorylated by IRK in the intact hBVR [KYCCSRK (Y291), KEDQYMKMTV (Y198), and GLKRNKYLSFHFK (Y228)] were all phosphorylated by IRK (Fig. 7A). Kinetic analysis of IRK with KEDQYMKMTV and GLKRNKYLSFHFK as substrates yielded a Km value for KEDQYMKMTV of 4.3 μM, and for GLKRNKYLSFHFK of 1.1 μM (Fig. 7B, C). To test the effect of these peptides on IRK activity in a physiological context, KEDQYMKMTV, which contains the SH2 binding motif YMXM, was included in IRK kinase reactions with the IRS-1 peptide substrate, and the effect of increasing concentrations of KEDQYMKMTV on IRK kinase activity was examined. KEDQYMKMTV was found to be a potent inhibitor of IRK (Fig. 7D) kinase activity. Fitting the data to a hyperbolic function indicated a Ki value of 8.9 μM, significantly lower than the Km value of ∼130 μM observed for the substrate (Fig. 2B). This peptide and KRNRYLSF that includes the YLSF motif were examined for their effects on glucose uptake in HepG2 cells. Neither activated glucose uptake in the absence of insulin; moreover, both effectively blocked insulin-mediated glucose uptake (Fig. 7E). It is intriguing that, on its own, KEDQYMKMTV is a substrate for IRK, but in the presence of the IRS-derived substrate, it is an inhibitor. It is possible that the inhibition is due to a slower dissociation rate of KEDQYMKMTV from the enzyme than that of the IRS peptide, thereby resulting in a competitive inhibition
Figure 7.

Identification of hBVR-based peptide inhibitors of IRK activity and glucose uptake. A) IRK phosphorylates hBVR-derived tyrosine-containing peptides. Noted hBVR-based peptides (20 μM) were used as substrates in IRK assays in vitro, and incorporation of [32P] into peptide was measured. B) KEDQYMKMTV is a high-affinity substrate for IRK. IRK was assayed using increasing concentrations of KEDQYMKMTV as substrate. Data were fitted to the Michaelis-Menten equation. C) GLKRNKYLSFHFK is a high-affinity substrate for IRK. Data were collected and analyzed as in B. D) Concentration dependence of KEDQYMKMTV on IRK activity. In vitro kinase reactions with IRK and the IRS-derived peptide substrate included the indicated concentrations of KEDQYMKMTV. Incorporation of [32P] was determined and expressed as percentage inhibition of kinase activity. Data were fitted to a hyperbolic function to estimate Ki. E) Inhibition of insulin-dependent glucose uptake in HepG2 cells by hBVR-based SH2 domain peptides. HepG2 cells were starved, treated with myristoylated peptides and insulin, and assayed for [3H]-deoxyglucose uptake as in Fig. 3.
Secondary structure changes in IRK induced by peptide binding
In order to investigate the mechanisms by which KYCCSRK activates and KEDQVYMKMTV inhibits IRK, we elected to use physical techniques to probe the secondary structure of the kinase. We cloned the sequence encoding IR residues E988 to L1263 (Fig. 8A), including the ATP binding site and the catalytic and activation loops. The sequence includes 4 cysteine residues, which were systematically mutated to yield constructs that each carried 3 C-A replacements. The WT His6-tagged protein was expressed in E. coli and purified from inclusion bodies. In the standard kinase assay, it had low activity and was activated, albeit weakly, by KYCCSRK (Fig. 8B; 100% represents incorporation of 0.25–0.3 pmol phosphate). The low activity was probably due to the harsh protein purification conditions, to bacterial cells that lack tyrosine kinase activity, and to impaired ability of the kinase to form dimers essential for maximum activity (32). Despite this, the protein cysteine mutants showed similar activity to WT protein. The R1164Q replacement in the activation loop allowed KYCCSRK to stimulate activity similar to that for the WT protein, whereas in the double replacement E1159TD–K1159TK mutant, the response to KYCCSRK was effectively abolished. Mutation of D1229QTDN to AATAA, however, did not alter the activation by KYCCSRK, suggesting the activation is specific to residues in the activation loop, possibly by E1159 and/or D1161 associating with one of the peptide lysines. The His-tagged open reading frame was also cloned into pcDNA3, and HEK cells were cotransfected with either the KYCCSRK or empty peptide expression vector; the peptide expression vector was cotransfected with pcDNA as a second control; transfection was verified by microscopy, with EGFP being visible in most cells (not shown). The soluble His-tagged IRK was immunoprecipitated and assayed for IRK kinase activity. Immunoprecipitates from cells transfected with a single expression plasmid, either for KYCCSRK or for his-tagged IRK plasmid, gave low activity in the kinase assay. The activity of the expressed protein was significantly enhanced by coexpression of KYCCSRK (Fig. 8C).
