

Abstract
Reactive oxygen species (ROS) contribute to the pathogenesis of many acute and chronic pulmonary disorders, including bronchopulmonary dysplasia (BPD), a respiratory condition that affects preterm infants. However, the mechanisms of susceptibility to oxidant stress in neonatal lungs are not completely understood. We evaluated the role of genetic background in response to oxidant stress in the neonatal lung by exposing mice from 36 inbred strains to hyperoxia (95% O2) for 72 h after birth. Hyperoxia-induced lung injury was evaluated by using bronchoalveolar lavage fluid (BALF) analysis and pathology. Statistically significant interstrain variation was found for BALF inflammatory cells and protein (heritability estimates range: 33.6–55.7%). Genome-wide association mapping using injury phenotypes identified quantitative trait loci (QTLs) on chromosomes 1, 2, 4, 6, and 7. Comparative mapping of the chromosome 6 QTLs identified Chrm2 (cholinergic receptor, muscarinic 2, cardiac) as a candidate susceptibility gene, and mouse strains with a nonsynonymous coding single-nucleotide polymorphism (SNP) in Chrm2 that causes an amino acid substitution (P265L) had significantly reduced hyperoxia-induced inflammation compared to strains without the SNP. Further, hyperoxia-induced lung injury was significantly reduced in neonatal mice with targeted deletion of Chrm2, relative to wild-type controls. This study has important implications for understanding the mechanisms of oxidative lung injury in neonates.—Nichols, J. L., Gladwell, W., Verhein, K. C., Cho, H.-Y., Wess, J., Suzuki, O., Wiltshire, T., Kleeberger, S. R. Genome-wide association mapping of acute lung injury in neonatal inbred mice.
Keywords: bronchopulmonary dysplasia, inflammation, quantitative trait locus, cholinergic receptor, muscarinic 2, cardiac
Bronchopulmonary dysplasia (BPD) is a chronic respiratory disease that is associated with prematurity and an underdeveloped pulmonary system (1, 2). Because the lung does not reach maturation until after birth, babies born prematurely are frequently incapable of adequate ventilation and gas exchange (1, 3). To compensate, respiratory support with supplemental oxygen and/or mechanical ventilation is used until respiration can be maintained independently (4). Despite the success of this strategy, subsets of infants are adversely affected and may develop BPD. Although the mechanisms are poorly understood (5, 6), the combination of developmental immaturity and ventilator support in these infants results in impaired alveolarization and abnormal vascularization, and decreased respiratory function has been reported beyond childhood (7, 8).
Initial efforts to understand differential susceptibility to BPD revealed a strong genetic component (9); clinical studies in twins reported heritability estimates ranging from 53 to 82% (10, 11). Although genetic studies have found associations between disease and genetic variants in pathological processes underlying BPD, including inflammation, antioxidant defense, lung development, and vascular development (7, 10, 12, 13), these studies are limited by small sample sizes, confounding variables (3), lack of reproducibility, and phenotypic heterogeneity and polygenicity (14, 15). Therefore, appropriate animal models are needed to better understand key events in late lung development during the transition from saccularization to alveolarization and the factors that contribute to BPD (2). Although models are reported in various species (16–18), mice are invaluable for genetic studies. In addition, there is concordance between BPD phenotypes and hyperoxic lung injury in neonatal mice, as hyperoxia augments reactive oxygen species (ROS) formation (19, 20), and neonatal exposure can produce similar impaired alveolarization and reduced lung function in adolescence (21–23).
Currently, genetic determinants of susceptibility to neonatal hyperoxic lung injury are not completely understood. Haplotype association mapping approaches in inbred strains of mice have yielded many important findings that have contributed significantly to our understanding of the genetic basis of susceptibility to several diseases (24). In the current study, our first objective was to establish a genetic neonatal model of hyperoxic lung injury in inbred strains of mice. The second objective was to use the injury phenotypes for genome-wide association (GWA) mapping to identify quantitative trait loci (QTLs) and candidate susceptibility genes that associate with neonatal hyperoxic lung injury.
MATERIALS AND METHODS
Animals and hyperoxia exposure
Male and female mice (4–10 wk) from 36 inbred strains were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice with targeted deletion of Chrm2 (cholinergic receptor, muscarinic 2, cardiac; 129S4/SvJaeJ X CF-1 F1-Chrm2−/−) and wild-type (129S4/SvJaeJ X CF-1 F1-Chrm2+/+) mice were obtained from the National Institute of Diabetes and Digestive and Kidney Diseases (Bethesda, MD, USA). Development of the Chrm2−/− mice and the effect of the deletion on expression in the lung have been described previously (25, 26). The mice were caged individually at the National Institute of Environmental Health Sciences (NIEHS) facility, maintained in a 12-h light-dark cycle at 72°F with 50% humidity, and provided food and water ad libitum. The animals were acclimated to these conditions for a week before mating. The males and females were housed together for 3–5 d; after cohabitation, the males were removed, and 1–3 of the females were housed per cage until pregnancy was confirmed. At 12–24 h after spontaneous birth, pups from multiple litters were pooled and randomly assigned to air or hyperoxia exposure groups. The litters from all strains were cross-fostered with Swiss Webster dams (Charles River, Wilmington, MA, USA) to ensure adequate and consistent maternal care. The litters with foster dams were maintained in room air or a chamber supplied with ≥95% oxygen (UHP grade, minimum purity 99.994%; National Welders, Durham, NC, USA) for 72 h. The chamber was opened for 30 min each morning for health checks, cage cleaning, and cross-fostering. After termination of exposure, the mice were weighed and euthanized by i.p. injection of pentobarbital sodium (104 mg/kg). Animal procedures were approved by the NIEHS Institutional Animal Care and Use Committee.
Bronchoalveolar lavage (BAL)
After the neonates were euthanized, the whole lung of each one was lavaged in situ 4 times with 100 μl Hanks' balanced salt solution (HBSS; pH 7.2–7.4), and the recovered BAL fluid (BALF) was immediately cooled to 4°C. For each mouse, the 4 BALF returns were centrifuged (500 g at 4°C), and the supernatant was removed for determination of total protein (an indicator of lung permeability; n≥5/group). Details of obtaining BALF differential cell counts and performing the Bradford protein assay have been published (27). Briefly, protein concentration was measured by the Bradford method (Bio-Rad, Hercules, CA, USA), according to the manufacturer's protocol. The cells were collected from the BALF by centrifugation and were resuspended in 0.5 ml of HBSS. The numbers of cells (per milliliter total BALF return) were counted with a hemocytometer as indicators of lung injury and inflammation. An aliquot (200 μl) of BALF cell suspension was cytocentrifuged (Shandon Southern Products, Pittsburgh, PA, USA) and stained with Wright-Giemsa stain (Diff-Quik; Baxter Scientific Products, McGaw Park, IL, USA) for differential cell analysis. Differential counts for epithelial cells, macrophages, and polymorphonuclear leukocytes (PMNs) were made by identifying 300 cells according to standard cytological techniques. Epithelial cells in particular were identified by the presence of cilia or as sheets of epithelium that are unique to neonatal BALF.
Lung histopathology
After euthanasia, the whole lung was fully inflated intratracheally in situ with zinc formalin (Fisher Scientific, Pittsburgh, PA, USA). The trachea was clamped and the lung removed and immersed in zinc formalin for 48 h. Fixed lungs were then placed in 70% ethanol until embedded, sectioned, and stained [hematoxylin and eosin (H&E)] for pathology assessment.
Heritability and correlation of phenotypes
Heritability estimates were calculated for each phenotype (28). The proportion of phenotype variation attributable to genetic background was determined by estimating the broad-sense heritability. Intrastrain correlations were estimated by the following formula:
where r1 is the intrastrain correlation estimate, MSB is the mean square of the between-strain comparison, MSW is the mean square of the within-strain correlation, and n is the number of animals per strain.
Correlation of phenotypes was determined using Pearson's correlation coefficients (Gene Network, University of Tennessee, Knoxville, TN, USA; http://www.genenetwork.org).
