

Abstract
The ability of cells to invade into the dermis is a critical event in the development of cutaneous melanoma and ultimately an indicator of poor prognosis. However, the molecular events surrounding the acquisition of this invasive phenotype remain incompletely understood. Mutations in B-RAF are frequent in melanoma and are known to regulate the invasive phenotype. In this study, we sought to determine the molecular mechanisms controlling melanoma invasion. We found that mutant B-RAF signaling regulates a cadherin switch. In melanoma cells expressing mutant B-RAF we observed high levels of N-cadherin and low levels of E-cadherin. Depletion of mutant B-RAF, by siRNA, caused a decrease in the levels of N-cadherin and an increase in the levels of E-cadherin. Mechanistically, we found that this cadherin switch required the activity of Rac1 and its GEF, Tiam1, both of which show suppressed activity in the presence of mutant B-RAF. Consistent with the work of others, we found that depletion of mutant B-RAF decreased the invasive capacity of the melanoma cells. However, simultaneous depletion of B-RAF and Rac or Tiam1 resulted in invasive capacity similar to that of control cells. Taken together, our results suggest that mutant B-RAF signaling downregulates Tiam1/Rac activity resulting in an increase in N-cadherin levels and a decrease in E-cadherin levels and ultimately enhanced invasion.
Keywords: B-RAF, melanoma, Rac, cadherin, Tiam1
Introduction
Melanoma is the deadliest form of skin cancer and its incidence rate has been rising rapidly (1). Cutaneous melanomas arise from transformations in the epidermal melanocytes, the pigment producing cells of the skin. A key event in melanoma progression is the transition from the radial growth phase, where cells grow only within the epidermis, to the vertical growth phase, where cells invade and grow into the dermis (2). The molecular mechanisms underlying the invasive phenotype acquired during the vertical growth phase remain poorly understood.
The serine/threonine kinase B-RAF is mutated in ∼60% of melanomas (3). The most common mutation encodes for a valine to glutamic acid substitution (V600E) within the activation loop of B-RAF, resulting in the constitutive activation of B-RAF and subsequently the downstream MEK/ERK pathway (3). Although a B-RAF v600E mutation is not sufficient to cause melanoma progression (4), B-RAF mutations do correlate with melanoma progression as they have been detected in 75% of vertical growth phase tumors and only 10% of radial growth phase tumors (5). Mutant B-RAF and its downstream activation of the MEK/ERK pathway have been implicated in regulating melanoma proliferation, migration and invasion (6), (7).
The acquisition of invasive ability is caused, in part, by the loss of cell-cell adhesion. Cadherins are a family of cell-surface glycoproteins that promote calcium-dependent homotypic and heterotypic cell-cell adhesion and serve as key components of adherens junctions (8). Cadherins are expressed in a cell-type specific manner. E-cadherin is present in polarized epithelial cells while N-cadherin is present in several cell types including, neurons, endothelial cells and fibroblasts. During melanoma progression there is a progressive loss of E-cadherin (9), (10). Although there is a loss in the expression of E-cadherin many melanoma cells express high levels of N-cadherin (11), (9). The switch from E-cadherin to N-cadherin allows communication of melanoma cells with fibroblast and endothelial cells (12), (13) and promotes both the survival and migration of the melanoma cells (14).
Rac1 is an important regulator of both the migratory and invasive phenotype of cells. Rac1 activity is required for both the formation and maintenance of adherens junctions in epithelial cells (15), (16) and is essential for the recruitment of cadherins to sites of cell-cell contact (17). Although rarely mutated, changes in the expression or activity of Rac1 are associated with changes in tumor progression. Rac1 has been well-established to induce invasion and metastasis in a number of cancers (18), (19). However, Rac1 is also known to inhibit the invasion and migration of renal cell carcinomas (20). This is due to the ability of Rac1 to upregulate cadherin-dependent adhesion.
In this study we sought to determine if mutant B-RAF regulates the invasive capacity of melanoma cells by regulating Rac1 activity and the subsequent effects on cadherin expression. We demonstrate that knock down of mutant B-RAF or inhibition of MEK/ERK signaling results in an increase in Rac1 activity due to the activation of the Rac1 GEF Tiam1. This increase in Rac1 activity is then responsible for a switch in the expression of N-cadherin to E-cadherin and a down regulation of the invasive capacity of melanoma cells.
