

ABSTRACT
Caspase-3 is an effector caspase that is activated downstream of mitochondrial outer-membrane permeabilization (MOMP) during apoptosis. However, previous work has demonstrated that caspase-3-deficient mouse embryonic fibroblasts (MEFs) are resistant to mitochondrially mediated cell death and display a delay in the mitochondrial events of apoptosis, including Bax activation, MOMP and release of cytochrome c. Here, we show that caspase-3 regulates fibronectin secretion and impacts on cell morphology, adhesion and migration. Surprisingly, the catalytic activity of caspase-3 is not required for these non-apoptotic functions. Moreover, we found that caspase-3-deficient MEFs are not resistant to death by anoikis and that exogenous fibronectin protects wild-type MEFs from cell death induced by serum withdrawal. Taken together, our data indicate that procaspase-3 has a non-apoptotic function; it regulates the secretion of fibronectin and influences morphology, adhesion and migration. Furthermore, this novel procaspase-3 function might alter the apoptotic threshold of the cell.
KEY WORDS: Caspase, Adhesion, Migration
INTRODUCTION
During the stimulation of intrinsic apoptosis, BH3-only proteins regulate the activation of Bax and Bak, which causes mitochondrial outer-membrane permeabilization (MOMP) and the release of cytochrome c. This results in the formation of the apoptosome, caspase-9 activation and the subsequent activation of effector caspases, including caspase-3 (Danial and Korsmeyer, 2004). The role of these effector caspases during the degradation phase of apoptosis is well characterized and includes the activation of DNA fragmentation, cell shrinkage and membrane blebbing (Shi, 2002; Woo et al., 1998). However, previous work shows that caspase-3-deficient mouse embryonic fibroblasts (MEFs) display a delay in the mitochondrial events of intrinsic apoptosis, including Bax activation, MOMP and cytochrome c release, which are thought to occur upstream of caspase-3 activation (Lakhani et al., 2006). These data suggest that caspase-3 either feeds back on the mitochondria after activation or has other unknown functions upstream of the mitochondria that influence the apoptotic threshold of a cell.
Classically, caspases are thought to act as cysteine proteases that cleave their substrates at specific aspartic acid residues (Shi, 2002). However, roles of these caspases that can be distinguished from their catalytic functions have recently been discovered. Caspase-8 has been shown to interact with the p85 subunit (also known as PIK3R2) of phosphatidylinositol 3-kinase (PI3K) to promote Rac activation during migration, and caspase-11 has been shown to interact with Aip1 (also known as WDR1) and promote actin depolymerization during cell migration (Li et al., 2007; Senft et al., 2007). Therefore, we hypothesized that caspase-3 also has a non-apoptotic function and that loss of this function could result in a change in the apoptotic threshold of the cell.
Here, we show that caspase-3 regulates cell morphology, adhesion and migration. Interestingly, we found that the catalytic activity of caspase-3 is not required for these novel non-apoptotic functions. Furthermore, we found that caspase-3 regulates fibronectin secretion and, by doing so, influences the apoptotic threshold of a cell. Taken together, our data indicate that procaspase-3 has a novel non-apoptotic function that is independent of its role as an executioner caspase, yet can influence the apoptotic threshold upstream of the mitochondrial events of apoptosis.
RESULTS
Caspase-3 regulates cell adhesion
While studying the role of caspase-3 in mitochondrial dysfunction during apoptosis, we made the empiric observation that caspase-3-deficient (Casp3−/−) MEFs were difficult to trypsinize from cell culture plates and displayed changes in morphology. This raised the possibility that, in addition to its role as an executioner caspase during apoptosis, caspase-3 could have other cellular functions, and these novel functions could influence the apoptotic threshold of a cell. Specifically, we hypothesized that caspase-3 was a negative regulator of adhesion and that loss of caspase-3 resulted in an increase in adhesion-mediated survival signaling. Therefore, we formally tested the role of caspase-3 on adhesion-associated cellular events. Initially, we measured the effects of loss of caspase-3 on cellular morphology and adhesion. Wild-type MEFs display a small rounded morphology with some cells having a distinct leading-edge lamellipodial structure (Fig. 1A). Casp3−/− MEFs share a similar overall size to wild-type MEFs (Fig. 1B); however, they have more of an elongated morphology (Fig. 1A). Wild-type MEFs have an average cell length of 42.2 µm±1.7 µm (±s.e.m.), whereas Casp3−/− MEFs are nearly two times longer (67.8 µm±3.6 µm) (Fig. 1C). These changes were not due to differences in overall size or cell volume of Casp3−/− MEFs (Fig. 1B,D), indicating that caspase-3 influences cell spreading and elongation, which are regulated in part through the binding of integrins to the extracellular matrix (ECM) (Gumbiner, 1996). Therefore, we next determined whether Casp3−/− MEFs are more adherent. We plated an equal number of cells into 96-well plates and allowed cells to adhere for 0.5, 1, 2 and 4 hours. The medium was then aspirated, and adherent cells were fixed and stained with Crystal Violet to analyze adhesion. Casp3−/− MEFs display a threefold increase in adhesion compared to wild-type MEFs after 30 minutes and at least a twofold increase in adhesion at 4 hours (Fig. 1E).
Fig. 1.
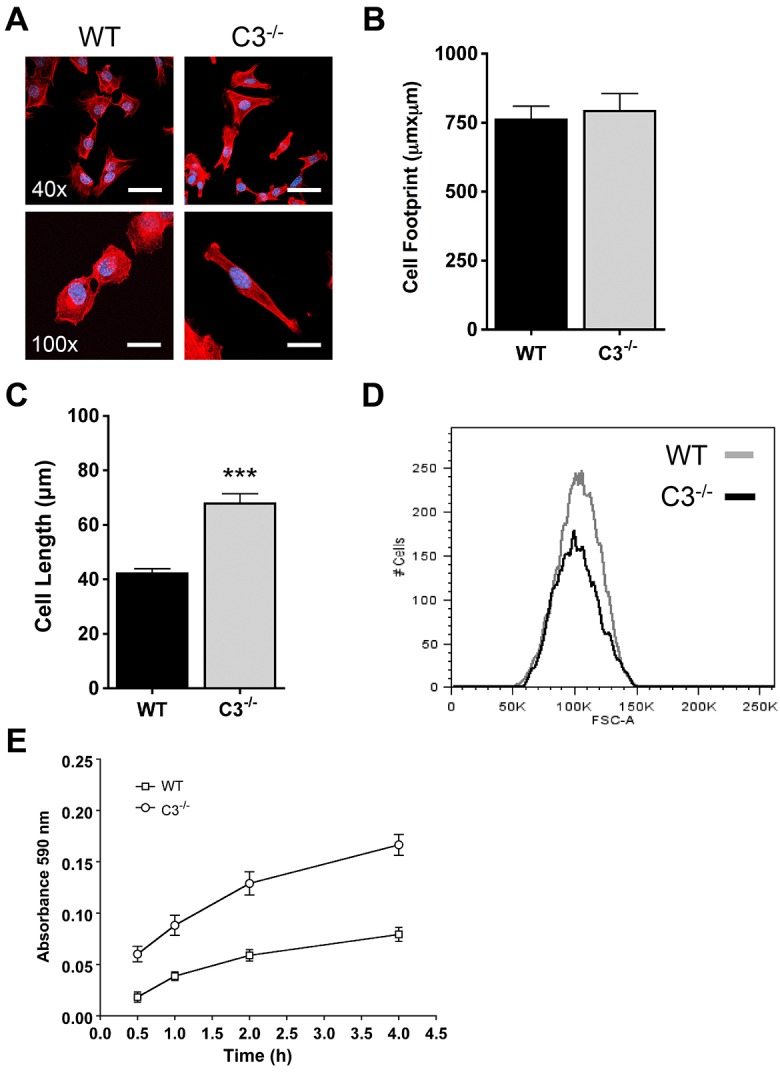
Caspase-3 regulates morphology and adhesion. (A–C) MEFs were grown on fibronectin-coated glass coverslips for 24 hours, fixed and stained with phalloidin (red) and DAPI (blue). (A) Casp3−/− MEFs display an elongated morphology. Cell morphology was determined by confocal fluorescence microscopy. WT, wild type; C3−/−, Casp3−/−. Scale bars: 50 µm (40×), 20 µm (100×). (B) Casp3−/− MEFs have a cell footprint in contact with the ECM that is comparable in size to that of wild-type MEFs. The area of contact was quantified using LSM 510 META software. Data are presented as mean±s.e.m. (>30 individual cells from at least three experiments). (C) Casp3−/− MEFs are longer than wild-type MEFs. Cell length was quantified using LSM 510 META software. Data are presented as mean±s.e.m. (>30 individual cells from at least three experiments); ***P<0.0001. (D) Casp3−/− MEFs have the same cell volume as wild-type MEFs. Cell volume was analyzed by assessing FSC by using flow cytometry. (E) Casp3−/− MEFs adhere at a faster rate than wild-type MEFs. MEFs were allowed to adhere for the indicated times and Crystal Violet staining was quantified as described in Materials and Methods. Data are presented as mean±s.e.m. All data are from at least three independent experiments.