Figure 8.

Expression and mutagenesis of IRK. A) Schematic representation of the IRK expressed in E. coli. Region includes the ATP binding site and catalytic and activation loops (29). Approximate positions of amino acids mutated in these studies are indicated. B) Kinase activity of bacterially expressed protein is stimulated by KYCCSRK. Kinase reactions included WT or mutant IRK, with or without added KYCCSRK. Reactions were carried out as described in Materials and Methods, using 1 μg of recombinant protein. C) Kinase activity of the IRK fragment in HEK cells. Cell extract containing expressed KYCCSRK, His6-IRK, or both was incubated with antibody to His tag, and the complex collected on proteinA/G-agarose, for assay with IRS peptide substrate.
A unique, suitably located fluorophore in a protein serves as a sensitive marker of secondary structure, as its emission spectrum is highly dependent on the immediate chemical environment. The mutant proteins, each retaining a single cysteine residue, were labeled with Cy3, purified, and renatured by stepwise dialysis. The fluorescence spectrum of each Cy3-labeled protein was determined in the presence and absence of peptides: the activator KYCCSRK and the substrate and competitive inhibitor KEDQYMKMTV. In the case of protein tagged at Cys1056, the spectra obtained in the presence of peptides were virtually identical to that obtained for the protein alone (Fig. 9A), suggesting that the addition of any of the peptides does not cause secondary structure changes in the largely β-sheet lobe of the kinase. For protein tagged at Cys1138, adjacent to the catalytic loop of the kinase (29), no change in fluorescence yield was observed with KYCCSRK (Fig. 9B). Addition of KEDQYMKMTV results in a 45% increase in quantum yield of the fluorophore, indicating that the secondary structure surrounding the fluorophore has changed on peptide binding. Since this peptide is phosphorylated by IRK and inhibits phosphorylation of the IRS-1 peptide substrate, binding to the IRK active site is expected. Despite the change in yield in the presence of KEDQYMKMTV, spectra normalized on the emission maxima of the Cys1138-tagged protein were virtually identical.
Figure 9.

Peptide binding induces specific secondary structure changes in IRK. The IRK expression construct was mutated at cysteine residues by serial site-directed mutagenesis until mutants that retained only a single cysteine residue were obtained. Proteins expressed in E. coli were labeled with Cy3 prior to dialysis and activation. Fluorescence spectra were determined on 0.1–0.25 nmol protein in the presence and absence of 10 μM peptide. A) Protein labeled at Cys1056. B) Protein labeled at Cys1138. C) Protein labeled at Cys1234. D, E) Activation site mutations, as used in Fig. 7, were introduced in the construct that retained only Cys1234: mutation at E1159TD, to KTK (D); R1164Q mutation (E).
A fluorophore tag on Cys1234, which is at some distance from the active site, shows increased quantum yield irrespective of the peptide added (Fig. 9C). Addition of KEDQYMKMTV stimulated emission by 55%; the activator peptide resulted in a 2.4-fold increase in yield. The increased quantum yield of the major emission peak was accompanied by a change in the shape of the normalized spectra, such that the relative height of the shoulder was reduced, albeit by only 10–15%. This region of the protein therefore appears to change structure on peptide binding. The activation loop mutations (E1159TD-KTK, R1164Q) were introduced into the protein that retains only Cys1234. Neither Cy3-tagged mutant protein showed increased quantum yield after addition of peptide (Fig. 9D, E); in fact, there was up to 20–40% quenching, depending on the peptide used. At a minimum, this suggests that these mutant proteins undergo different structural changes in response to peptide. However, each of these mutants also had nearly identical normalized spectra in the presence or absence of peptide.