Results
To determine whether B-RAF had any effect on cadherin expression in WM793 cells, a human vertical growth phase melanoma harboring a constitutively active B-RAFV600E allele, we utilized RNA interference. Two siRNAs targeting different sequences in B-RAF were used: one targeted a sequence present in both mutant and wild type B-RAF (B-RAF siRNA) and the other targeted the region containing the V600E mutation (V600E siRNA) (21). Transfection with either B-RAF or V600E siRNA ablated B-RAF protein levels when compared with control transfected cells (Fig 1A). Concomitant with decreased B-RAF protein expression, phosphorylation of its downstream target, ERK1/2, was also attenuated. Cadherin expression was determined in control and B-RAF knockdown cells by Western blotting. In control WM793 cells N-cadherin was expressed while there was no detectable level of E-cadherin observed (Fig 1A). In contrast, B-RAF knockdown with either of the two B-RAF targeting siRNAs showed decreased levels of N-cadherin when compared to control cells. In addition, the B-RAF knockdown cells also showed detectable levels of E-cadherin. PLX4032 is a potent and selective inhibitor of mutant B-RAF signaling (22). We analyzed cadherin expression following inhibition of mutant B-RAF signaling using PLX4720, the non-clinical equivalent of PLX4032. PLX4720 acts in a manner that is indistinguishable from PLX4032 (7), (22). When cells were treated with PLX4720 we observed a decrease in the expression of N-cadherin and an increase in the expression of E-cadherin when compared to control cells (Fig 1B). B-RAF is a potent activator of the MEK/ERK1/2 signaling pathway; therefore, we sought to determine if MEK activity was important for changes in cadherin expression in melanoma cells. WM793 cells were treated with or without the MEK inhibitor, U0126, and cadherin expression was examined by Western blotting. When cells were treated with U0126 we observed a decrease in the expression of N-cadherin and an increase in the expression of E-cadherin when compared to control cells (Fig 1C). To determine if the effects of B-RAF knockdown caused a cadherin switch in more than one cell line, we performed similar experiments in WM115 cells (another VGP cell line that expresses an activating mutation in B-RAF, V600D (3)). Importantly, we found that knockdown of B-RAF or inhibition of MEK with U0126 caused a decrease in N-cadherin expression and an increase in E-cadherin expression in the WM115 cells as well (Fig 1D). Taken together, these results suggest that mutant B-RAF and its downstream MEK/ERK1/2 signaling regulates cadherin expression in WM793 and WM115 melanoma cells.
Figure 1.

BRAF knockdown causes a cadherin switch. A) WM793 cells were transfected with control, total B-RAF (BRAF) or mutant B-RAF (V600E) siRNA. Cell lysates were analyzed by Western blotting for BRAF, P-ERK, ERK, E-cadherin, N-cadherin and actin (as a loading control). B) WM793 cells were treated with5uM PLX4720 for 24 hours. N-cadherin and E-cadherin expression as well as P-Erk levels were analyzed by Western blotting. Actin was used as a loading control. C) WM793 cells were treated with 1uM U0126 for 24 hours. N-cadherin and E-cadherin expression as well as P-Erk levels were analyzed by Western blotting. Actin was used as a loading control. D) WM115 cells were transfected with control, total B-RAF (BRAF) siRNA or 1uM U0126. Cell lysates were analyzed by Western blotting for P-Erk, N-cadherin and E-cadherin levels. Actin was used as a loading control.