Caspase-3 regulates migration velocity
The regulation of the organization of the actin cytoskeleton and focal adhesion assembly and disassembly are essential for cell migration (Ballestrem et al., 2000; Krause et al., 2003; Ridley et al., 2003). Because caspase-3 was regulating morphology and adhesion, we next determined its role in cell motility. In vitro wound-healing assays were performed with wild-type and Casp3−/− MEFs and the percentage of wound closure was analyzed by using time-lapse microscopy. Casp3−/− MEFs were unable to close wounds as efficiently as wild-type MEFs, showing 37.8%±8.2 and 50.5%±9.4 (±s.e.m.) wound closure at 9 and 12 hours, respectively, whereas wild-type MEFs display 63.8%±4.9 and 84.0%±7.2 wound closure at these time-points (Fig. 2). Wound closure can be accomplished through the activation of cell migration and/or cell proliferation (Chera et al., 2009; Li et al., 2010; Witte and Barbul, 1997; Tseng et al., 2007). Therefore, we determined the cell proliferation rate in wild-type and Casp3−/− MEFs through analysis of cell cycle and cell-doubling time. In a standard cell cycle assay, the percentage of cells in G1, S or G2 phases of the cell cycle was not significantly different between Casp3−/− and wild-type MEFs (Fig. 3A). However, this did not represent a wound-healing situation where cells are at confluency and then are released from contact inhibition. Therefore, we determined cell cycle distribution while simulating wound healing, by growing cells to confluency and then scratching the plates with 8 parallel scratches or a grid of 16 scratches. At 12 hours after scratching, analysis indicated no difference in cell cycle distribution under conditions of 8 scratches or 16 scratches (Fig. 3B). Wild-type and Casp3−/− MEFs were also seeded and counted over time to analyze cell proliferation and doubling time. There is no significant difference in the fold change in cell number over time between Casp3−/− and wild-type MEFs (Fig. 3C). Thus, the differences in wound closure are not due to changes in cell proliferation, indicating that caspase-3 regulates cell motility.
Fig. 2.
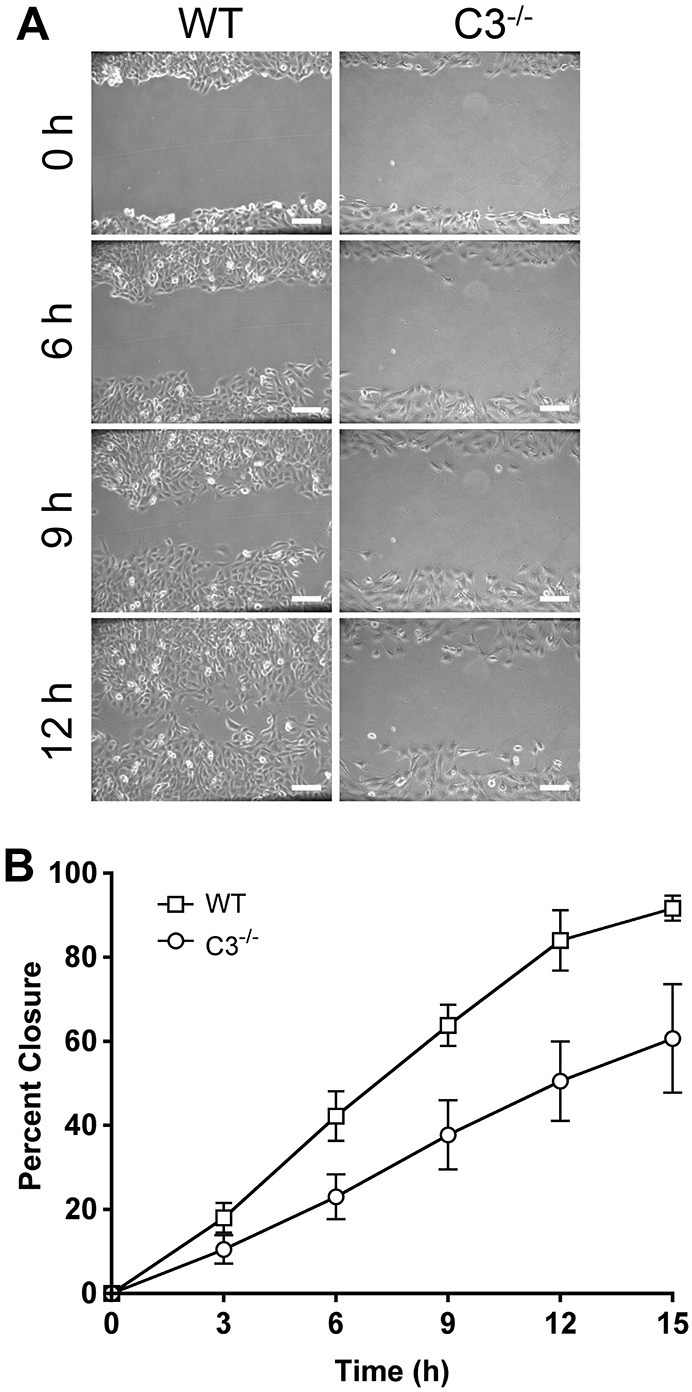
Caspase-3 regulates migration. (A,B) MEFs were grown to confluency, a wound was created and then analyzed by time-lapse microscopy for at least 15 hours. (A) Casp3−/− MEFs display defective wound closure. WT, wild type; C3−/−, Casp3−/−. (B) Data were quantified using Volocity software and are presented as mean±s.e.m. All data are from at least three independent experiments. Scale bars: 100 µm.