DISCUSSION
Diabetes, whether caused by failure of the pancreas to secrete insulin or by diminished response of the target tissues to the hormone, is defined by high levels of circulating glucose levels (33). The initial step that triggers insulin response, activates overlapping signaling pathways, and has dramatic effects on cellular growth and metabolism, is its binding to the extracellular α subunits of the transmembrane glycoprotein, IR, with tyrosine kinase activity (34). Insulin, IGF-1, and IGF-2 are the high-affinity, high-specificity ligands for A isoforms of α subunits (9–11). Insulin binding results in autophosphorylation of specific tyrosines in the cytoplasmic β chains (4). The kinase domain of the insulin receptor has been examined by X-ray crystallography and appears to have 2 distinct lobes that form the faces of the active site (29). ATP is bound by a glycine-rich GXGX2G motif (aa 1003–1010) in the smaller of the lobes, which consists primarily of β strands. The larger lobe includes both the catalytic and activation loops (aa 1130–1137 and 1149–1170, respectively); the latter prevents access of substrate and the cofactor to the active site prior to ligand binding. The autophosphorylation-induced conformational change in the activation loop initiates insulin and IGF-1 signaling and glucose uptake (4, 29). Presently, we report on characterization of peptides that stimulate or inhibit IRK and glucose uptake by binding and changing the conformation of the kinase domain, the local solvent environment, or both, as is evident from the changes in response, on addition of peptides, of fluorophores introduced into the kinase domain. It is noted that KYCCSRK has a very narrow spectrum of activity, because prior to this study, we had found only 1 other role could be attributed to it, namely, activation of hBVR (27). Therefore, in cells treated with the peptide, there could be 2 parallel events: interaction with, and activation of, IRK and IGFRK, and activation of BVR. The outcome of both activation events is stimulation of the insulin-dependent signaling pathways.
To our knowledge this is an unprecedented finding that a small peptide can, on its own, be a highly effective activator of IRK and the closely related IGFRK and stimulate glucose uptake. That this response occurs in both skeletal muscle and smooth muscle cells suggests that the peptide might be of use in ameliorating hyperglycemia in intact animals. This finding became more intriguing by finding that the sequence of the peptide, except for containing the Cys-Cys dipeptide, bore no resemblance to any of known activators of IRK, including insulin, IGF-1, TGF, and PDGF-α; the dipeptide participates in divalent metal-ATP (Mg2+ or Mn2+) binding, as well as coordinating Zn2+. Cys292Cys293 (in KYCCSRK) in the helical structure of the C-terminal 29 residues of hBVR is involved in Zn2+ coordination (16) and potentially is involved in stabilizing the secondary structure of the hBVR protein. This possibility may explain the finding that deletion of the sequence disrupts the ability of hBVR to autophosphorylate, accept phosphate from IRK, or potentiate IRK autophosphorylation (Fig. 1). It is noteworthy that the conformation of the C-terminal helix (15) and accessibility of the KYCCSRK sequence may well account for hBVR being an activator of IRK. Support for this possibility is that the IRK-substrate-scaffold proteins bind through their phosphotyrosine binding (PTB) domain, adjacent to the pleckstrin (PH) domain to NPEY motifs of activated IRK (7). The PTB domain contains the YMXM motif; this domain and pleckstrin homology (PH) domains are not conserved at the primary structure level but, rather, are structurally conserved. The formation of the postulated hBVR PH domain depends on the final helix that terminates with KYCCSRK (20).
Because of the highly unprecedented ability of KYCCSRK to activate IRK, we searched whether there are other proteins that have a sequence similar to KYCCSRK. A GenBank Blast search, however, identified partial alignment with 3 cytosolic proteins and a cell surface sulfate proteoglycan (Glypican 3 isoform 3). The proteoglycan and phospholipase A2 share the CCSRK sequence with the hBVR-based peptide. The CREB-binding protein has the sequence YCCSGK, and the cysteine- and histidine-rich domain containing protein 1 has KYWSCCRRK. None of these proteins, to our knowledge, have been reported to activate IRK. Whether isolated peptides derived from these proteins display biological activity remains to be examined.