GWA mapping
GWA mapping was performed on quantitative phenotype data by using 3 different algorithms: SNPster, a haplotype-based approach (Genomics Institute, Novartis Research Foundation, San Diego, CA, USA); efficient mixed-model association (EMMA), using individual single-nucleotide polymorphism (SNP) associations (University of California, Los Angeles, CA, USA); and FastMap (Carolina Environmental Bioinformatics Center, University of North Carolina, Chapel Hill, NC, USA). The algorithms underlying these approaches are described elsewhere (29–32). SNPster was initially used to determine the association of phenotype (mean BAL cell count or protein level for each strain) with haplotype structure throughout the mouse genome, because its algorithm is very conservative and usually generates lower −log10 P values relative to other haplotype association methods for identical associations because it includes family-wise error rates in the determination of −log10 P values for haplotype/phenotype associations. We then used the FastMap algorithm to determine whether we could replicate the findings of SNPster, because FastMap uses a similar haplotype structure approach. Furthermore, it performs permutation tests to calculate levels of genome-wide statistical significance for each 3-SNP window and associated analysis of variance (ANOVA) F statistic. EMMA was also used, because it is based on single SNP associations, rather than the 3-SNP haplotype, for lung response phenotypes of each mouse for each strain. It also corrects for genetic relatedness and population structure. We reasoned that if we identified the same QTLs with more than one of the algorithms, we would have greater confidence in our findings than if we used either of them alone. Only those QTLs identified with SNPster and one or both of the other approaches were included for further consideration. This procedure may have led to the exclusion of important associations, but it provided a strong rationale for moving forward on gene candidates. We used the conservative Bonferroni correction in Table 3 to indicate whether the identified QTLs are significant or suggestive. −Log10 P values were considered significant if they exceeded 6.60 (genome-wide significance at P<0.05) and suggestive if they exceeded 6.06 (genome-wide suggestive at P<0.20). All of the strains have been genotyped to a level of ∼630,000 SNPs. However, only a subset of the SNPs (between 200,000 and 230,000) were informative for haplotype association mapping with the strains of mice that were used in our analyses (i.e., each haplotype had to be present in ≥10% of strains). Bonferroni corrections were calculated based on the 230,000 comparisons. Genes found within the QTLs were prioritized on the basis of whether nonsynonymous coding SNPs or promoter SNPs associate with differential responsiveness to hyperoxia exposure and biological plausibility.
Table 3.
Candidate susceptibility genes in QTLs for hyperoxia-induced inflammation in neonatal mouse lungs
| QTL | Gene symbol | SNP |
|||||
|---|---|---|---|---|---|---|---|
| rs ID | Location (Mb)a | AA change | Major allele | Minor allele | P | ||
| Hsnl1 | Cyp2j11 | 28105291 | 95.961592 | I 476 V | T, 21.6 ± 1.8a | C, 7.7 ± 2.6a | 0.0005 |
| Cyp2j6 | 28120055 | 96.202243 | C 192 F | C, 21.6 ± 1.8a | A, 7.7 ± 2.6a | 0.0005 | |
| Cyp2j9 | 28119885 | 96.252622 | I 90 V | T, 22.1 ± 1.8a | C, 7.6 ± 2.0a | 0.0003 | |
| 28119867 | 96.257924 | R 37 L | C, 21.8 ± 1.8a | A, 6.0 ± 1.6a | 0.0003 | ||
| Cyp2j5 | 28119831 | 96.300844 | P 394 T | C, 21.8 ± 1.8a | A, 6.1 ± 1.6a | 0.0003 | |
| Hsnl2 | Mgmt | 33644808 | 144.143061 | L 32 F | G, 22.1 ± 1.8a | T, 7.6 ± 2.0a | <0.0001 |
| 33669425 | 144.277666 | G 53 C | G, 24.8 ± 2.6a | T, 14.1 ± 1.5a | 0.0030 | ||
| Hsnl4 | Chrm2 | 30378838 | 36.474006 | P 265 L | C, 9.4 ± 0.9b | T, 4.0 ± 0.6b | <0.0001 |
Candidate susceptibility genes are indicated according to QTL. Locations are from build 37. All SNPs resulting in an amino acid (AA) change are listed for each gene. Phenotypic means ± se for the respective QTL phenotype for strains having the major or minor allele are reported; P values indicate significant differences between allelic groups.
Macrophages ×103.
Percentage PMNs.
We obtained the Chrm2 haplotype from the Mouse Phenome Database (Jackson Laboratory; http://phenome.jax.org), and noninformative SNPs were removed (i.e., no heterozygosity between strains or the minor allele was found in <10% of the strains). The strains were then arranged based on SNP similarity across the entire gene. The 3 haplotypes that were identified had ≥3 members (strains). Haplotypes with <3 members were not considered for statistical consideration, because a sample size of ≤2 is not appropriate. There are three 5′ and twenty-six 3′ untranslated region (UTR) SNPs in Chrm2, but none of them are associated with differential responses to hyperoxia among inbred strains, as we found with the nonsynonymous coding SNP, and they were not informative.
Statistics
Two-way ANOVA was used to evaluate the effects of exposure (air or hyperoxia) and strain (genotype) on disease phenotypes. Student-Newman-Keuls tests were used for a posteriori comparisons of the means. One-way ANOVA was used to compare the haplotype means, and Student's t test was used to compare the allelic group means in candidate genes. Statistical significance was accepted at P < 0.05.
RESULTS
Variation in hyperoxia-induced lung inflammation across strains of neonatal mice
Strain-specific responses were found in the hyperoxia-induced BALF phenotypes compared with the air-exposed controls (Table 1). For example, significant differences in mean BALF protein or PMNs were not found between the air- and hyperoxia-exposed 129S1/SvImJ mice (Fig. 1A, B and Table 1). In contrast, relative to their respective air-exposed controls, hyperoxia induced significantly greater increases in mean BALF protein and PMNs in the C3H/HeJ and PWD/PhJ neonatal mice than in the 129S1/SvImJ mice (Fig. 1A, B and Table 1).