Once we had observed that E-cadherin expression was upregulated upon B-RAF knockdown we investigated whether the E-cadherin was functional. We performed immunofluorescence on WM793 cells subjected to control siRNA or B-RAF siRNA treatment and looked at the localization of E-cadherin staining (Fig 2A). We found that in the control cells there was no detectable staining of E-cadherin, while in the B-RAF knockdown cells staining for E-cadherin is visible at discrete areas of cell-cell contact. We next performed a co-immunoprecipitation analysis to determine if E-cadherin was able to bind β-catenin, a known binding partner. As demonstrated in Fig 2B, under control conditions immunoprecipitating β-catenin brought down no E-cadherin (likely because there is no E-cadherin present). However, upon B-RAF knockdown we find that E-cadherin does co-immunoprecipitate with β-catenin. While normal epidermal melanocytes form cell-cell adhesions to neighboring keratinocytes, melanoma cells do not and instead form interactions with neighboring fibroblasts or endothelial cells (12), (13). To analyze the effect of B-RAF knockdown on cell-cell adhesion partners a co-culture model was used. WM793 melanoma cells subjected to control siRNA treatment or B-RAF siRNA treatment were cultured with either fibroblasts (MRC5 cells) or keratinocytes (HaCat cells) and transepithelial electrical resistance (TER) was measured. We found that B-RAF knockdown in the melanoma cells decreases fibroblast/melanoma TER when compared to control melanoma cells (Fig 2C). Conversely, B-RAF knockdown in the melanoma cells increased keratinocyte/melanoma TER. Taken together with earlier data, these data suggest that B-RAF knockdown causes a functional switch in cadherins.
Figure 2.

BRAF knockdown results in the expression of a functional E-cadherin. A) WM793 cells were transfected with BRAF siRNA for 72h. Cells were then stained for the presence of E-cadherin. B) WM793 cells were transfected with control or BRAF siRNA. Cells were then lysed with immunoprecipitation buffer and β-catenin was immunoprecipitated from cells. Immunoprecipitates were then blotted for the presence of E-cadherin. C) WM793 cells were transfected with control siRNA or BRAF siRNA and co-cultured with either MRC5 cells (fibroblasts) or HaCat cells (keratinocytes) on transwell filters. After 72h TER readings were taken with a voltohmeter. * indicates statistical significance when compared to control treatment (p<0.05, Student's t-test).
Rac1 activity is known to regulate key components within the adherens junction (15). To determine if mutant B-RAF expression has any effect on Rac1 activity, control and B-RAF knockdown WM793 cells were analyzed for Rac1 GTP-loading, measured by pull-down assays with glutathione S-transferase-p21-activating kinase. As shown in Fig 3A, knockdown of B-RAF with either siRNA resulted in an increase in Rac1 activity. To investigate a role for mutant B-RAF signaling in Rac1 activity, we again used PLX4720, the inhibitor of mutant B-RAF signaling. We found that WM793 cells treated with PLX4720 had increased levels of Rac1 activity when compared to control cells (Fig 3B). To determine if the effect of B-RAF knockdown on Rac1 activity was due to the ability of B-RAF to signal downstream to MEK the U0126 compound was used (Fig 3C). In WM793 cells treated with U0126 an increase in the levels of active Rac1 was observed when compared to control treated cells. Similar to the WM793 cells, we found that B-RAF knockdown or MEK inhibition in the WM115 cells also resulted in an increase in Rac activity (Fig 3D). These results are consistent with the idea that mutant B-RAF inhibits Rac1 activity in WM793 and WM115 melanoma cells.
Figure 3.
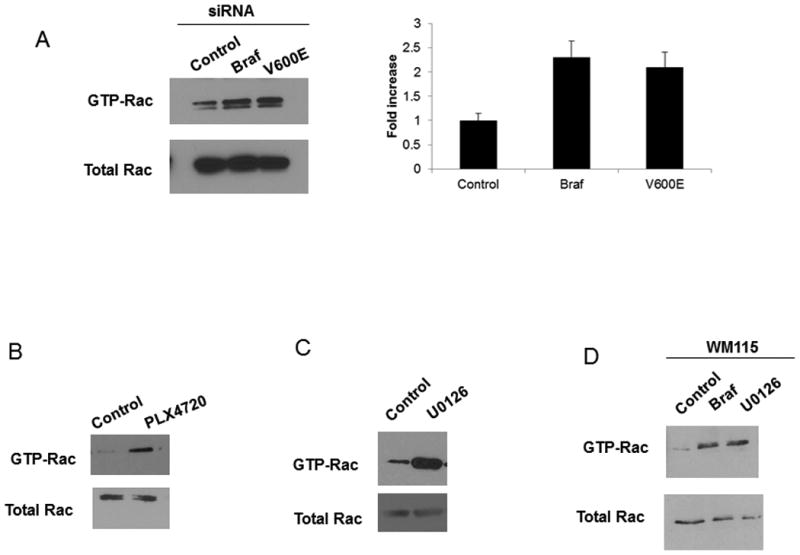
BRAF knockdown results in the activation of Rac1. A) WM793 cells were transfected with control, total B-RAF (BRAF) or mutant B-RAF (V600E) siRNA. The cells were lysed and the activation of Rac was measured through a GST-PBD pulldown assay as described in the Methods. B and C) WM793 cells were treated with 5uM PLX4720 (B) or 1uM U0126 (C) for 24 hours. The cells were lysed and the activation of Rac was measured through a GST-PBD pulldown assay as described in the Methods. D) WM115 cells were transfected with control, total B-RAF (BRAF) siRNA or 1uM U0126. The cells were lysed and the activation of Rac was measured through a GST-PBD pulldown assay as described in the Methods.