Fig. 3.

Wild-type and Casp3−/− MEFs have comparable rates of proliferation. (A) MEFs were grown for 24 hours and cell cycle was analyzed by PI staining. WT, wild type; C3−/−, Casp3−/−. Data are presented as mean±s.e.m. (B) Casp3−/− and wild-type MEFs display comparable amounts of proliferation when migration is stimulated. MEFs were grown to confluency and scratched with eight parallel scratches or a grid of 16 scratches. After 12 hours of migration, cell cycle was analyzed by PI staining. Data are presented as mean±s.e.m. (C) Casp3−/− and wild-type MEFs proliferate at the same rate. Doubling time was analyzed by cell counting at the indicated times. Data are presented as mean±s.e.m. All data are from at least three independent experiments.
Because no differences in proliferation were detected, the two most likely explanations for a defect in wound healing are a decrease in migration velocity or a loss of directional persistence. Therefore, we performed single-cell tracking to identify changes in migration that result in inefficient wound closure in Casp3−/− MEFs. Cell tracks showed that wild-type MEFs moved further into the wound than Casp3−/− MEFs (Fig. 4A). The cell tracks were analyzed for average cell velocity (distance/time) and meandering index (displacement/distance). Wild-type MEFs have an average velocity of 37.9 µm/h±1.7 µm/h (±s.e.m.), whereas a significant decrease in the average velocity of Casp3−/− MEFs was observed (21.7 µm/h±1.2 µm/h) (Fig. 4B). Wild-type MEFs have a meandering index of 0.79±0.02, whereas Casp3−/− MEFs display a statistically significant, albeit marginal, decrease in their meandering index (0.74±0.02) (Fig. 4C). Taken together, our data indicate that caspase-3 regulates adhesion and is required for efficient migration during in vitro wound healing.
Fig. 4.
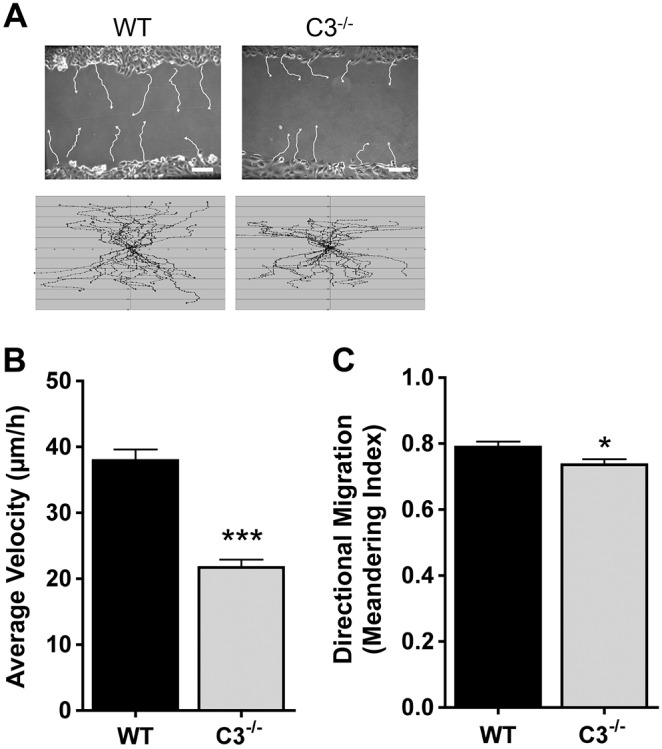
Casp3−/− MEFs display a decrease in average velocity and directional migration. (A) Single-cell tracks formed over a period of ∼10 hours were analyzed using Volocity software. Representative (upper panels) and total (lower panels) tracks are shown. WT, wild type; C3−/−, Casp3−/−. (B,C) Data were quantified and are presented as mean±s.e.m. of ≥36 individual cells from at least three independent experiments. *P = 0.03, ***P<0.0001. Scale bars: 100 µm.
Control of morphology and migration is independent of caspase-3 catalytic activity
Our data demonstrate that caspase-3 has non-apoptotic functions in regulating cell morphology, adhesion and migration. Because these MEFs developed in the absence of caspase-3, we next determined whether these effects were a direct consequence of the absence of caspase-3 or were due to changes in development. Additionally, the changes in morphology and migration are displayed when there is no exogenous apoptotic stimulation, suggesting that there is either localized and controlled activation of caspase-3 or that these functions are independent of the catalytic activity of caspase-3. In order to test these possibilities, we introduced caspase-3 (Casp3) or a catalytically inactive caspase-3 (Casp3C163S) into the Casp3−/− MEFs (thus producing Casp3−/− C3 and Casp3−/− C3C163S MEFs, respectively) and determined whether the reconstituted MEFs reverted to a wild-type phenotype. Casp3 and Casp3C163S were expressed at similar levels when compared with endogenous expression in wild-type MEFs (Fig. 5A). Introduction of Casp3 into the Casp3−/− MEFs restored DEVDase activity (the amount of cleavage of the caspase-3 target site, DEVD) to a level comparable to that observed in wild-type MEFs following serum withdrawal, whereas introduction of Casp3C163S did not (Fig. 5B). Remarkably, we found that both Casp3 and Casp3C163S reverted Casp3−/− MEFs to a wild-type morphology (Fig. 5C,D) and to a wild-type migration phenotype. Casp3−/− C3 and Casp3−/− C3C163S MEFs closed wounds as efficiently as wild-type MEFs (Fig. 5E), primarily resulting from an increase in average velocity, which allowed cells to move further into the wound during the assay (Fig. 5F,G). The modest change in directional persistence that was observed in Casp3−/− MEFs was also corrected by the introduction of Casp3 or Casp3C163S (Fig. 5H). Taken together, these data indicate that caspase-3 regulates morphology and migration through a mechanism that is independent of its catalytic activity.
Fig. 5.

Caspase-3 regulates morphology and migration through a mechanism that is independent of its catalytic activity. (A) Casp3−/− MEFs were reconstituted with Casp3 or Casp3C163S, and caspase-3 expression was analyzed by western blotting. WT, wild type; C3−/−, Casp3−/−. (B) Casp3C163S does not function as a catalytic caspase. DEVDase activity was measured after 48 hours of serum withdrawal. Data are presented as mean±s.e.m. (C,D) Casp3−/− C3 and Casp3−/− C3C163S MEFs revert to a wild-type morphology. MEFs were fixed, stained with phalloidin (red) and DAPI (blue). Scale bars: 20 µm. Data were quantified using LSM 510 software and are presented as mean±s.e.m. (≥30 individual cells from at least three experiments). (E) Casp3−/− C3 and Casp3−/− C3C163S MEFs revert to a wild-type migration phenotype. MEFs were grown to confluency, wounds were created and then samples were analyzed by time-lapse microscopy for at least 15 hours. Data were quantified using Volocity software and are presented as mean±s.e.m. (F–H) Casp3−/− C3 and Casp3−/− C3C163S MEFs revert to a wild-type average velocity and directional migration. Single-cell tracks formed over a period of ∼10 hours were analyzed using Volocity software and total tracks are shown (G). Data are presented as mean±s.e.m. (≥36 individual cells from at least three experiments). **P<0.01, ***P<0.0001. Data in B–H are from at least three independent experiments.