We noted two earlier studies that had explored either synthetic chemical structures or derivatives of naturally occurring fungal metabolites on the IR and demonstrated activation of IRK in cells and efficacy in reducing hyperglycemia in db/db mice (35–37). However, in only one of those studies was the structure of the active molecule given, which bears no resemblance to KYCCSRK. In addition, little work in those studies addressed the mechanism of action of the activators. A crude IR preparation was analyzed in one study (35) and increased autophosphorylation was observed; however, in the same study, a 2-fold activation of recombinant IRK was observed only at a concentration some 50-fold greater than the EC50 of the activator in cells. Another study used an IR mutant defective in signal transmission between the α and β subunits to demonstrate that a “sensitizer” molecule did not stimulate the mutant receptor (36). The same study examined a second small molecule, described as an “insulin agonist” (36), that nonetheless was capable of activating IRK independently of insulin.
Activation of IRK by hBVR does not extend to enhancing glucose uptake by insulin- and IGF-1-activated IRK (20), because the activated hBVR is a regulator of IRS activity. IRS proteins (≥3 in humans) lack intrinsic kinase activity, but when tyrosine phosphorylated by IRK form a platform for assembly of signaling complexes that control organ-dependent glucose uptake, transport, and metabolism. Cessation of glucose uptake is dependent on serine phosphorylation and subsequent degradation of IRS-1 (38); IRS-1 is a substrate for hBVR kinase activity. This process depends on tyrosine phosphorylation of YMKM motif of hBVR (20); this is not an autophosphorylation site. Consequently hBVR plays a significant role in regulating glucose uptake. The specificity of IRK for its targets is the likely explanation for the inhibition of IRK kinase activity by KEDQYMKMTV (Fig. 7D). The fact that the Y198 mutant hBVR protein disrupts the controlled uptake of glucose (20) is consistent with the idea that in the intact protein, the YMKM and the KYCCSRK motifs act independently in interaction with IRK and in regulating glucose uptake, while as isolated peptides, they have opposing effects on the uptake.
There are clear differences in the mechanisms by which insulin and KYCCSRK stimulate glucose uptake. Binding of insulin to the extracellular α subunit of the receptor results in activation of the kinase, whereas the evidence gathered from both the in vitro and in cell experiments suggests that KYCCSRK acts predominantly on the intracellular kinase domain in the β subunits. In cells, only myristoylated peptides that are capable of crossing the plasma membrane can modulate glucose uptake. This argument is also supported by the results obtained with the expression vector (Figs. 4B and 5D), where glucose uptake was clearly augmented in cells transfected with the plasmid. Peptides synthesized from this vector complex lack a signal sequence for extracellular export of the translation products and, hence, are highly unlikely to be secreted and subsequently to interact with the insulin binding sites. The augmentation of insulin-stimulated glucose uptake by KYCCSRK indicates that there is cooperation between the two stimuli, supporting the likelihood of their acting at different sites. Because IGFs, particularly IGF-2, bind to and stimulate the insulin receptor (9–11), it is possible that the effects of KYCCSRK on glucose uptake in response to IGF-1 are mediated by peptide interaction with the IRK domain. The kinase domains of the IR and IGFR are highly similar (>82% sequence similarity). We have demonstrated that the peptide activates IGFRK in vitro (Fig. 5E) and therefore we cannot eliminate the possibility that IGF-1 is acting primarily through the IGFR to stimulate uptake.