Table 1.
Differential hyperoxic lung injury phenotypes in neonates from 36 strains of inbred mice
| Strain | Parameter | Protein (μg/ml) |
Total cells |
Neutrophils (%) |
Macrophages |
Epithelial cells |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Air | O2 | Air | O2 | Air | O2 | Air | O2 | Air | O2 | ||
| 129 × 1/SvJ | Mean | 70.7 | 98.6* | 33,333 | 35,833 | 0.08 | 2.48* | 21,894 | 24,362 | 5,578 | 4,557* |
| sem | 10.6 | 9.9 | 4,278 | 4,345 | 0.08 | 0.28 | 2,547 | 1,681 | 1,568 | 423 | |
| n | 6 | 9 | 6 | 9 | 4 | 7 | 4 | 7 | 4 | 7 | |
| 129S1/SvImJ | Mean | 71.8 | 109.3 | 36,806 | 53,194# | 0.19 | 2.72* | 27,608 | 40,730# | 5,367 | 7,523# |
| sem | 9.7 | 10.4 | 4,654 | 4,349 | 0.12 | 0.44 | 3,749 | 4,645 | 795 | 1,054 | |
| n | 9 | 18 | 9 | 18 | 7 | 13 | 7 | 13 | 7 | 13 | |
| A/J | Mean | 46.4 | 95.6* | 17,500 | 30,769 | 1.18 | 3.11* | 13,978 | 34,097 | 2,395 | 11,001# |
| sem | 2.7 | 12.3 | 1,887 | 5,134 | 0.64 | 0.77 | 1,446 | 3,409 | 446 | 2,467 | |
| n | 9 | 14 | 9 | 13 | 4 | 6 | 4 | 6 | 4 | 6 | |
| AKR/J | Mean | 60.2 | 89.4* | 20,673 | 24,688 | 5.44 | 5.38* | 14,621 | 19,674 | 2,703 | 5,651* |
| sem | 4.4 | 6.8 | 1,881 | 1,405 | 0.73 | 0.82 | 1,588 | 1,504 | 468 | 756 | |
| n | 13 | 16 | 13 | 16 | 3 | 7 | 3 | 7 | 3 | 7 | |
| BALB/cBy | Mean | 35.2 | 64.3* | 24,063 | 24,853 | 1.46 | 5.11* | 21,766 | 2,4932 | 3,282 | 3,994* |
| sem | 8.0 | 5.4 | 1,866 | 1,991 | 0.59 | 1.74 | 1,073 | 2,592 | 146 | 1,026 | |
| n | 4 | 17 | 4 | 17 | 5 | 6 | 5 | 6 | 5 | 6 | |
| BALB/cJ | Mean | 28.4 | 80.6* | 39,375 | 39,643 | 0.47 | 4.62* | 33,061 | 43,929# | 6,292 | 7,167 |
| sem | 1.7 | 6.3 | 3,576 | 1,864 | 0.08 | 0.67 | 4,076 | 4,913 | 843 | 774 | |
| n | 6 | 8 | 6 | 7 | 5 | 7 | 5 | 7 | 5 | 7 | |
| BTBR T+tf/J | Mean | 76.2 | 111.1 | 31,750 | 28,456 | 0.00 | 0.41* | 24,981 | 16,904* | 7,019 | 7,334# |
| sem | 8.3 | 9.0 | 5,327 | 3,350 | 0.00 | 0.16 | 6,753 | 4,676 | 1,184 | 1,447 | |
| n | 13 | 16 | 10 | 17 | 5 | 9 | 5 | 9 | 5 | 9 | |
| BUB/BnJ | Mean | 54.6 | 98.0* | 10,313 | 26,111 | 0.17 | 3.54* | 7,398 | 19,767 | 2,893 | 3,769* |
| sem | 8.4 | 6.4 | 1,288 | 2,119 | 0.09 | 0.67 | 1,053 | 2,531 | 485 | 667 | |
| n | 8 | 8 | 8 | 9 | 9 | 8 | 8 | 8 | 8 | 8 | |
| C3H/HeJ | Mean | 43.1 | 249.1# | 12,692 | 37,938 | 1.74 | 13.30# | 11,185 | 23,870 | 2,257 | 5,802 |
| sem | 8.0 | 16.5 | 1,216 | 4,227 | 0.33 | 1.02 | 1,670 | 3,047 | 373 | 570 | |
| n | 13 | 25 | 13 | 20 | 9 | 18 | 9 | 18 | 9 | 18 | |
| C57BL/10J | Mean | 46.2 | 79.6* | 62,292 | 61,136# | 1.56 | 8.24 | 53,483 | 42,492# | 6,635 | 13,647# |
| sem | 6.1 | 10.0 | 6,385 | 3,897 | 0.53 | 1.21 | 5,875 | 3,540 | 981 | 1,389 | |
| n | 6 | 11 | 6 | 11 | 6 | 11 | 6 | 11 | 6 | 11 | |
| C57BL/6J | Mean | 48.3 | 135.9 | 23,875 | 43,355 | 1.10 | 8.60 | 19,138 | 31,573 | 4,464 | 7,381# |
| sem | 7.4 | 14.1 | 843 | 4,626 | 0.40 | 1.12 | 679 | 4,449 | 772 | 913 | |
| n | 12 | 19 | 10 | 19 | 10 | 19 | 10 | 19 | 10 | 19 | |
| C57BR/cdJ | Mean | 72.8 | 92.1* | 15,583 | 16,250* | 0.53 | 5.07* | 11,551 | 10,305* | 4,940 | 3,769* |
| sem | 7.5 | 8.6 | 1,156 | 1,021 | 0.17 | 0.63 | 1,113 | 916 | 676 | 373 | |
| n | 13 | 22 | 15 | 22 | 10 | 15 | 10 | 15 | 10 | 15 | |
| C57L/J | Mean | 46.3 | 183.9# | 14,896 | 27,679 | 0.13 | 7.50 | 11,342 | 21,072 | 3,317 | 5,381* |
| sem | 9.3 | 25.1 | 1,527 | 1,925 | 0.06 | 3.29 | 2,044 | 2,581 | 521 | 884 | |
| n | 12 | 14 | 12 | 14 | 7 | 8 | 7 | 8 | 7 | 8 | |
| C58/J | Mean | 63.4 | 154.3 | 34,554 | 34,803 | 1.56 | 11.44 | 24,859 | 25,410 | 9,191 | 7,765# |
| sem | 3.8 | 10.5 | 4,599 | 3,031 | 0.57 | 1.42 | 6,688 | 2,558 | 1,470 | 766 | |
| n | 14 | 19 | 14 | 19 | 8 | 14 | 8 | 14 | 8 | 14 | |
| CAST/EiJ | Mean | 85.1 | 104.3* | 11,000 | 11,458* | 4.80 | 8.52 | 7,584 | 7,064* | 2,680 | 3,154* |
| sem | 17.1 | 26.4 | 1,000 | 1,528 | 1.78 | 3.05 | 1,068 | 1,049 | 286 | 736 | |
| n | 7 | 6 | 5 | 6 | 4 | 5 | 4 | 5 | 4 | 5 | |
| CBA/J | Mean | 36.3 | 102.1* | 23,000 | 26,375 | 0.09 | 9.88 | 17,851 | 23,320 | 2,663 | 4,373* |
| sem | 4.6 | 13.2 | 2,682 | 2,792 | 0.09 | 1.98 | 2,374 | 2,540 | 199 | 837 | |
| n | 10 | 9 | 10 | 10 | 7 | 5 | 7 | 5 | 7 | 5 | |
| CE/J | Mean | 82.0 | 157.5 | 17,404 | 24,438 | 0.34 | 5.76 | 6,424 | 14,326* | 2,271 | 5,265* |
| sem | 5.1 | 7.3 | 2,353 | 1,588 | 0.21 | 0.56 | 730 | 1,625 | 218 | 394 | |
| n | 12 | 19 | 13 | 20 | 5 | 11 | 5 | 11 | 5 | 11 | |
| DBA/2J | Mean | 68.3 | 181.1# | 18,750 | 25,703 | 1.00 | 5.42 | 14,412 | 18,159 | 3,920 | 4,531* |
| sem | 10.6 | 22.9 | 1,518 | 2,848 | 0.38 | 0.95 | 1,800 | 2,400 | 451 | 497 | |
| n | 16 | 17 | 16 | 16 | 10 | 12 | 10 | 12 | 10 | 12 | |
| FVB/NJ | Mean | 101.2 | 202.1# | 15,000 | 37,981 | 2.38 | 8.71 | 13,037 | 29,737 | 3,225 | 7,271 |
| sem | 16.3 | 19.3 | 1,869 | 3,332 | 1.19 | 1.12 | 2,533 | 2,863 | 517 | 1,344 | |
| n | 11 | 13 | 11 | 13 | 7 | 11 | 7 | 11 | 7 | 11 | |
| I/LnJ | Mean | 39.4 | 181.8# | 8,125 | 7,505* | 1.40 | 13.20# | 5,481 | 3,970* | 23,856 | 2,128 |
| sem | 3.7 | 19.9 | 360 | 319 | 0.21 | 2.16 | 454 | 218 | 193 | 557 | |
| n | 4 | 6 | 4 | 6 | 4 | 6 | 4 | 6 | 4 | 6 | |
| KK/HlJ | Mean | 68.3 | 142.1 | 15,625 | 22,353 | 0.00 | 5.58* | 10,725 | 12,729* | 3,025 | 5,619* |
| sem | 12.6 | 11.5 | 2,113 | 1,711 | 0 | 2.86 | 1,475 | 2,366 | 225 | 190 | |
| n | 8 | 17 | 8 | 17 | 3 | 4 | 3 | 4 | 3 | 4 | |
| MA/MyJ | Mean | 108.3 | 184.7# | 19,167 | 17,763* | 2.27 | 5.43* | 14,369 | 11,501* | 4,698 | 4,356* |
| sem | 11.8 | 22.6 | 1,758 | 1,561 | 1.22 | 1.21 | 1,562 | 1,176 | 527 | 548 | |
| n | 9 | 19 | 6 | 19 | 5 | 11 | 5 | 11 | 5 | 11 | |
| MOLF/EiJ | Mean | 89.0 | 116.3 | 16,250 | 9,722* | 0.58 | 5.00* | 10,806 | 2,996* | 5348 | 1,783* |
| sem | 33 | 21 | 2,339 | 1,455 | 0.38 | 1.69 | 1,682 | 454 | 926 | 777 | |
| n | 4 | 9 | 4 | 9 | 4 | 3 | 4 | 3 | 4 | 3 | |
| MRL/MpJ | Mean | 77.6 | 137.5 | 25,078 | 24,196 | 1.07 | 6.55 | 25,813 | 16,049* | 7,131 | 5,967* |
| sem | 8.4 | 9.0 | 1,994 | 1,270 | 0.30 | 1.01 | 6,899 | 1,791 | 1,464 | 712 | |
| n | 17 | 28 | 16 | 28 | 6 | 7 | 6 | 7 | 6 | 7 | |