As Rac1 activity has been shown to be a critical regulator of E-cadherin within the adherens junction (17) we next sought to determine if the ability of B-RAF to modulate cadherin expression was dependent on the B-RAF mediated regulation of Rac1 activity. To test this idea we simultaneously knocked down both B-RAF and Rac1 in WM793 cells and WM115 cells and examined the levels of N-cadherin and E-cadherin by Western blotting (Fig 4A and C). As demonstrated earlier in Fig 1A, knockdown of mutant B-RAF resulted in the downregulation of N-cadherin. However, when both B-RAF and Rac1 are ablated with siRNA, N-cadherin levels remain similar to that of control. Conversely, when mutant B-RAF alone is knocked down E-cadherin levels increase. However, when both B-RAF and Rac1 are knocked down E-cadherin levels remain undetectable. To further examine whether the effects of B-RAF knockdown on cadherin expression were dependent on increased Rac1 activity, we used NSC, an inhibitor of Rac1 activity (23), (24) (Fig 4B). WM793 cells that had been subjected to either control or B-RAF knockdown were treated with NSC, to inhibit Rac1 activity, and the relative levels of N-cadherin and E-cadherin were analyzed. We found that in B-RAF knockdown cells inhibition of Rac1 activity with NSC caused N-cadherin levels to remain elevated and E-cadherin levels to remain undetectable, similar to control siRNA treated cells. In contrast, when B-RAF was knocked down in the absence of NSC N-cadherin levels decreased while E-cadherin levels increased. Collectively, these results suggest that Rac1 is required for the B-RAF regulated cadherin switch.
Figure 4.

The cadherin switch induced upon BRAF knockdown requires Rac. A) WM793 cells were transfected with control, total B-RAF (BRAF) or mutant B-RAF (V600E) siRNA and Rac siRNA. Cell lysates were analyzed by Western blotting for E-cadherin, N-cadherin or actin (loading control). The levels of B-RAF and Rac were also analyzed to confirm knockdown. B) Cells were treated overnight with NSC. Cell lysates were analyzed by Western blotting for E-cadherin, N-cadherin or actin (loading control). To confirm the activity of the NSC compound cells were lysed and the activation of Rac was measured through a GST-PBD pulldown assay as described in the Methods. C) WM115 cells were transfected with control, total B-RAF (BRAF) siRNA and Rac siRNA. Cell lysates were analyzed by Western blotting for E-cadherin, N-cadherin or actin (loading control). The levels of B-RAF and Rac were also analyzed to confirm knockdown.
Once we had determined that knockdown of mutant B-RAF in melanoma cells resulted in a Rac-dependent cadherin switch we wondered what the effect would be on Rac activity and cadherin status if we transfected normal melanocytes with mutant B-RAF? We found that melanocytes expressing mutant B-RAF show decreased levels of E-cadherin and increased levels of N-cadherin when compared to their control counterparts (Fig 5). Additionally, we found that Rac activity was decreased in the mutant B-RAF expressing melanocytes.
Figure 5.