Caspase-3 inhibits fibronectin secretion
Casp3−/− MEFs displayed increased cell adhesion and elongation along the ECM, and a slower average velocity compared with wild-type MEFs. These phenotypes could be explained by an increased number of mature focal adhesion complexes, which are required for adhesion and have been shown to be detrimental to migration (Beningo et al., 2001; Gardel et al., 2010; Totsukawa et al., 2004). However, we were unable to detect differences in integrin β-1 protein expression on the cell surface, integrin β-1 mRNA levels or integrin β-3 protein expression on the cell surface (Fig. 6A; data not shown). Alternatively, these phenotypes could be the result of the increased production and secretion of ECM, which is required for adhesion (Gumbiner, 1996; Pankov and Yamada, 2002). The previous adhesion experiments were performed in serum-containing medium and/or on fibronectin-coated glass, both of which are exogenous sources of components of the ECM. Therefore, to determine whether the adhesion phenotype of Casp3−/− MEFs was due to endogenous production of ECM, exogenous factors were removed through a 4-hour serum withdrawal, and then an adhesion assay was performed for 1 hour on glass coverslips with or without fibronectin coating. Additionally, to determine the role of secretion of endogenous ECM, assays were also performed in the presence of the ER–Golgi transport inhibitor brefeldin A. Both Casp3−/− and wild-type MEFs adhere to and spread on fibronectin with some localized FAK activation on the cellular edge; however, only Casp3−/− MEFs spread in the absence of a supplied ECM at this time-point (Fig. 6B), and the addition of brefeldin A blocks the spreading of Casp3−/− MEFs. Introduction of Casp3 or Casp3C163S reverts Casp3−/− MEFs to a wild-type phenotype (Fig. 6B). These data suggest that Casp3−/− MEFs are able to produce and/or secrete their own ECM more efficiently than wild-type MEFs, indicating that procaspase-3 regulates the secretion of ECM through a mechanism that is independent of its catalytic activity. To analyze the production and secretion of ECM, we repeated the adhesion assay; however, cells were fixed without permeabilization and were stained with phalloidin (to visualize actin) and DAPI (to visualize nuclei), as well as with a fibronectin-specific antibody. Casp3−/− MEFs spread faster than wild-type MEFs and have prominent extracellular fibronectin staining after 1 hour of adhesion (Fig. 6C). Some cells were also fixed without permeabilization and stained with phalloidin, DAPI and an antibody specific for an intracellular protein, P-FAK, to demonstrate that, under these conditions, antibodies cannot access the intracellular space (compare P-FAK staining in Fig. 6C). To demonstrate this in a more quantitative fashion, we also analyzed fibronectin secretion by using an enzyme-linked immunosorbent assay (ELISA) after performing an adhesion assay, and we found that, when compared with wild-type MEFs, Casp3−/− MEFs had a significantly higher level of soluble fibronectin in the medium when normalized to cell number (Fig. 6D). However, intracellular levels of fibronectin were not significantly different between wild-type and Casp3−/− MEFs (Fig. 6D). Moreover, steady-state levels of fibronectin mRNA were not different between these cells (data not shown). Taken together, these data suggest that fibronectin production alone cannot account for the increased adhesion of Casp3−/− MEFs. To determine whether fibronectin secretion was altered by the loss of procaspase-3, we investigated the effects of brefeldin A on fibronectin levels in the culture medium and in cells. Consistent with the cell-spreading data (Fig. 6B), we found that brefeldin A significantly reduced the amount of fibronectin in the culture medium of Casp3−/− MEFs, returning it to the level of wild-type MEF secretion (Fig. 6D). To directly test the role of fibronectin in the Casp3−/− adhesion phenotype, we next determined the effects of silencing fibronectin in Casp3−/− MEFs. Consistent with an important role for cell adhesion in the survival of these cells (see below) we were unable to stably silence fibronectin in a constitutive fashion (data not shown). Therefore, we developed stable lines carrying an inducible fibronectin shRNA (Tet-On). In the absence of doxycycline, these cells adhered in a manner similar to that of cells infected with empty vector (pLKO) (Fig. 6E) or parental Casp3−/− cells (Fig. 6B). However, treatment with doxycycline for 72 hours prior to performing the adhesion assay resulted in a significant decrease in cellular fibronectin and a reversion to an adhesion phenotype similar to that of wild-type cells (Fig. 6E). Importantly, the addition of exogenous fibronectin rescued the adhesion phenotype, demonstrating that neither the shRNA nor doxycycline had off-target effects. Taken together, these data demonstrate that fibronectin secretion is necessary for the Casp3−/− adhesion phenotype.
Fig. 6.

Caspase-3 regulates the secretion of fibronectin. (A) Wild-type and Casp3−/− MEFs have similar levels of cell-surface integrin β-1 protein expression. Cells were collected and stained with a FITC-conjugated anti-integrin-β-1 antibody or a FITC-conjugated IgG isotype control and analyzed by flow cytometry. Black, wild-type MEFs isotype control; green, wild-type MEFs anti-integrin; grey, Casp3−/− MEFs isotype control; red, Casp3−/− MEFs anti-integrin. (B,C) Casp3−/− MEFs adhere to and spread on uncoated glass coverslips at a faster rate than wild-type MEFs. (B) MEFs were subjected to serum withdrawal in the presence or absence of brefeldin A (BA) for 4 hours and were then allowed to adhere in the presence or absence of brefeldin A for 1 hour. Cells were fixed, permeabilized and stained with phalloidin (red), DAPI (blue) and antibodies against P-FAK (green). WT, wild type; C3−/−, Casp3−/−. Scale bars: 20 µm. (C) MEFs were subjected to serum withdrawal for 4 hours and then allowed to adhere for 1 hour. Cells were fixed without permeabilization and stained with phalloidin, DAPI and antibodies for either fibronectin (green) or P-FAK. Scale bars: 20 µm. (D) Casp3−/− MEFs secrete an increased amount of fibronectin. MEFs were subjected to serum withdrawal for 4 hours in the presence or absence of brefeldin A and then allowed to adhere for 1 hour in the presence or absence of brefeldin A. Supernatants and cells were collected and analyzed for fibronectin by ELISA. Data are presented as mean±s.e.m. (E) Knockdown of fibronectin blocks the ability of Casp3−/− MEFs to spread. Casp3−/− pLKO or fibronectin-depleted Casp3−/− (shFN) MEFs were treated for 72 hours with or without doxycycline. Cells were then trypsinized, subjected to serum withdrawal for 4 hours and then allowed to adhere for 1 hour with or without fibronectin coating (upper panel, scale bars: 20 µm) or were collected and analyzed for fibronectin by ELISA (lower panel). Data are presented as mean±s.e.m. *P<0.05, **P<0.01. All data are from at least three independent experiments.