| NOR/LtJ | Mean | 68.4 | 161.9# | 19,083 | 23,529 | 0.85 | 6.02* | 19,861 | 15,041* | 3,946 | 7,284 |
| sem | 5.7 | 9.0 | 1,995 | 1,658 | 0.30 | 1.31 | 2,279 | 1,637 | 387 | 1,050 | |
| n | 15 | 17 | 15 | 17 | 4 | 5 | 4 | 5 | 4 | 5 | |
| NZO/HlLtJ | Mean | 67.1 | 157.5# | 19,583 | 19,844* | 2.23 | 8.90 | 15,703 | 13,301* | 3,432 | 5,059* |
| sem | 4.6 | 7.1 | 2,732 | 3,138 | 0.29 | 4.08 | 1,742 | 2,756 | 878 | 1,249 | |
| n | 3 | 7 | 3 | 8 | 3 | 4 | 3 | 4 | 3 | 4 | |
| NZW/LacJ | Mean | 79.7 | 148.1 | 37,188 | 52,250# | 0.25 | 7.28 | 32,965 | 46,611# | 6,359 | 6,413 |
| sem | 7.0 | 8.7 | 3,673 | 6,957 | 0.17 | 2.94 | 4,250 | 7,970 | 1,301 | 762 | |
| n | 8 | 10 | 8 | 10 | 4 | 7 | 4 | 7 | 4 | 7 | |
| P/J | Mean | 92.1 | 136.7 | 8,875 | 20,987 | 3.67 | 18.07# | 8,824 | 12,417* | 4,492 | 11,754# |
| sem | 12.5 | 15.5 | 1,723 | 4,579 | 0.79 | 5.30 | 2,382 | 3,944 | 1,223 | 4123 | |
| n | 11 | 21 | 10 | 19 | 7 | 7 | 7 | 7 | 7 | 7 | |
| PL/J | Mean | 86.3 | 173.9# | 24,531 | 25,417 | 6.00 | 6.15 | 21,213 | 16,614* | 5,568 | 2,849* |
| sem | 14.7 | 28.8 | 2,302 | 1,916 | 5.02 | 1.29 | 318 | 2,710 | 1,331 | 622 | |
| n | 8 | 15 | 8 | 15 | 3 | 9 | 3 | 9 | 3 | 9 | |
| PWD/PhJ | Mean | 58.6 | 166.1# | 12,375 | 24,375 | 5.48 | 17.85# | 8,555 | 13,834* | 3,379 | 5,638* |
| sem | 13.4 | 24.7 | 1,782 | 2,767 | 2.75 | 2.80 | 1,591 | 2,648 | 858 | 443 | |
| n | 10 | 14 | 10 | 12 | 4 | 10 | 4 | 10 | 4 | 10 | |
| PWK/PhJ | Mean | 55.8 | 167.9# | 10,446 | 17,188* | 0.14 | 20.07# | 6,595 | 9,996* | 4,286 | 4,821* |
| sem | 5.9 | 17.8 | 968 | 1,306 | 0.10 | 2.08 | 1,066 | 1,352 | 337 | 589 | |
| n | 15 | 27 | 14 | 28 | 7 | 16 | 7 | 16 | 7 | 16 | |
| RIIIS/J | Mean | 84.4 | 88.8* | 22,031 | 20,909 | 0.96 | 1.71* | 13,610 | 14,098* | 4,862 | 5,391* |
| sem | 7.2 | 9.5 | 1,844 | 1,328 | 0.19 | 0.40 | 664 | 2,078 | 1,374 | 206 | |
| n | 8 | 10 | 8 | 11 | 5 | 7 | 5 | 7 | 5 | 7 | |
| SJL/J | Mean | 57.1 | 104.6* | 13,000 | 18,125* | 2.26 | 8.96 | 8,890 | 9,304* | 3,780 | 6,832 |
| sem | 7.1 | 15.5 | 1,090 | 3,143 | 0.73 | 1.47 | 773 | 1,982 | 432 | 148 | |
| n | 5 | 7 | 5 | 8 | 5 | 7 | 5 | 7 | 5 | 7 | |
| SM/J | Mean | 37.3 | 112.9 | 14,688 | 17,143* | 1.57 | 7.13 | 10,167 | 11,627* | 2,665 | 3,016* |
| sem | 3.0 | 16.1 | 1,386 | 1,630 | 0.47 | 0.99 | 546 | 1,041 | 205 | 155 | |
| n | 4 | 10 | 4 | 7 | 3 | 6 | 3 | 6 | 3 | 6 | |
| SWR/J | Mean | 47.0 | 80.7* | 30,938 | 51,618# | 0.50 | 1.64 | 27,227 | 38,336# | 3,560 | 8,980# |
| sem | 7.5 | 11.6 | 2,571 | 6,033 | 0.22 | 0.46 | 2,666 | 6,036 | 200 | 1,072 | |
| n | 7 | 17 | 4 | 17 | 4 | 13 | 4 | 13 | 4 | 13 | |
| WSB/EiJ | Mean | 35.9 | 85.0* | 16,250 | 22,292 | 0.77 | 4.89* | 15,061 | 16,485* | 3,631 | 4,674 |
| sem | 3.1 | 13.3 | 4,705 | 2,509 | 0.77 | 1.42 | 5,012 | 1,515 | 687 | 806 | |
| n | 4 | 8 | 4 | 6 | 3 | 7 | 3 | 6 | 3 | 6 | |
For each strain, the phenotypic mean, sem, and sample size (n) for the phenotypes are listed. Air, filtered room air exposure for 4 d; O2, 3 d of hyperoxia exposure.
P < 0.05 vs. highest-responding strain;
P < 0.05 vs. lowest-responding strain.
Figure 1.

Histopathologic and BALF responses to hyperoxia exposure in 129S1/SvImJ, C3H/HeJ, and PWD/PhJ neonates. A) BALF protein concentration. B) PMNs. C) Representative H&E-stained histopathology specimens from formalin-fixed neonatal lungs. Arrows show differences in peribronchiolar edema, vascular leakage, cell infiltrate, and alveolar septal thickness. AV, alveoli; BR, bronchus or bronchiole; BV, blood vessel. *P < 0.05 vs. indicated strain; #P < 0.05 vs. air-exposed control; Student-Newman-Keuls test.
Across all strains, significant interstrain variation was found for hyperoxia-induced differences in the mean number of BALF total cells relative to the air-exposed controls. Significantly more total cells were found after hyperoxia in a subset of strains (e.g., 129S1/SvImJ, C57BL/10J, and NZW/LacJ) than in the air-exposed controls, whereas the other strains had reduced or unchanged BALF total cells (e.g., I/LnJ, MOLF/EiJ, and CAST/EiJ; Table 1). The heritability of this phenotype was 47.6%.
Similar to BALF total cells, mean counts of hyperoxia-induced BALF macrophages, epithelial cells, and PMNs were significantly different across the strains of neonates (Table 1 and Fig. 2). Compared to counts in the respective air-exposed controls, the greatest increases in PMNs were found in the PWD/PhJ, P/J, and PWK/PhJ neonates, and the fewest PMNs were found in the BTBR T+tf/J, SWR/J, and RIIIS/J neonates (Fig. 2A and Table 1). The greatest mean counts of hyperoxia-induced BALF macrophages were found in the NZW/LacJ, BALB/cJ, and C57BL/10J neonates, and the lowest were found in the MOLF/EiJ, I/LnJ, and CAST/EiJ neonates (Fig. 2B and Table 1). Similarly, the greatest mean counts of hyperoxia-induced epithelial cells were found in the A/J, P/J, and C57BL/10J neonates, whereas the lowest were found in the MOLF/EiJ and I/LnJ neonates (Fig. 2C and Table 1). Heritability estimates of the BALF cell types were 39.8% (epithelial cells), 48.7% (PMNs), and 55.7% (macrophages).
Figure 2.

Strain distribution patterns of lung injury phenotypes from BALF after 72 h exposure to hyperoxia. A) PMNs as a percentage of BALF total cells. B) Macrophages per milliliter BALF. C) Epithelial cells per milliliter BALF. D) Protein concentration in BALF). Bars represent means ± sem (n=5–28/strain).
Compared to levels in the respective air-exposed controls, hyperoxia caused significant increases in mean BAL protein concentration in all strains (Table 1). However, the amount of hyperoxia-induced increase in BAL protein varied significantly between strains, ranging from 64.3 ± 5.4 μg/ml in the BALB/cByJ to 249.1 ± 16.5 μg/ml in the C3H/HeJ (Fig. 2D and Table 1) strain. The intra- and interstrain variations in the BAL protein response to room air and hyperoxia exposure was greater than those in the other BAL phenotypes. Consequently, the heritability estimate for BAL protein was the lowest (33.6%) among all phenotypes.