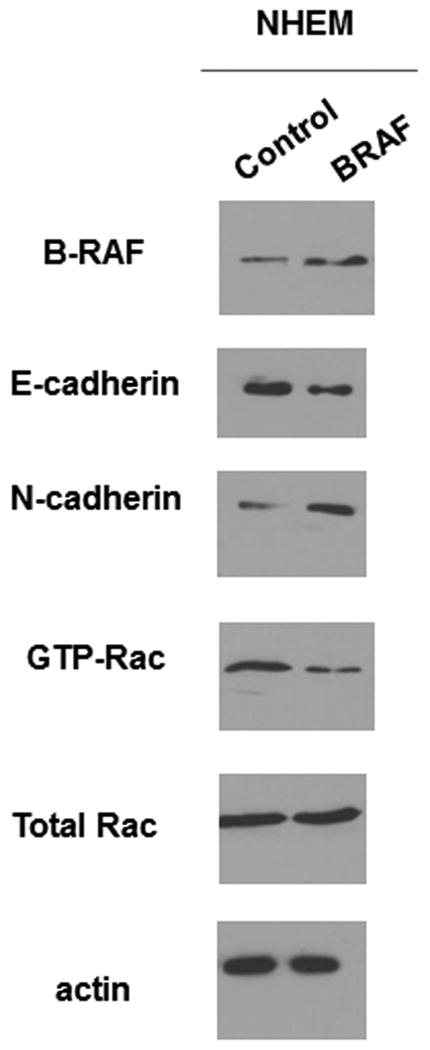
Melanocytes experience a cadherin switch upon mutant B-Raf expression. NHEM were transfected with B-Raf V600E for 24 hours. Cells were lysed and the lysates were analyzed for the presence of B-Raf, E-cadherin, N-cadherin, GTP-Rac, total Rac and actin (loading control).
While approximately 60% of melanomas harbor B-RAF mutations an additional 15% of melanomas express mutant N-Ras (25), (3). We wondered if melanoma cells that expressed N-Ras would show a similar Rac-dependent cadherin switch to the B-RAF mutant melanomas. In order to test this idea we used the VMM39 cell line, a melanoma cell line that expresses oncogenic N-Ras but has WT B-RAF. In Fig 6A we compared the basal levels of p-Erk and active Rac in the B-RAF mutant cell lines, WM793 and WM115, to the N-Ras mutant cell line, VMM39. We found that the mutant N-Ras cell line, VMM39, had lower levels of p-ERK and higher levels of active Rac than the mutant B-RAF cell lines, WM793 and WM115. We earlier demonstrated that in mutant B-RAF cells MEK inhibition with the U0126 compound raised the levels of active Rac and caused a switch in the expression of N-cadherin and E-cadherin (Fig 1 and 3). To determine if MEK inhibition had similar affects in the VMM39 cells we treated the cells with U0126 and analyzed levels of active Rac, N-cadherin and E-cadherin (Fig 6B). In contrast to the mutant B-RAF cell lines we found that MEK inhibition had no effect on the levels of active Rac, N-cadherin or E-cadherin in the VMM39 cells. Furthermore, we found that knockdown of Rac expression with siRNA also had no effect on the levels of N-cadherin or E-cadherin in the VMM39 cells. Collectively, these data suggest that a Rac-dependent cadherin switch is not present in the VMM39 N-Ras mutant cell line.
Figure 6.
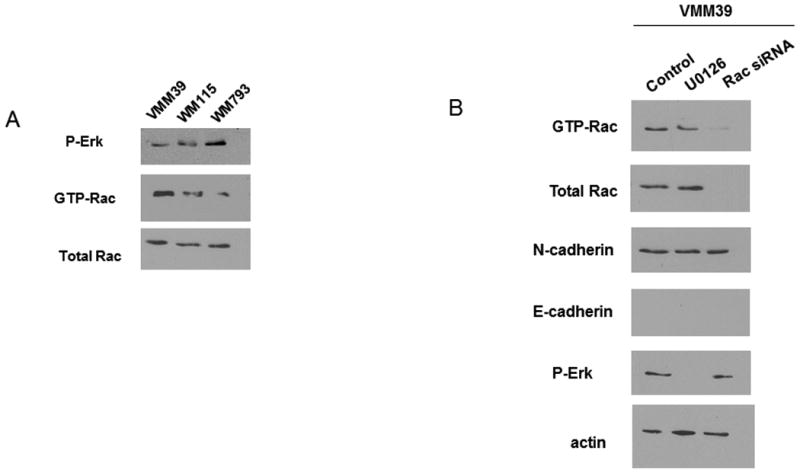
The BRAF- induced cadherin switch does not occur in N-Ras mutant melanoma cells. A) VMM39, WM115 and WM793 cell lysates were blotted to compare relative levels of p-Erk, Rac and active Rac. Actin was used as a loading control. B) VMM39 cells were transfected with control siRNA, BRAF siRNA or treated with 1uM U0126. Cells were lysed and samples were blotted for N-cadherin, E-cadherin, active Rac, total Rac and p-Erk levels. Actin was used as a loading control.