Increased adhesion is part of the Casp3−/− survival phenotype
Previous work has demonstrated that, during stimulation of cell death, effector-caspase-deficient MEFs display delayed Bax activation, MOMP and cytochrome c release (Lakhani et al., 2006). Based on the current findings, it is possible that the delay in the mitochondrial events of apoptosis in Casp3−/− MEFs might be explained by the ability of these cells to adhere and signal more efficiently, resulting in an increased apoptotic threshold. To test this possibility, we examined the role of adhesion in the cell death of wild-type and Casp3−/− MEFs. Cells were seeded in the presence or absence of serum on plates coated with or without polyHEMA, which blocks adhesion and results in the induction of anoikis (Munoz et al., 2010). Consistent with previous findings, Casp3−/− MEFs were significantly protected from serum-withdrawal-induced cell death when compared with wild-type MEFs at all time-points analyzed (Fig. 7A). After 24 hours of serum withdrawal, wild-type MEFs were 38.3%±6.6% (±s.e.m.) annexin-V-positive, whereas Casp3−/− MEFs were only 7.7%±1.2% annexin-V-positive (Fig. 7A, P = 0.01). However, Casp3−/− MEFs were not completely resistant to serum-withdrawal-induced cell death; after 96 hours of serum withdrawal, wild-type MEFs were 88.0%±2.5% annexin-V-positive, whereas Casp3−/− MEFs were still significantly protected yet displayed 55.7%±4.5% annexin-V-positive cells (Fig. 7A, P = 0.003). In contrast to serum withdrawal, Casp3−/− MEFs were significantly less resistant to cell death induced by anoikis. After 24 hours of anoikis, Casp3−/− MEFs were 29.3%±1.8% annexin-V-positive and by 72 hours loss of caspase-3 offers no significant protection (Fig. 7A). Thus, the protective effects of caspase-3 deficiency require cell adhesion. Interestingly, at 24 and 48 hours, the combination of serum withdrawal and anoikis had an additive killing effect on wild-type MEFs. By contrast, this combination was antagonistic during the first 48 hours in Casp3−/− MEFs. This suggests that serum withdrawal is protective against anoikis when cells lack caspase-3. We hypothesize that this initial protection is from the activation of autophagy by serum withdrawal, and this is currently under investigation. Consistent with this possibility, we observed a similar effect of anoikis and the topoisomerase II inhibitor etoposide on the death of wild-type MEFs. However, etoposide does not confer protection against anoikis in Casp3−/− MEFs (data not shown). Caspase-3 is the primary executioner caspase during serum withdrawal (Brentnall et al., 2013) and, consistent with this observation, the effects of serum withdrawal and anoikis on Casp3 and Casp7 double knockout MEFs are identical to that seen in Casp3−/− MEFs (Fig. 7B). Taken together, these data indicate that procaspase-3 is regulating adhesion-dependent survival signaling upstream of the mitochondria, which alters the apoptotic threshold of the cell and allows the mitochondrial events of apoptosis to happen quickly and efficiently upon stimulation of cell death.
Fig. 7.
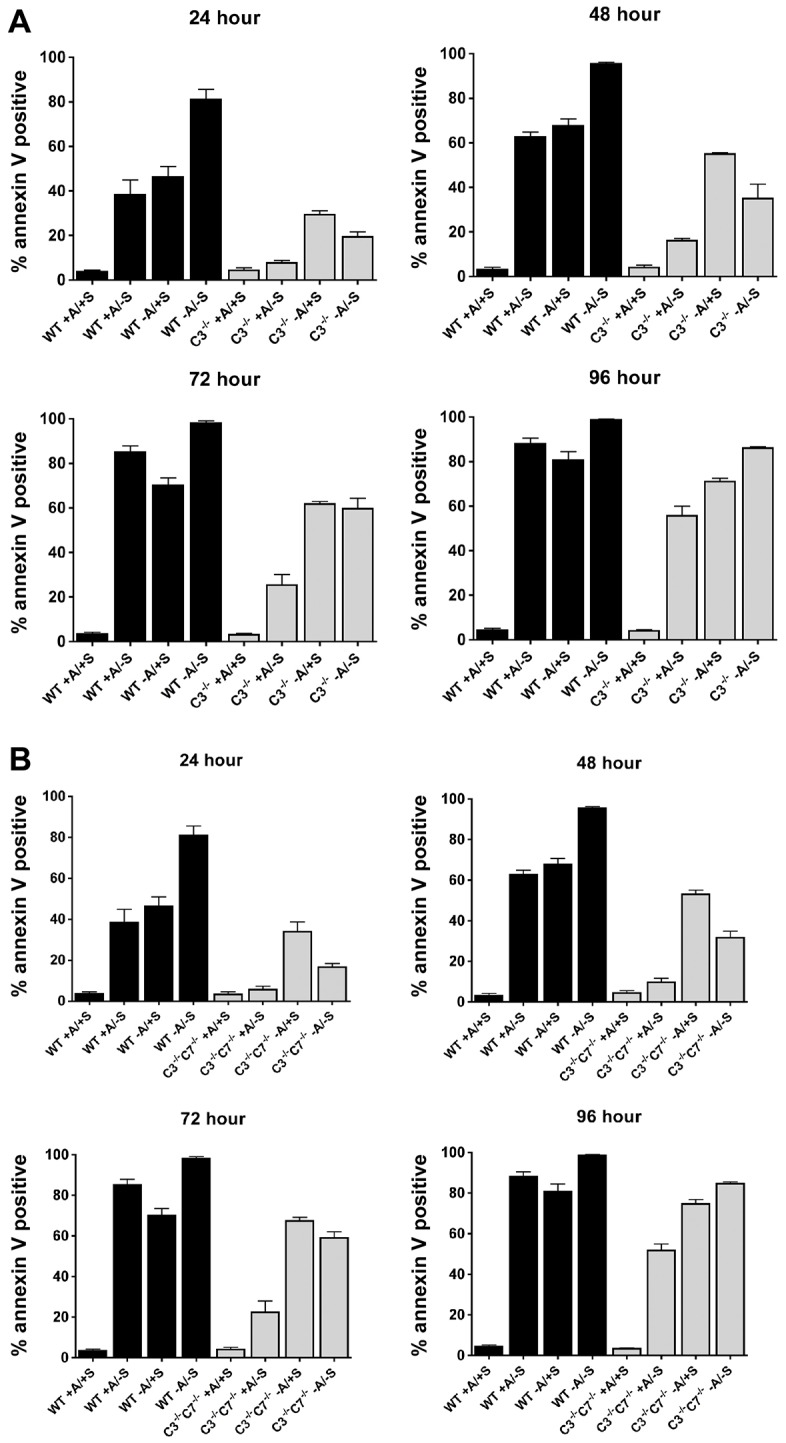
Caspase-3 regulates the apoptotic threshold of cells through adhesion, independently of caspase-7. (A,B) MEFs were seeded onto plates with or without polyHEMA coating (+/− A) in medium with or without serum (+/− S). At the indicated times, cell death was analyzed by annexin V staining. Data are presented as mean±s.e.m. (of at least three independent experiments). WT, wild type; C3−/−, Casp3−/−. (A) Casp3−/− MEFs are protected from serum-withdrawal-induced cell death; however, they are not protected from death induced by anoikis. (B) Caspase-3 regulates the apoptotic threshold of the cell through adhesion, independently of caspase-7. C3−/−C7−/− MEFs die in a similar manner to Casp3−/− MEFs during serum withdrawal and/or anoikis.