Correlation of hyperoxic lung injury phenotypes
We then asked whether disease phenotypes correlate across strains, to provide a better understanding of disease pathogenesis and potential mechanisms of injury. Mean BALF total, macrophage, and epithelial cell counts, but not PMN counts or protein level, correlated significantly with one another after air exposure (Pearson's R correlation coefficients; Supplemental Fig. S1). After exposure to hyperoxia, a weak but statistically significant correlation was found between PMNs and protein; total, macrophage, and epithelial cell counts also correlated significantly with one another (Supplemental Fig. S1).
Histopathology of hyperoxic lung injury in neonatal mice
Lung maturation and injury were compared by histopathology in the hyperoxia-nonresponsive 129S1/SvImJ neonates and the hyperoxia-responsive C3H/HeJ and PWD/PhJ neonates. Similar lung development (branched septae and multilobular alveoli) was found in each of the air-exposed strains by postnatal d 4 (Fig. 1C). However, strain-specific variation in lung injury was found after 72 h of hyperoxia. Minimal inflammatory cells and peribronchiolar edema were found in the hyperoxia-exposed 129S1/SvImJ neonates compared to those in the air-exposed controls. However, more severe injury, characterized by vascular leakage and inflammatory cell influx, was found in the C3H/HeJ lungs, whereas alveolar edema and thickened alveolar septae were found in the PWD/PhJ lungs.
Haplotype association mapping with disease phenotypes
To better understand the genetic basis of differential susceptibility to hyperoxia exposure in neonates, we used GWA mapping for each injury phenotype. Mapping of BALF macrophages by SNPster after hyperoxia exposure identified loci on chromosomes 4 and 7 with −log10 P > 5 (Table 2). These same 2 loci were also identified by FastMap and EMMA and have been named hyperoxia susceptibility in neonatal lungs 1 (Hsnl1) and Hsnl2, respectively. Mapping of hyperoxia-induced increases in BALF PMNs with SNPster identified a peak on chromosome 6, with −log10 P=4.60, and peaks on chromosomes 2 and 7, with −log10 P=3.97 and 4.33, respectively (Fig. 3 and Table 2). These 3 loci for the PMN phenotype were also identified by FastMap, with corresponding significant −log10 P = 7.48, 6.47, and 7.28, respectively (Table 2), and they have been named Hsnl4, Hsnl3, and Hsnl5. GWA analyses of other hyperoxia response phenotypes with SNPster did not identify peaks greater than −log10 P = 3.70 or were not confirmed with FastMap or EMMA.
Table 2.
QTLs and −log10 P values for hyperoxia-induced inflammation identified by haplotype association mapping in 36 neonatal inbred mouse strains
| Phenotype and chromosome | QTL | QTL type | Location (Mb) | −Log10
P |
|
|---|---|---|---|---|---|
| SNPster | FastMap | ||||
| Macrophages | |||||
| 4 | Hsnl1 | Significant | 95.963320–97.095051 | 5.95 | 6.88 |
| 7 | Hsnl2 | Suggestive | 143.452455–145.619658 | 5.38 | 6.30 |
| Neutrophils | |||||
| 2 | Hsnl3 | Suggestive | 125.157670–129.353319 | 3.97 | 6.47 |
| 6 | Hsnl4 | Significant | 35.750448–36.712935 | 4.60 | 7.48 |
| 7 | Hsnl5 | Significant | 103.108764–109.235511 | 4.33 | 7.28 |
QTLs were given the name hyperoxia susceptibility in neonatal lungs (Hsnl). Genome-wide significant or suggestive association was determined by Bonferroni corrections for multiple testing (see Materials and Methods for details).
Figure 3.

Genome-wide haplotype association map of BALF PMNs in 36 inbred strains of neonatal mice exposed to hyperoxia. A) Manhattan plot for hyperoxia-induced BALF PMNs (percentage of BALF total cells). Cumulative genomic position is on the x axis; −log10 P values are on the y axis. Circled rectangle (chromosome 6) identifies a locus with a −log10 P value of 4.60, indicative of association with hyperoxia-induced PMN infiltration. B) Enlarged view of the chromosome 6 locus identified by GWA of the PMN response to hyperoxia in inbred strains of mice and the underlying gene Chrm2.
We next queried the Mouse Phenome Database for informative SNPs in genes located within the QTLs, to determine whether any of them were associated with susceptibility to hyperoxia-induced lung injury. The minor allele SNPs must have been present in ≥10% of inbred strains to be considered for further analysis. Four cytochrome P450, family 2, subfamily j (Cyp2j) polypeptide genes, Cyp2j11, Cyp2j6, Cyp2j9, and Cyp2j5), were found in Hsnl1. Hyperoxia-induced changes in BALF macrophages were significant in strains homozygous for the minor allele SNPs, when compared to strains homozygous for the major allele (Table 3). In Hsnl2, nonsynonymous coding SNPs were found in O-6-methylguanine-DNA methyltransferase (Mgmt), and they significantly associated with differential susceptibility to hyperoxia among the 36 strains.
We used the same search strategy to query Hsnl4 for hyperoxia susceptibility genes, and Chrm2 was found directly beneath the peak of Hsnl4 (Fig. 3B). The haplotype structure for synonymous and nonsynonymous coding SNPs was investigated, and 3 Chrm2 haplotypes were identified (Fig. 4A). Significantly greater hyperoxia-induced increases in BALF PMNs were found in the mice with Chrm2 haplotype 1 than in those with haplotypes 2 or 3, and the PMN response to hyperoxia was significantly greater in the mice with Chrm2 haplotype 2 than in those with haplotype 3 (Fig. 4B). Furthermore, the nonsynonymous C→T coding SNP (rs30378838; 36.474006 Mb) in exon 1 causes replacement of the proline residue at position 265 with leucine. Significantly reduced BALF PMN counts were found in the mice homozygous for T allele (9.6±0.9% PMNs/μl) compared to those in the strains homozygous for the C allele (4.2±0.6% PMNs/μl; Fig. 4C and Table 3).
Figure 4.

Haplotype structure of Chrm2 and the association with susceptibility to hyperoxia-induced neutrophilia in BALF in inbred neonatal mice. A) Truncated haplotype structure of Chrm2 with synonymous (Cs) and nonsynonymous (Cn) coding SNPs in strains of mice with similar haplotype. Uppercase symbols represent validated nucleotides. Lowercase symbols represent imputed nucleotides. B) Mean number of PMNs in strains of mice with the Chrm2 haplotypes 1 (CCT; n=3 strains), 2 (GCC; n=17 strains), and 3 (CTT; n=14 strains). *P < 0.05, Student-Newman-Keuls test. C) Mean number of BALF PMNs for strains (n=21) homozygous for the rs30378838 C allele and strains (n=15) homozygous for the T allele. *P < 0.05; t test.
We prioritized the subsequent investigation of these candidate genes on the basis of biological plausibility and strength of association of putative gene SNPs with responses to hyperoxia among the inbred strains (Table 3) and selected Chrm2. Moreover, the Chrm2−/− mouse had been developed (26) and was available for investigation in this model.
Targeted disruption of Chrm2 reduces hyperoxia-induced lung injury
We further investigated Chrm2 by comparing the pulmonary response to hyperoxia exposure in the Chrm2+/+ and Chrm2−/− mice. We found statistically significant genotype, exposure, and interaction effects on BALF protein concentration (Fig. 5A). Relative to respective air-exposed controls, mean BALF protein concentration was significantly increased in the Chrm2+/+ neonates, but no increase was found in the Chrm2−/− neonatal lungs. Further, hyperoxia-induced BALF protein concentration was significantly greater in the Chrm2+/+ mice than in the Chrm2−/− mice. Moreover, increased alveolar edema, thickening of the bronchial epithelium, and alveolar septae proliferation were found in the Chrm2+/+ compared with the Chrm2−/− neonates after hyperoxia (Fig. 5B). Hyperoxic effects were found for other phenotypes (e.g., BALF PMNs), but no genotype effects were detected.
Figure 5.