Considering that activation of IRK is known to reflect changes in conformation of the kinase domain (4), together with our data obtained with fluorescence spectrometry, it is likely that the activator and the inhibitor peptides bind to different regions of the kinase domain to perturb its secondary structure. The emission spectrum and quantum yield of the fluorophore are highly dependent on the chemical environment of the fluorophore. Both peptides (KYCCSRK and KEDQYMKMTV) likely induced changes in the structure of the sequence located C-terminal to the catalytic loop in the expressed kinase domain, without affecting the spectrum or quantum yield of a fluorophore located in the N-terminal lobe (at C1056) that includes the ATP-binding loop (Fig. 8A). The quantum yield of a fluorophore located immediately adjacent to the catalytic loop (C1138) was not altered by association with KYCCSRK, whereas a fluorophore in the C-terminal lobe (C1234) showed a more than doubled quantum yield in the presence of the peptide; this increase was abolished by mutations in the activation loop, indicating a role for this region, specifically E1159 and D1161 in binding of the peptide and subsequent structure changes. KEDQYMKMTV, on the other hand, likely binds at the substrate site. It is a high-affinity substrate with a significantly lower Km value (4.3 μM) than that of the IRS-1 peptide substrate (∼130 μM, Fig. 2B). Binding resulted in increased quantum yield of fluorophores located at the catalytic loop and in the C-terminal lobe. The predicted binding, using the program PepSite (39), for the peptide to the active IRK structure is virtually identical to that of a substrate peptide determined in the crystal structure (4). The increased yield from the C1234 fluorophore in response to the inhibitor peptide was sensitive to activation loop mutations, suggesting that the peptide maintains the activated conformation. It is noted that the C1234 fluorophore is some distance from the activation loop; in the 1IR3 structure (4), the α carbon of C1234 is 34.3 Å from that of R1164. This suggests that binding of either peptide triggers long-range changes in protein secondary structure, as revealed by the altered chemical environment of the fluorophore.
The observation that the peptide KEDQYMKMTV is an inhibitor of IRK is not unprecedented. Two crystallographic studies with IRK inhibitors have addressed the mode of interaction of peptide based inhibitors with IRK. In the first, a well-characterized substrate peptide was coupled with an ATP derivative to yield a transition state analog (40). The peptide moiety of this molecule was more highly ordered in the crystal structure than was the substrate peptide (4). Moreover, the IRS-2 kinase regulatory-loop binding region interacts with the IRK active site and inhibits the kinase. Crystallographic studies with a 15-residue peptide encompassing the putative binding region revealed a highly ordered peptide structure in the active site (41). In both studies, the higher degree of order in the peptide structure was interpreted as tighter binding to the kinase. The Km and Ki values of KEDQYMKMTV are consistent with tight binding to the active site.
The significance of our study should be evaluated in the context of the prevalence of disorders of glucose metabolism, in particular diabetes, that afflict a large segment of the population. Diabetes is a major cause of heart disease, stroke, kidney failure, nontraumatic lower limb amputation, and blindness. Currently, treatment options for dysfunction of glucose metabolism and the associated pathophysiology are limited, and new treatment approaches are lagging behind the rapidly increasing global incidence of the disorders. We believe that the findings discussed here may offer a path to novel therapeutic approach to controlling abnormal glucose metabolism.
Acknowledgments
This work is supported by U.S. National Institutes of Health grant ES004066.
The authors thank Dr. Tihomir Miralem for his assistance with the Akt activation analyses.
Footnotes
- BVR
- biliverdin reductase
- Cy3-MAL
- Cy3 maleimide derivative
- hBVR
- human biliverdin reductase
- HEK
- human embryonic kidney
- HO
- heme oxygenase
- IGF
- insulin-like growth factor
- IGFR
- insulin-like growth factor receptor
- IGFRK
- IGFR kinase
- IR
- insulin receptor
- IRK
- insulin receptor kinase
- IRS
- insulin receptor substrate