Our data indicate that loss of caspase-3 protects cells in an adhesion-dependent manner, and that these cells secrete approximately twice as much fibronectin as wild-type MEFs. This would suggest that relatively modest changes in fibronectin secretion could have profound effects on the inhibition of cell death. To test this possibility, we performed a serum withdrawal experiment with wild-type, Casp3−/−, Casp3−/− C3 and Casp3−/− C3C163S MEFs on plates coated with a range of fibronectin densities (0–10 µg/cm2 fibronectin). Wild-type MEFs displayed 48.7%±4.4% (±s.e.m.) cell death in the absence of a supplied ECM; however, even at the lowest density of fibronectin tested (0.6 µg/cm2) a trend toward cell protection was observed, and significantly less cell death was observed when cells were plated on fibronectin at densities of 2.5 µg/cm2 or greater (Fig. 8). Interestingly, the amount of protection was not statistically different at densities >2.5 µg/cm2. These data suggest that a minimal increase in the density of fibronectin is needed to confer protection from serum-withdrawal-induced cell death. Casp3−/− MEFs did not show significant cell death during serum withdrawal and, therefore, the addition of fibronectin did not protect these cells. However, Casp3−/− C3 MEFs died in a similar fashion to wild-type MEFs during serum withdrawal, and the addition of fibronectin at densities of 2.5 µg/cm2 or greater protected cells (Fig. 8). Introduction of C3C163S did not result in a significant increase in cell death. This is consistent with the lack of DEVDase activity observed in these cells (Fig. 5B) and with our previous finding that caspase-3 is the primary executioner during serum withdrawal (Brentnall et al., 2013). Taken together, these data suggest that, in the presence of fibronectin, caspase-3 activity is still required for serum-withdrawal-induced death.
Fig. 8.

Fibronectin protects wild-type and Casp3−/− C3 MEFs from serum-withdrawal-induced cell death. MEFs were seeded onto plates coated with increasing densities of fibronectin in serum-free medium. After 24 hours, cell death was analyzed by annexin V staining. WT, wild type; C3−/−, Casp3−/−. Data are presented as mean±s.e.m. (of at least three independent experiments). *P<0.01, compared with serum withdrawal.
DISCUSSION
Cells constitutively express many, if not most, of the pro-apoptotic proteins required to initiate and execute programmed cell death. This has led to the concept that all cells are poised to die if and when they encounter a stress signal, such as loss of trophic factors (Ishizaki et al., 1995). However, several studies have now demonstrated that pro-death molecules have additional unrelated functions, which they perform in living cells. Importantly, these additional functions often do not require the same activity or domains necessary for these proteins to participate in cell death. The first example of a protein having two distinct functions is cytochrome c, where the region required for binding to Apaf-1 in the formation of the apoptosome during apoptosis is dispensable for its role in electron transport (Kluck et al., 2000; Yu et al., 2001). More recently, the BH3-only protein Bad was shown to be important in the regulation of glucokinase and insulin secretion (Danial et al., 2003; Danial et al., 2008). Interestingly, the BH3 domain is important for both its pro-apoptotic and metabolic functions; however, in the latter case it must be phosphorylated. In these two cases, neither protein has enzymatic activity, therefore separation of the non-apoptotic activity from the pro-apoptotic activity must occur in part through inhibition of the latter. In the case of cytochrome c this occurs through compartmentalization, whereas for Bad the post-translational modification that is required for regulation of glucokinase inhibits its ability to function as a Bcl-2 antagonist (Danial et al., 2003; Danial et al., 2008; Kluck et al., 2000; Yu et al., 2001). In contrast to these examples, the non-apoptotic functions of caspases might or might not involve the same catalytic activity involved in apoptosis. For example, caspase-8 has been shown to increase cell motility and metastasis in neuroblastoma cancer cells through Rab5 activation that is independent of its catalytic activity (Torres et al., 2010). The inflammatory caspase, caspase-11, regulates macrophage migration by regulating actin dynamics through an interaction with Aip1 (Li et al., 2007). However, caspase-8 and -10 can also regulate cell survival through their catalytic activity. Caspase-8 prevents RIPK-mediated necrosis, whereas a recent report demonstrated that caspase-10 prevents autophagic death of myeloma cells by cleaving the Bcl2-interacting protein BCLAF1 (Green et al., 2011; Kaiser et al., 2011; Lamy et al., 2013; Oberst et al., 2011).
Caspase-3 has also recently been shown to have non-apoptotic roles. Low levels of caspase-3 activation have been shown to regulate cell differentiation through activation of a DNase. During this process, the level of caspase-3 activation is controlled by NF-κB signaling to avoid apoptosis (Basu et al., 2012; Larsen et al., 2010). In addition, during wound healing, caspase-3 activation in apoptotic cells promotes the proliferation of surrounding cells by activating growth signals through the release of prostaglandin in a process termed ‘phoenix rising’ (Li et al., 2010). Another form of controlled and localized activation of caspase-3 is needed for the removal of neuronal dendrites during pruning (Williams et al., 2006). Although these functions are non-apoptotic roles of caspase-3, they are dependent on the catalytic activity of the protein and often resemble specialized forms of cellular degradation. We now provide evidence that procaspase-3 also has a non-apoptotic role in controlling cell adhesion that is not related to its function in apoptosis or its catalytic activity. Our data demonstrate that procaspase-3 is regulating fibronectin secretion and that fibronectin secretion is required for the cell adhesion phenotype observed in Casp3−/− cells. However, we have not ruled out the possibility that the secretion or cell-surface expression of additional molecules also contributes to this phenotype. Our data suggest that procaspase-3 plays a role in ER–Golgi transport or vesicle trafficking, and this is currently under investigation. However, by regulating cellular adhesion, procaspase-3 can indirectly regulate the apoptotic threshold of the cell. Thus, a re-evaluation of survival data in Casp3−/− cells might be appropriate. Finally, these data also provide new insight as to why caspase-3 is rarely lost or mutated in cancer (Jäger and Zwacka, 2010). Previously, it was believed that this was due to the lack of selective pressure to lose a protein that functioned ‘past the point of no return’ in cell death (Ghavami et al., 2009). However, these new findings raise the possibility that caspase-3 is maintained in cancer cells so that they have a motility or migration advantage.
MATERIALS AND METHODS
Cell culture
Immortalized mouse embryonic fibroblasts (MEFs) were generated and grown in Dulbecco's Modification of Eagle's Medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum (FBS, Cellgro), 1% penicillin-streptomycin (Cellgro), 1% l-glutamine (Cellgro), 1% non-essential amino acids (Cellgro), 1% sodium pyruvate (Cellgro) and 0.001% 2-mercaptoethanol (Gibco) at 37°C in a humid incubator under 5% CO2. ΦNX-Ecotropic packaging cell lines (Nolan lab, Stanford University) and 293T cells were grown in DMEM medium supplemented with 10% FBS, 1% penicillin-streptomycin, 1% l-Glutamine, 1% non-essential amino acids and 1% sodium pyruvate at 37°C in a humid incubator under 5% CO2. Serum-free medium for death assays was made as described above without FBS. Splitting and harvesting of adherent cells was conducted by washing cells with phosphate buffered saline (PBS, Cellgro) and applying 0.25% trypsin (Cellgro) for 5–10 minutes.
Morphological studies
Cells were grown on glass coverslips (Fisher) coated with 5 µg/cm2 fibronectin unless otherwise specified (Chemicon International) and were fixed with PHEMO buffer (68 mM PIPES, 25 mM HEPES, 15 mM EGTA-Na2, 3 mM MgCl2•6H2O, 10% DMSO, pH 6.8) supplemented with 3.7% formaldehyde (Fisher), 0.05% glutaraldehyde (Fisher) and 0.5% Triton X-100 (Fisher) for permeabilization when indicated. Actin was labeled by staining with Alexa-Fluor-555–phalloidin (Invitrogen) and DNA was stained with 300 nM 4′-6-diamidino-2-phenylindole dilactate (DAPI, Invitrogen) for 5–10 minutes in dH2O. Coverslips were then mounted on microslides (Fisher) by using polyvinyl alcohol mounting medium with DABCO anti-fade (Fluka). Cell morphology was determined by visualizing cells using a point-scanning laser confocal microscope (LSM 510 META) and analyzing cell length and cell-footprint area using Ziess Image Browser's region of interest (ROI) tool.