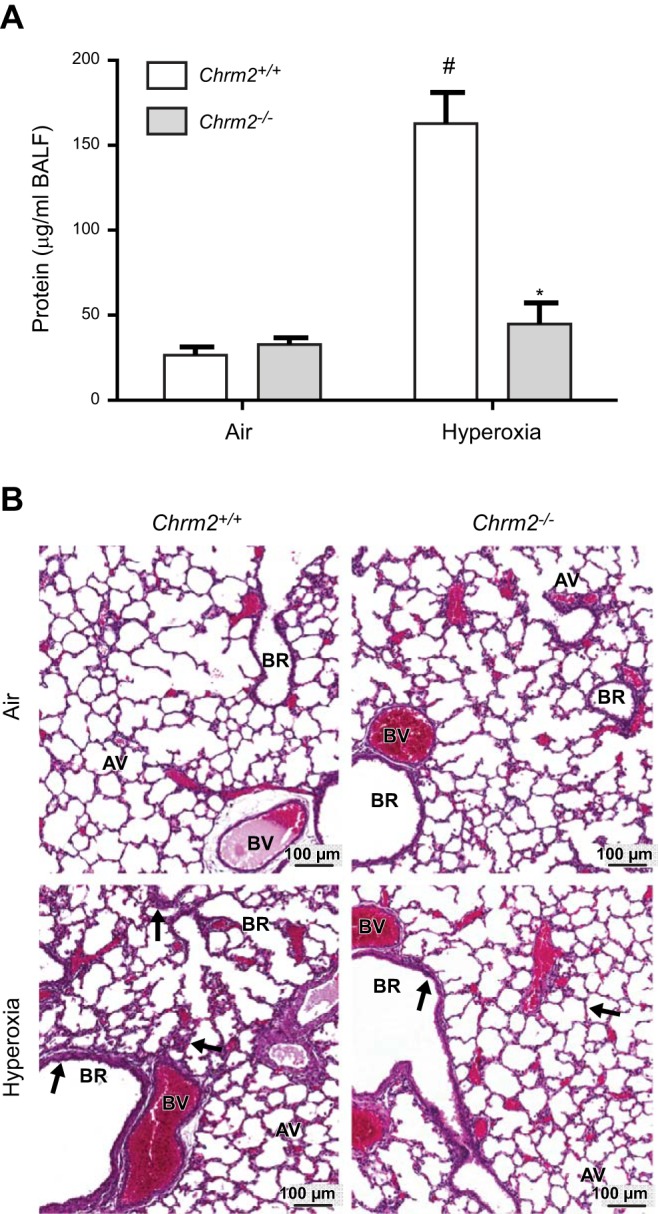
Differential pulmonary responses to hyperoxia exposure in Chrm2+/+ and Chrm2−/− neonates. A) BALF protein concentrations. B) Representative H&E-stained histopathology images. Arrows indicate differences in alveolar edema and thickening of the bronchial epithelium and alveolar septae. AV, alveoli; BR, bronchus or bronchiole; BV, blood vessel. Bars represent means ± sem (n=6–9/group). *P < 0.05 vs. Chrm2+/+; #P < 0.05 vs. air; 2-way ANOVA.
DISCUSSION
Despite decades of research on the role of oxidant injury in neonatal lung disease, differential susceptibility remains poorly understood. This is the first study to evaluate hyperoxia-induced lung injury phenotypes in neonates across a genetically diverse panel of inbred strains of mice. Notably, strain distribution patterns of injury phenotypes in neonates did not replicate those in adult inbred mice exposed to hyperoxia. For example, neonatal C3H/HeJ mice were characterized as hyperoxia-responsive for inflammatory reactions in the present study, but C3H/HeJ mice were resistant to hyperoxic lung injury in a previous study of 6 strains of adult male mice (33). Similarly, hyperoxia-induced hyperpermeability was significantly increased in adult male BALB/cJ and C57BL/10J mice (33), whereas increases in BALF protein were minimal in neonates. Increased survival times were also found in adult female C3H/HeJ mice compared with other strains, such as 129S1/SvImJ, which we found to be resistant as neonates (34). Vancza et al. (35) also found age-dependent effects after ozone (O3) exposure: C3H/HeJ neonates were very susceptible to O3 as neonates but resistant as adults. These observations suggest that susceptibility mechanisms differ between adults and neonates.
The neonatal mouse lung at full term is undergoing saccular lung development that is comparable to that of the preterm human lung and very different from that of a mature adult lung. This study was designed to identify genetic factors that may have a role in neonatal lung injury, and we have demonstrated that it is important to consider the interaction between hyperoxia exposure with lung growth and development in the neonates as an important cofactor for hyperoxic lung injury. We also found that few strains (e.g., PWD/PhJ and C3H/HeJ) had severe lung injury, and most of the strains had moderate, yet different, responses to the measured phenotypes. This finding is similar to clinical observations, as few preterm infants develop chronic lung disease or BPD after treatment with oxygen and respiratory support.
We further demonstrated that the strain distribution pattern for each of the quantitative phenotypes was continuous, which suggests that multiple genes contribute to the injury phenotypes. The large and genetically diverse panel of inbred strains phenotyped in the present study enables leverage of a rich and expanding SNP database for genetic analyses. Our analyses in an age-relevant animal model identified novel QTLs with several candidate susceptibility genes that associate with disease phenotypes. Candidate genes were limited to those having promoter or nonsynonymous coding SNPs as a means for prioritization; however, this does not imply that intronic SNPs are not important. Among the candidate susceptibility genes of interest are Cyp2j6, Mgmt, and Chrm2 because of their biological plausibility in oxidative- and immune-mediated stress responses. Cyp2j6 has been implicated as an anti-inflammatory mediator in acute allergic airway inflammation in ovalbumin-challenged mice and has a human orthologue, CYP2J2 (36, 37). Furthermore, studies of human CYP2J2 in vitro have demonstrated that its activity associated with peroxisome proliferator-activated receptor (PPAR)α and PPARγ activation and NF-κB and matrix metalloproteinase 9 inhibition (38–40).
Another candidate gene, Mgmt, encodes O-6-methylguanine-DNA methyltransferase, which is involved in DNA repair, and polymorphisms have been described in various cancers and contribute to epigenetic modifications (41). A specific role for Mgmt in development has not been elucidated, but it may have important implications, as epigenetic alterations play a critical part in developmental origins of lung disease (42). Joss-Moore et al. (42) demonstrated that mechanical ventilation with supplemental oxygen in neonates results in epigenetic changes that lead to altered expression of developmental genes, including elastin, vascular endothelial growth factor (VEGF), and PPARγ. Thus, further investigation of Mgmt polymorphisms may yield a novel epigenetic role for the gene in neonatal hyperoxic lung injury.
Chrm2 encodes the M2 muscarinic acetylcholine receptor, a member of the muscarinic receptor family (M1–M5). All members of this receptor family belong to the superfamily of G-protein-coupled receptors. Muscarinic receptors modulate airway reactivity and have been studied in the pathogenesis of allergies and asthma (43). More recently, a new role of muscarinic receptors has been found in nonneuronal cholinergic signaling (44). Of particular interest are studies that reported the influence of muscarinic receptor activation on the release of proinflammatory cytokines in smooth muscle cells (45), bronchial epithelial cells (46), and alveolar macrophages (47). Specifically, human airway smooth muscle cells were found to secrete interleukin 8 when exposed in vitro to a muscarinic agonist and cigarette smoke extract (46). M2 muscarinic receptor expression differences have also been described in sputum samples from subjects with chronic obstructive pulmonary disease (45, 46). Although the effect of muscarinic receptor expression on pulmonary inflammatory cells is still unknown, neuroimmune crosstalk has recently been suggested as a regulatory mechanism of inflammation and infection (48, 49). For example, Rosas-Ballina et al. (48) found that acetylcholine acts on splenic macrophages to inhibit the production of proinflammatory tumor necrosis factor-α.