- PASM
- pulmonary artery smooth muscle
- PH
- pleckstrin homology
- PI3K
- phosphatidyl inositol 3-kinase
- PKC
- protein kinase C
- PTB
- phosphotyrosine binding
- SH2
- src-homology 2
- WT
- wild type
REFERENCES
- 1. Ward C. W., Menting J. G., Lawrence M. C. (2013) The insulin receptor changes conformation in unforeseen ways on ligand binding: sharpening the picture of insulin receptor activation. BioEssays 35, 945–954 [DOI] [PubMed] [Google Scholar]
- 2. Cobb M. H., Sang B. C., Gonzalez R., Goldsmith E., Ellis L. (1989) Autophosphorylation activates the soluble cytoplasmic domain of the insulin receptor in an intermolecular reaction. J. Biol. Chem. 264, 18701–18706 [PubMed] [Google Scholar]
- 3. Ellis L., Clauser E., Morgan D. O., Edery M., Roth R. A., Rutter W. J. (1986) Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell 45, 721–732 [DOI] [PubMed] [Google Scholar]
- 4. Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 6. Shoelson S. E., Chatterjee S., Chaudhuri M., White M. F. (1992) YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc. Natl. Acad. Sci. U.S.A. 89, 2027–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Songyang Z., Shoelson S. E., McGlade J., Olivier P., Pawson T., Bustelo X. R., Barbacid M., Sabe H., Hanafusa H., Yi T. (1994) Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 14, 2777–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Favelyukis S., Till J. H., Hubbard S. R., Miller W. T. (2001) Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 8, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 9. Morrione A., Valentinis B., Xu S. Q., Yumet G., Louvi A., Efstratiadis A., Baserga R. (1997) Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 94, 3777–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vella V., Pandini G., Sciacca L., Mineo R., Vigneri R., Pezzino V., Belfiore A. (2002) A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 87, 245–254 [DOI] [PubMed] [Google Scholar]
- 11. Sciacca L., Mineo R., Pandini G., Murabito A., Vigneri R., Belfiore A. (2002) In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 21, 8240–8250 [DOI] [PubMed] [Google Scholar]
- 12. Ciaraldi T. P., Carter L., Rehman N., Mohideen P., Mudaliar S., Henry R. R. (2002) Insulin and insulin-like growth factor-1 action on human skeletal muscle: preferential effects of insulin-like growth factor-1 in type 2 diabetic subjects. Metabolism 51, 1171–1179 [DOI] [PubMed] [Google Scholar]
- 13. Stocker R. (2004) Antioxidant activities of bile pigments. Antioxid. Redox Signal. 6, 841–849 [DOI] [PubMed] [Google Scholar]
- 14. Kapitulnik J., Maines M. D. (2009) Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 30, 129–137 [DOI] [PubMed] [Google Scholar]
- 15. Whitby F. G., Phillips J. D., Hill C. P., McCoubrey W., Maines M. D. (2002) Crystal structure of a biliverdin IXalpha reductase enzyme-cofactor complex. J. Mol. Biol. 319, 1199–1210 [DOI] [PubMed] [Google Scholar]
- 16. Maines M. D., Polevoda B. V., Huang T. J., McCoubrey W. K., Jr. (1996) Human biliverdin IXalpha reductase is a zinc-metalloprotein: characterization of purified and Escherichia coli expressed enzymes. Eur. J. Biochem. 235, 372–381 [DOI] [PubMed] [Google Scholar]
- 17. Maines M. D., Miralem T., Lerner-Marmarosh N., Shen J., Gibbs P. E. (2007) Human biliverdin reductase, a previously unknown activator of protein kinase CβII. J. Biol. Chem. 282, 8110–8122 [DOI] [PubMed] [Google Scholar]
- 18. Tudor C., Lerner-Marmarosh N., Engelborghs Y., Gibbs P. E., Maines M. D. (2008) Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 413, 405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gibbs P. E., Miralem T., Lerner-Marmarosh N., Tudor C., Maines M. D. (2012) Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS). J. Biol. Chem. 287, 1066–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lerner-Marmarosh N., Shen J., Torno M. D., Kravets A., Hu Z., Maines M. D. (2005) Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 7109–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Standaert M. L., Bandyopadhyay G., Galloway L., Soto J., Ono Y., Kikkawa U., Farese R. V., Leitges M. (1999) Effects of knockout of the protein kinase C beta gene on glucose transport and glucose homeostasis. Endocrinology 140, 4470–4477 [DOI] [PubMed] [Google Scholar]