Cell adhesion
5×103 cells were seeded in 96-well plates in triplicate and allowed to adhere for indicated times. Cells were then fixed with PHEMO fixative and stained with Crystal Violet (Fisher Scientific) in 2% ethanol for 10 minutes. Plates were washed three times in H2O and then 2% SDS was added for 30 minutes to reconstitute the remaining Crystal Violet. Absorbance was measured on a 96-well-plate reader at 590 nM (Humphries, 2009).
Cell volume analysis
Cells were harvested by trypsinization and resuspended in FACS buffer (PBS with 1% bovine serum albumin and 0.01% sodium azide), and the cell volume was measured by recording forward scatter (FSC) on a FACSCanto II flow cytometer and analyzing using FlowJo software.
Motility analysis
Cells were seeded into 35 mm×10 mm cell culture dishes (Corning) and allowed to grow until confluent. Initiation of migration was achieved by scratching confluent cells with a P10 pipette tip. Motility was measured on a Zeiss Axiovert 200M microscope mounted with a Perkin Elmer Ultraview ERS enclosed in a heated chamber (37°C), with 5% CO2 injection. Images were acquired every 10 minutes for at least 15 hours (Liang et al., 2007). After image acquisition, Volocity software was used to determine the percentage of wound closure at designated time-points and to analyze single-cell tracks over time to obtain velocity and meandering index. The meandering index is a direct measure of how straight a cell moves on a scale from 0 to 1, where a meandering index of 1 is a straight line (displacement is equal to distance).
Cell cycle analysis
Cells were seeded on six-well plates, allowed to grow for 24 hours, harvested and fixed. Cell cycle analysis was performed on a FACSCanto II flow cytometer after staining with PI/RNase staining buffer (BD Pharmingen) for 30 minutes at room temperature (Krishan, 1975).
Motility cell cycle analysis
Cells were seeded on six-well plates and allowed to grow to confluency. A P10 pipette tip was used to make eight parallel scratches or an eight-by-eight scratch grid in order to mimic motility-assay conditions and the release of cells from contact inhibition. After 12 hours of migration in a 37°C incubator, the cells were harvested and cell cycle was analyzed as above.
Doubling-time analysis
5×104 cells were seeded per well of a six-well plate, and the cells were trypsinized and counted on a hemocytometer at the indicated time points.
Retroviral transduction
ΦNX-Ecotropic packaging cell lines (Nolan lab, Stanford University) were transfected with a plasmid (pBabe-puro, Casp3 pBabe-puro or Casp3C163S pBabe-puro) using Lipofectamine (Invitrogen). Target MEFs were seeded on six-well plates and allowed to grow for 24 hours. Viral supernatants were collected and filtered through 0.45-µm syringe filters (Pall) at 24, 28 and 32 hours and then replaced with fresh medium. At each time point, viral supernatants were applied directly to target cells for infection using Polybrene Infection/Transfection Reagent (Millipore). After 24 hours, viral supernatants were removed from the target cells and replaced with fresh medium for 24–72 hours. Once cells recovered from infection they were selected with 2.5 µg/ml puromycin (Sigma).
Western blot analysis
Cells were harvested, lysed and subjected to western blotting as described previously (Johnson and Boise, 1999). Primary antibody against caspase-3 (Cell Signaling Technology) was used. Actin (Sigma) was used as a loading control and was visualized as described previously (Johnson and Boise, 1999).
Caspase-activity assay
Cells were seeded and allowed to grow for 24 hours. Then the medium was removed and replaced with complete medium or serum (FBS)-free medium, and cells were collected after 48 hours. Caspase activity was determined using a colorimetric caspase-activity assay kit (Invitrogen).
Flow cytometry for integrin expression
Cells were collected and stained with a FITC-conjugated integrin β-1 antibody (Molecular Probes) or a FITC-conjugated IgG isotype control (BioLegend) in FACS buffer for 15 minutes at 4°C. Cells were then washed, resuspended in FACS buffer and analyzed by flow cytometry.
Cell spreading
Cells were harvested, washed and incubated under serum-free conditions with or without brefeldin A at 37°C for 4 hours. Cells were resuspended in Hanks' Balanced Salt Solution (HBSS, Cellgro) with or without brefeldin A and were seeded onto glass coverslips with or without fibronectin coating. Plates were incubated at 37°C for 1 hour and then cells were fixed, permeabilized when indicated, and stained with phalloidin, DAPI and an antibody against either P-FAK (Sigma) or fibronectin (Sigma). Cells were then mounted and visualized as described above.
Fibronectin secretion and production analysis
Cell-spreading assays were performed as described above with equal cell numbers that were plated on plastic Petri dishes. Medium was collected for the analysis of soluble fibronectin secretion, which was performed by using a fibronectin ELISA kit (Abcam), and cells were collected and lysed for analysis of intracellular fibronectin production by ELISA.
Lentiviral transduction and fibronectin knockdown
293T packaging cell lines were co-transfected with a plasmid (pLKO-TET-on or pLKO-TET-on-shFibronectin) and two helper plasmids using Lipofectamine (Invitrogen). Infections were performed as described above. Fibronectin knockdown was accomplished by growing cells in 5 µg/ml doxycycline (Sigma) for 72 hours prior to experiments.
Cell death assays
Cells were seeded in six-well plates and treated as indicated. At the indicated time-points, the medium and adherent cells were harvested, washed with PBS, and stained with annexin-V–FITC (Biovision) and propidium iodide (PI, Sigma) as described previously (Johnson and Boise, 1999). Cell death was determined by flow cytometry on a BD FACSCanto II system with FACSDiva software, and data was analyzed with FlowJo.
Statistics
Student's t-test was performed using GraphPad Prism software, and the data was determined to be statistically significant when P<0.05.
Acknowledgments
The authors thank Leslie de Armas (University of Miami, Miami, FL) for technical help, David Jaye (Emory University, Atlanta, GA) for feedback on fibronectin staining, Oskar Laur of the Emory Custom Cloning Core and the Integrated Cell Imaging Core at Emory University.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
M.B. designed and performed experiments and prepared the manuscript. D.B.W. designed and performed experiments. A.R. and A.I.M. helped with data analysis. L.H.B. oversaw the project, designed experiments and prepared the manuscript.
Funding
The work was funded by the National Institutes of Health [grant number R01 CA127910 to L.H.B.]. Additional support was provided by the Georgia Research Alliance and the TJ Martell Foundation (to L.H.B.). Deposited in PMC for release after 12 months.