A potential role for Chrm2 in differential susceptibility to hyperoxia-induced neonatal lung injury was supported by 2 lines of evidence. We initially found that both the haplotype structure of Chrm2 and a nonsynonymous SNP (P265L) were associated with differential susceptibility to hyperoxia-induced inflammation and lung injury. The P265L substitution is found within the third intracellular loop of the receptor, a region known to be involved in receptor/G-protein coupling and regulation of receptor activity (50). Additional investigation is needed to validate the functional prediction. The second line of supportive evidence for Chrm2 in hyperoxia susceptibility is that, relative to wild-type mice, targeted deletion of Chrm2 significantly reduced hyperoxia-induced lung injury. Targeted deletion of Chrm2 modulated hyperoxia-induced hyperpermeability rather than PMNs, the phenotype used to identify Chrm2 by GWA mapping. We found only moderate hyperoxia-induced neutrophilia in wild-type and Chrm2−/− mice, and the difference between the strains was not significant. The lack of a genotype effect on the neutrophilia induced by hyperoxia may be attributable to the genetic background of the mice. Although hyperoxia-induced inflammation correlated with increased BALF protein concentrations in some strains, the 2 phenotypes did not correlate in others. Overall, among all strains, we have shown that these phenotypes have only a weak correlation (Fig. 2 and Supplemental Fig. S1). The role of strain background on the effects of gene mutation has been described in many other models (51–56). Investigators have found that the specific phenotypes attributed to a gene alteration in 1 strain of mouse were reduced or that additional subphenotypes were found in another strain of mouse due to different strain-specific gene penetrance. It is also possible that inflammation is sufficient for BAL protein changes in some strains, but not in others. Additional studies with Chrm2 back-crossed to different inbred backgrounds are needed to address this question. Nevertheless, together these studies support the hypothesis that Chrm2 has a role in the neonatal lung injury and inflammation phenotypes induced by hyperoxia. More studies are necessary to identify additional phenotypic effects of Chrm2 deletion and the mechanisms through which Chrm2 modulates hyperoxia response.
In summary, we have developed a genetic model of hyperoxia-induced lung injury in neonatal mice, with phenotypes that resemble some of those found in BPD. Furthermore, using GWA, we identified QTLs for inflammation phenotypes, which demonstrated that genetic background is an important determinant of susceptibility to hyperoxia-induced injury of the neonatal mouse lung and is likely to be age-dependent. Candidate susceptibility genes, including Chrm2, were identified in the QTLs. Results of studies with neonatal mice deficient in Chrm2 were consistent with the hypothesis that Chrm2 contributes to the pathogenesis of hyperoxic lung injury. Further investigation is needed to elucidate the role of other candidate susceptibility genes and to better understand the exact mechanism by which Chrm2 modulates hyperoxia response and its relevance in human cohorts. Results of these studies have important implications for identifying the population at risk of neonatal lung injury and may provide therapeutic targets through which injury can be prevented.
Supplementary Material
Acknowledgments
The authors thank L. M. DeGraff [National Institute of Environmental Health Sciences (NIEHS)], L. Perrow (NIEHS), and H. Price (Alion Science and Technology, McLean, VA, USA) for assistance with animal breeding and exposures; the NIEHS Histology Core for tissue processing and staining; and Drs. D. Cook and M. Fessler for critical review of the manuscript.
This work was supported by a Director's Challenge Program and the Intramural Research Program, NIEHS, National Institutes of Health, U.S. Department of Health and Human Services.
Author contributions: J.L.N., W.G., H.-Y.C., and S.R.K. conceived and designed the experiments; J.L.N., W.G., and K.C.V. performed the experiments; J.L.N., O.S., J.W., T.W., and K.C.V. analyzed the data; J.L.N. and S.R.K. wrote the paper; J.W. contributed animals.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ANOVA
- analysis of variance
- BAL
- bronchoalveolar lavage
- BALF
- bronchoalveolar lavage fluid
- BPD
- bronchopulmonary dysplasia
- Cyp2j
- cytochrome P450
- family 2
- subfamily j
- Chrm2
- cholinergic receptor, muscarinic 2, cardiac
- EMMA
- efficient mixed-model association
- GWA
- genome-wide association
- H&E
- hematoxylin and eosin
- HBSS
- Hanks' balanced salt solution
- Hsnl
- hyperoxia susceptibility in neonatal lungs
- Mgmt
- O-6-methylguanine-DNA methyltransferase
- PMN
- polymorphonuclear leukocyte
- PPAR
- peroxisome proliferator-activated receptor
- QTL
- quantitative trait locus
- ROS
- reactive oxygen species
- SNP
- single-nucleotide polymorphism
REFERENCES
- 1. Northway W. H., Jr., Rosan R. C., Porter D. Y. (1967) Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N. Engl. J. Med. 276, 357–368 [DOI] [PubMed] [Google Scholar]
- 2. Jobe A. H., Bancalari E. (2001) Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 163, 1723–1729 [DOI] [PubMed] [Google Scholar]
- 3. Bhandari A., Bhandari V. (2009) Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics 123, 1562–1573 [DOI] [PubMed] [Google Scholar]
- 4. Jobe A. H. (2011) The new bronchopulmonary dysplasia. Curr. Opin. Pediatr. 23, 167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parton L. A., Strassberg S. S., Qian D., Galvin-Parton P. A., Cristea I. A. (2006) The genetic basis for bronchopulmonary dysplasia. Front. Biosci. 11, 1854–1860 [DOI] [PubMed] [Google Scholar]
- 6. Hallman M., Haataja R. (2003) Genetic influences and neonatal lung disease. Semin. Neonatol. 8, 19–27 [DOI] [PubMed] [Google Scholar]
- 7. Bose C. L., Dammann C. E., Laughon M. M. (2008) Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch. Dis. Child. Fetal Neonatal Ed. 93, F455–F461 [DOI] [PubMed] [Google Scholar]
- 8. Baraldi E., Carraro S., Filippone M. (2009) Bronchopulmonary dysplasia: definitions and long-term respiratory outcome. Early Hum. Dev. 85, S1–S3 [DOI] [PubMed] [Google Scholar]
- 9. Parker R. A., Lindstrom D. P., Cotton R. B. (1996) Evidence from twin study implies possible genetic susceptibility to bronchopulmonary dysplasia. Semin. Perinatol. 20, 206–209 [DOI] [PubMed] [Google Scholar]
- 10. Bhandari V., Gruen J. R. (2006) The genetics of bronchopulmonary dysplasia. Semin. Perinatol. 30, 185–191 [DOI] [PubMed] [Google Scholar]
- 11. Lavoie P. M., Pham C., Jang K. L. (2008) Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics 122, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson A., Bhandari V. (2008) Pulmonary biomarkers of bronchopulmonary dysplasia. Biomark. Insights 3, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bokodi G., Treszl A., Kovacs L., Tulassay T., Vasarhelyi B. (2007) Dysplasia: a review. Pediatr. Pulmonol. 42, 952–961 [DOI] [PubMed] [Google Scholar]
- 14. Newton-Cheh C., Hirschhorn J. N. (2005) Genetic association studies of complex traits: design and analysis issues. Mutat. Res. 573, 54–69 [DOI] [PubMed] [Google Scholar]
- 15. Ohashi J., Tokunaga K. (2001) The power of genome-wide association studies of complex disease genes: statistical limitations of indirect approaches using SNP markers. J. Hum. Genet. 46, 478–482 [DOI] [PubMed] [Google Scholar]
- 16. Coalson J. J., Winter V. T., Siler-Khodr T., Yoder B. A. (1999) Neonatal chronic lung disease in extremely immature baboons. Am. J. Respir. Crit. Care Med. 160, 1333–1346 [DOI] [PubMed] [Google Scholar]
- 17. Hillman N. H., Polglase G. R., Pillow J. J., Saito M., Kallapur S. G., Jobe A. H. (2011) Inflammation and lung maturation from stretch injury in preterm fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 300, L232–L241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng H., Mason S. N., Auten R. L., Jr (2000) Lung inflammation in hyperoxia can be prevented by antichemokine treatment in newborn rats. Am. J. Respir. Crit. Care Med. 162, 2316–2323 [DOI] [PubMed] [Google Scholar]
- 19. Saugstad O. D. (2003) Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin. Neonatol. 8, 39–49 [DOI] [PubMed] [Google Scholar]