- 22. Lerner-Marmarosh N., Miralem T., Gibbs P. E., Maines M. D. (2008) Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 6870–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gogg S., Smith U., Jansson P. A. (2009) Increased MAPK activation and impaired insulin signaling in subcutaneous microvascular endothelial cells in type 2 diabetes: the role of endothelin-1. Diabetes 58, 2238–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Won J. S., Im Y. B., Key L., Singh I., Singh A. K. (2003) The involvement of glucose metabolism in the regulation of inducible nitric oxide synthase gene expression in glial cells: possible role of glucose-6-phosphate dehydrogenase and CCAAT/enhancing binding protein. J. Neurosci. 23, 7470–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Talior I., Yarkoni M., Bashan N., Eldar-Finkelman H. (2003) Increased glucose uptake promotes oxidative stress and PKC-delta activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol. 285, E295–E302 [DOI] [PubMed] [Google Scholar]
- 26. Kawauchi K., Araki K., Tobiume K., Tanaka N. (2008) p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 10, 611–618 [DOI] [PubMed] [Google Scholar]
- 27. Lerner-Marmarosh N., Miralem T., Gibbs P. E., Maines M. D. (2007) Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. FASEB J. 21, 3949–3962 [DOI] [PubMed] [Google Scholar]
- 28. Provost E., Rhee J., Leach S. D. (2007) Viral 2A peptides allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis 45, 625–629 [DOI] [PubMed] [Google Scholar]
- 29. Hubbard S. R., Wei L., Ellis L., Hendrickson W. A. (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754 [DOI] [PubMed] [Google Scholar]
- 30. Hickerson R., Majumdar Z. K., Baucom A., Clegg R. M., Noller H. F. (2005) Measurement of internal movements within the 30 S ribosomal subunit using Forster resonance energy transfer. J. Mol. Biol. 354, 459–472 [DOI] [PubMed] [Google Scholar]
- 31. Holst J., Vignali K. M., Burton A. R., Vignali D. A. (2006) Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat. Methods 3, 191–197 [DOI] [PubMed] [Google Scholar]
- 32. Baer K., Al-Hasani H., Parvaresch S., Corona T., Rufer A., Nolle V., Bergschneider E., Klein H. W. (2001) Dimerization-induced activation of soluble insulin/IGF-1 receptor kinases: an alternative mechanism of activation. Biochemistry 40, 14268–14278 [DOI] [PubMed] [Google Scholar]
- 33. White M. F. (1997) The insulin signalling system and the IRS proteins. Diabetologia 40, S2–S17 [DOI] [PubMed] [Google Scholar]
- 34. Ullrich A., Bell J. R., Chen E. Y., Herrera R., Petruzzelli L. M., Dull T. J., Gray A., Coussens L., Liao Y. C., Tsubokawa M., Mason A., Seeburg P. H., Grunfeld C., Rosen O. M., Ramachandran J. (1985) Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761 [DOI] [PubMed] [Google Scholar]
- 35. Qureshi S. A., Ding V., Li Z., Szalkowski D., Biazzo-Ashnault D. E., Xie D., Saperstein R., Brady E., Huskey S., Shen X., Liu K., Xu L., Salituro G. M., Heck J. V., Moller D. E., Jones A. B., Zhang B. B. (2000) Activation of insulin signal transduction pathway and anti-diabetic activity of small molecule insulin receptor activators. J. Biol. Chem. 275, 36590–36595 [DOI] [PubMed] [Google Scholar]
- 36. Li M., Youngren J. F., Manchem V. P., Kozlowski M., Zhang B. B., Maddux B. A., Goldfine I. D. (2001) Small molecule insulin receptor activators potentiate insulin action in insulin-resistant cells. Diabetes 50, 2323–2328 [DOI] [PubMed] [Google Scholar]
- 37. Ding V. D., Qureshi S. A., Szalkowski D., Li Z., Biazzo-Ashnault D. E., Xie D., Liu K., Jones A. B., Moller D. E., Zhang B. B. (2002) Regulation of insulin signal transduction pathway by a small-molecule insulin receptor activator. Biochem. J. 367, 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pederson T. M., Kramer D. L., Rondinone C. M. (2001) Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes 50, 24–31 [DOI] [PubMed] [Google Scholar]
- 39. Trabuco L. G., Lise S., Petsalaki E., Russell R. B. (2012) PepSite: prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 40, W423–W427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parang K., Till J. H., Ablooglu A. J., Kohanski R. A., Hubbard S. R., Cole P. A. (2001) Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol. 8, 37–41 [DOI] [PubMed] [Google Scholar]
- 41. Wu J., Tseng Y. D., Xu C. F., Neubert T. A., White M. F., Hubbard S. R. (2008) Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat. Struct. Mol. Biol. 15, 251–258 [DOI] [PubMed] [Google Scholar]