References
- Ballestrem C., Wehrle-Haller B., Hinz B., Imhof B. A. (2000). Actin-dependent lamellipodia formation and microtubule-dependent tail retraction control-directed cell migration. Mol. Biol. Cell 11, 2999–3012 10.1091/mbc.11.9.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Rajakaruna S., Menko A. S. (2012). Insulin-like growth factor receptor-1 and nuclear factor κB are crucial survival signals that regulate caspase-3-mediated lens epithelial cell differentiation initiation. J. Biol. Chem. 287, 8384–8397 10.1074/jbc.M112.341586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo K. A., Dembo M., Kaverina I., Small J. V., Wang Y. L. (2001). Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881–888 10.1083/jcb.153.4.881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentnall M., Rodriguez-Menocal L., De Guevara R. L., Cepero E., Boise L. H. (2013). Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 14, 32 10.1186/1471--2121--14--32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S., Ghila L., Dobretz K., Wenger Y., Bauer C., Buzgariu W., Martinou J. C., Galliot B. (2009). Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 17, 279–289 10.1016/j.devcel.2009.07.014 [DOI] [PubMed] [Google Scholar]
- Danial N. N., Korsmeyer S. J. (2004). Cell death: critical control points. Cell 116, 205–219 10.1016/S0092--8674(04)00046--7 [DOI] [PubMed] [Google Scholar]
- Danial N. N., Gramm C. F., Scorrano L., Zhang C. Y., Krauss S., Ranger A. M., Datta S. R., Greenberg M. E., Licklider L. J., Lowell B. B. et al. (2003). BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424, 952–956 10.1038/nature01825 [DOI] [PubMed] [Google Scholar]
- Danial N. N., Walensky L. D., Zhang C. Y., Choi C. S., Fisher J. K., Molina A. J., Datta S. R., Pitter K. L., Bird G. H., Wikstrom J. D. et al. (2008). Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 14, 144–153 10.1038/nm1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 10.1146/annurev.cellbio.011209.122036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S., Hashemi M., Ande S. R., Yeganeh B., Xiao W., Eshraghi M., Bus C. J., Kadkhoda K., Wiechec E., Halayko A. J. et al. (2009). Apoptosis and cancer: mutations within caspase genes. J. Med. Genet. 46, 497–510 10.1136/jmg.2009.066944 [DOI] [PubMed] [Google Scholar]
- Green D. R., Oberst A., Dillon C. P., Weinlich R., Salvesen G. S. (2011). RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol. Cell 44, 9–16 10.1016/j.molcel.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. M. (1996). Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84, 345–357 10.1016/S0092--8674(00)81279--9 [DOI] [PubMed] [Google Scholar]
- Humphries M. J. (2009). Cell adhesion assays. Methods Mol. Biol. 522, 203–210 10.1007/978--1--59745--413--1_14 [DOI] [PubMed] [Google Scholar]
- Ishizaki Y., Cheng L., Mudge A. W., Raff M. C. (1995). Programmed cell death by default in embryonic cells, fibroblasts, and cancer cells. Mol. Biol. Cell 6, 1443–1458 10.1091/mbc.6.11.1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger R., Zwacka R. M. (2010). The enigmatic roles of caspases in tumor development. Cancers 2, 1952–1979 10.3390/cancers2041952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. W., Boise L. H. (1999). Bcl-2 and caspase inhibition cooperate to inhibit tumor necrosis factor-alpha-induced cell death in a Bcl-2 cleavage-independent fashion. J. Biol. Chem. 274, 18552–18558 10.1074/jbc.274.26.18552 [DOI] [PubMed] [Google Scholar]
- Kaiser W. J., Upton J. W., Long A. B., Livingston-Rosanoff D., Daley-Bauer L. P., Hakem R., Caspary T., Mocarski E. S. (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 10.1038/nature09857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R. M., Ellerby L. M., Ellerby H. M., Naiem S., Yaffe M. P., Margoliash E., Bredesen D., Mauk A. G., Sherman F., Newmeyer D. D. (2000). Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J. Biol. Chem. 275, 16127–16133 10.1074/jbc.275.21.16127 [DOI] [PubMed] [Google Scholar]
- Krause M., Dent E. W., Bear J. E., Loureiro J. J., Gertler F. B. (2003). Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19, 541–564 10.1146/annurev.cellbio.19.050103.103356 [DOI] [PubMed] [Google Scholar]
- Krishan A. (1975). Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 66, 188–193 10.1083/jcb.66.1.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhani S. A., Masud A., Kuida K., Porter G. A., Jr, Booth C. J., Mehal W. Z., Inayat I., Flavell R. A. (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851 10.1126/science.1115035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy L., Ngo V. N., Emre N. C., Shaffer A. L., III, Yang Y., Tian E., Nair V., Kruhlak M. J., Zingone A., Landgren O. et al. (2013). Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 23, 435–449 10.1016/j.ccr.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen B. D., Rampalli S., Burns L. E., Brunette S., Dilworth F. J., Megeney L. A. (2010). Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. USA 107, 4230–4235 10.1073/pnas.0913089107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Brieher W. M., Scimone M. L., Kang S. J., Zhu H., Yin H., von Andrian U. H., Mitchison T., Yuan J. (2007). Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat. Cell Biol. 9, 276–286 10.1038/ncb1541 [DOI] [PubMed] [Google Scholar]
- Li F., Huang Q., Chen J., Peng Y., Roop D. R., Bedford J. S., Li C. Y. (2010). Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13 10.1126/scisignal.2000634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C. C., Park A. Y., Guan J. L. (2007). In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 10.1038/nprot.2007.30 [DOI] [PubMed] [Google Scholar]
- Munoz J., Zhou Y., Jarrett H. W. (2010). LG4-5 domains of laminin-211 binds alpha-dystroglycan to allow myotube attachment and prevent anoikis. J. Cell. Physiol. 222, 111–119 10.1002/jcp.21927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 10.1038/nature09852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R., Yamada K. M. (2002). Fibronectin at a glance. J. Cell Sci. 115, 3861–3863 10.1242/jcs.00059 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704–1709 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- Senft J., Helfer B., Frisch S. M. (2007). Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 67, 11505–11509 10.1158/0008--5472.CAN--07--5755 [DOI] [PubMed] [Google Scholar]
- Shi Y. (2002). Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9, 459–470 10.1016/S1097--2765(02)00482--3 [DOI] [PubMed] [Google Scholar]
- Torres V. A., Mielgo A., Barbero S., Hsiao R., Wilkins J. A., Stupack D. G. (2010). Rab5 mediates caspase-8-promoted cell motility and metastasis. Mol. Biol. Cell 21, 369–376 10.1091/mbc.E09--09--0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsukawa G., Wu Y., Sasaki Y., Hartshorne D. J., Yamakita Y., Yamashiro S., Matsumura F. (2004). Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164, 427–439 10.1083/jcb.200306172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng A. S., Adams D. S., Qiu D., Koustubhan P., Levin M. (2007). Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev. Biol. 301, 62–69 10.1016/j.ydbio.2006.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. W., Kondo S., Krzyzanowska A., Hiromi Y., Truman J. W. (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236 10.1038/nn1774 [DOI] [PubMed] [Google Scholar]
- Witte M. B., Barbul A. (1997). General principles of wound healing. Surg. Clin. North Am. 77, 509–528 [DOI] [PubMed] [Google Scholar]
- Woo M., Hakem R., Soengas M. S., Duncan G. S., Shahinian A., Kägi D., Hakem A., McCurrach M., Khoo W., Kaufman S. A. et al. (1998). Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 12, 806–819 10.1101/gad.12.6.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Wang X., Purring-Koch C., Wei Y., McLendon G. L. (2001). A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J. Biol. Chem. 276, 13034–13038 10.1074/jbc.M009773200 [DOI] [PubMed] [Google Scholar]