- 20. Collard K. J., Godeck S., Holley J. E., Quinn M. W. (2004) Pulmonary antioxidant concentrations and oxidative damage in ventilated premature babies. Arch. Dis. Child. Fetal Neonatal Ed. 89, F412–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Londhe V. A., Sundar I. K., Lopez B., Maisonet T. M., Yu Y., Aghai Z. H., Rahman I. (2011) Hyperoxia impairs alveolar formation and induces senescence through decreased histone deacetylase activity and up-regulation of p21 in neonatal mouse lung. Pediatr. Res. 69, 371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Warner B. B., Stuart L. A., Papes R. A., Wispe J. R. (1998) Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am. J. Physiol. 275, L110–L117 [DOI] [PubMed] [Google Scholar]
- 23. Dauger S., Ferkdadji L., Saumon G., Vardon G., Peuchmaur M., Gaultier C., Gallego J. (2003) Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest 123, 530–538 [DOI] [PubMed] [Google Scholar]
- 24. Flint J., Eskin E. (2012) Genome-wide association studies in mice. Nat. Rev. Genet. 13, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gomeza J., Shannon H., Kostenis E., Felder C., Zhang L., Brodkin J., Grinberg A., Sheng H., Wess J. (1999) Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 1692–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Struckmann N., Schwering S., Wiegand S., Gschnell A., Yamada M., Kummer W., Wess J., Haberberger R. V. (2003) Role of muscarinic receptor subtypes in the constriction of peripheral airways: studies on receptor-deficient mice. Mol. Pharmacol. 64, 1444–1451 [DOI] [PubMed] [Google Scholar]
- 27. Cho H. Y., van Houten B., Wang X., Miller-Degraff L., Fostel J., Gladwell W., Perrow L., Panduri V., Kobzik L., Yamamoto M., Bell D. A., Kleeberger S. R. (2012) Targeted deletion of Nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid. Redox Signal. 17, 1066–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lightfoot J. T., Turner M. J., Daves M., Vordermark A., Kleeberger S. R. (2004) Genetic influence on daily wheel running activity level. Physiol. Genomics 19, 270–276 [DOI] [PubMed] [Google Scholar]
- 29. Pletcher M. T., McClurg P., Batalov S., Su A. I., Barnes S. W., Lagler E., Korstanje R., Wang X., Nusskern D., Bogue M. A., Mural R. J., Paigen B., Wiltshire T. (2004) Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2, e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McClurg P., Pletcher M. T., Wiltshire T., Su A. I. (2006) Comparative analysis of haplotype association mapping algorithms. BMC Bioinformatics 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gatti D. M., Shabalin A. A., Lam T. C., Wright F. A., Rusyn I., Nobel A. B. (2009) FastMap: fast eQTL mapping in homozygous populations. Bioinformatics 25, 482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang H. M., Zaitlen N. A., Wade C. M., Kirby A., Heckerman D., Daly M. J., Eskin E. (2008) Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hudak B. B., Zhang L. Y., Kleeberger S. R. (1993) Inter-strain variation in susceptibility to hyperoxic injury of murine airways. Pharmacogenetics 3, 135–143 [DOI] [PubMed] [Google Scholar]
- 34. Prows D. R., Hafertepen A. P., Gibbons W. J., Jr., Winterberg A. V., Nick T. G. (2007) A genetic mouse model to investigate hyperoxic acute lung injury survival. Physiol. Genomics 30, 262–270 [DOI] [PubMed] [Google Scholar]
- 35. Vancza E. M., Galdanes K., Gunnison A., Hatch G., Gordon T. (2009) Age, strain, and gender as factors for increased sensitivity of the mouse lung to inhaled ozone. Toxicol. Sci. 107, 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stoilov I., Krueger W., Mankowski D., Guernsey L., Kaur A., Glynn J., Thrall R. S. (2006) The cytochromes P450 (CYP) response to allergic inflammation of the lung. Arch. Biochem. Biophys. 456, 30–38 [DOI] [PubMed] [Google Scholar]
- 37. Ma J., Bradbury J. A., King L., Maronpot R., Davis L. S., Breyer M. D., Zeldin D. C. (2002) Molecular cloning and characterization of mouse CYP2J6, an unstable cytochrome P450 isoform. Biochem. Pharmacol. 64, 1447–1460 [DOI] [PubMed] [Google Scholar]
- 38. Wray J. A., Sugden M. C., Zeldin D. C., Greenwood G. K., Samsuddin S., Miller-Degraff L., Bradbury J. A., Holness M. J., Warner T. D., Bishop-Bailey D. (2009) The epoxygenases CYP2J2 activates the nuclear receptor PPARalpha in vitro and in vivo. PLoS One 4, e7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moshal K. S., Zeldin D. C., Sithu S. D., Sen U., Tyagi N., Kumar M., Hughes W. M., Jr., Metreveli N., Rosenberger D. S., Singh M., Vacek T. P., Rodriguez W. E., Ayotunde A., Tyagi S. C. (2008) Cytochrome P450 (CYP) 2J2 gene transfection attenuates MMP-9 via inhibition of NF-kappabeta in hyperhomocysteinemia. J. Cell. Physiol. 215, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cho H. Y., Gladwell W., Wang X., Chorley B., Bell D., Reddy S. P., Kleeberger S. R. (2010) Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 182, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gu F., Qureshi A. A., Kraft P., Guo Q., Hunter D. J., Han J. (2009) Polymorphisms in genes involved in DNA repair, cell growth, oxidative stress and inflammatory response, and melanoma risk. Br. J. Dermatol. 161, 209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joss-Moore L. A., Albertine K. H., Lane R. H. (2011) Epigenetics and the developmental origins of lung disease. Mol. Genet. Metab. 104, 61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verhein K. C., Fryer A. D., Jacoby D. B. (2009) Neural control of airway inflammation. Curr. Allergy Asthma Rep. 9, 484–490 [DOI] [PubMed] [Google Scholar]
- 44. Gwilt C. R., Donnelly L. E., Rogers D. F. (2007) The non-neuronal cholinergic system in the airways: an unappreciated regulatory role in pulmonary inflammation? Pharmacol. Ther. 115, 208–222 [DOI] [PubMed] [Google Scholar]
- 45. Oenema T. A., Kolahian S., Nanninga J. E., Rieks D., Hiemstra P. S., Zuyderduyn S., Halayko A. J., Meurs H., Gosens R. (2010) Pro-inflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir. Res. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Profita M., Bonanno A., Montalbano A. M., Ferraro M., Siena L., Bruno A., Girbino S., Albano G. D., Casarosa P., Pieper M. P., Gjomarkaj M. (2011) Cigarette smoke extract activates human bronchial epithelial cells affecting non-neuronal cholinergic system signalling in vitro. Life Sci. 89, 36–43 [DOI] [PubMed] [Google Scholar]
- 47. Sato E., Koyama S., Okubo Y., Kubo K., Sekiguchi M. (1998) Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am. J. Physiol. 274, L970–L979 [DOI] [PubMed] [Google Scholar]
- 48. Rosas-Ballina M., Olofsson P. S., Ochani M., Valdes-Ferrer S. I., Levine Y. A., Reardon C., Tusche M. W., Pavlov V. A., Andersson U., Chavan S., Mak T. W., Tracey K. J. (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Trakhtenberg E. F., Goldberg J. L. (2011) Immunology: neuroimmune communication. Science 334, 47–48 [DOI] [PubMed] [Google Scholar]
- 50. Wess J. (1996) Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 10, 69–99 [DOI] [PubMed] [Google Scholar]
- 51. Baleato R. M., Guthrie P. L., Gubler M. C., Ashman L. K., Roselli S. (2008) Deletion of CD151 results in a strain-dependent glomerular disease due to severe alterations of the glomerular basement membrane. Am. J. Pathol. 173, 927–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leontyev D., Katsman Y., Branch D. R. (2012) Mouse background and IVIG dosage are critical in establishing the role of inhibitory Fcgamma receptor for the amelioration of experimental ITP. Blood 119, 5261–5264 [DOI] [PubMed] [Google Scholar]
- 53. Tiozzo C., Danopoulos S., Lavarreda-Pearce M., Baptista S., Varimezova R., Al Alam D., Warburton D., Rehan V., De Langhe S., Di Cristofano A., Bellusci S., Minoo P. (2012) Embryonic epithelial Pten deletion through Nkx2.1-cre leads to thyroid tumorigenesis in a strain-dependent manner. Endocr. Relat. Cancer 19, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van den Buuse M., Martin S., Holgate J., Matthaei K., Hendry I. (2007) Mice deficient in the alpha subunit of G(z) show changes in pre-pulse inhibition, anxiety and responses to 5-HT(1A) receptor stimulation, which are strongly dependent on the genetic background. Psychopharmacology 195, 273–283 [DOI] [PubMed] [Google Scholar]
- 55. Wallace J. M., Golcuk K., Morris M. D., Kohn D. H. (2009) Inbred strain-specific response to biglycan deficiency in the cortical bone of C57BL6/129 and C3H/He mice. J. Bone Miner. Res. 24, 1002–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu J., Gontier G., Chaker Z., Lacube P., Dupont J., Holzenberger M. (2014) Longevity effect of IGF-1R mutation depends on genetic background-specific receptor activation. Aging Cell 13, 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.