The activity of Rac1 is regulated in part by the activity of GEFs. To identify the GEF(s) responsible for Rac1 activation in response to mutant B-RAF knockdown, we performed pull-down assays with the nucleotide free Rac1 mutant Rac1 (15A) (26). WM793 cells were subjected to knockdown of B-RAF by 2 different siRNAs and GEF activity was analyzed. The association of Rac1-15A with the GEF Tiam1 was increased upon B-RAF knock down, indicative of an increase in the activity of the GEF (Fig 7A). The association of Rac1-15A with the GEF Vav2 remains unchanged after B-RAF knockdown (Fig 7B). Activation of Tiam1 is known to change its cellular localization (27). To determine if there was any change in the cellular localization of Tiam1 upon its activation via BRAF knockdown, WM793 cells were fractionated in to cytosolic and membrane fractions. We found that BRAF knockdown caused a shift of Tiam1 from the cytosolic to the membrane fraction (Fig 5C).
Figure 7.
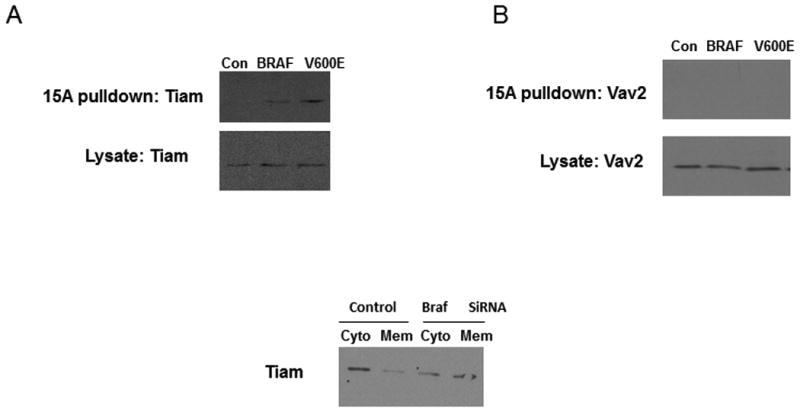
BRAF knockdown activates Tiam1. WM793 cells were transfected with control, total B-RAF (BRAF) or mutant B-RAF (V600E) siRNA and Rac siRNA. Pulldowns were then performed with GST-Rac(15A) and samples blotted with antibodies against Tiam1 (A) and Vav2 (B). B) WM793 cells were transfected with control or BRAF siRNA. Cells were then lysed, scraped and lystates homogenized. The lysates were then centrifuged to separate cytolsolic and membrane fractions. The fractions were then analyzed by Western blotting for the Tiam1.
To determine if Tiam1 was the GEF responsible for Rac1 activation upon B-RAF ablation siRNA was employed. WM-793 and WM115 cells were subjected to simultaneous knockdown of both B-RAF and Tiam1 and Rac1 activity was analyzed (Fig 8A and B). In the absence of Tiam1 siRNA the knockdown of B-RAF resulted in increased Rac1 activity. However, when both Tiam1 and B-RAF were knocked down Rac1 activity remained unchanged. Similar experiments were performed using Vav siRNA; however, no difference in Rac1 activity was observed in the presence or absence of Vav (Fig 8C). Taken together these results suggest that Tiam1 is the GEF responsible for the increase in Rac1 activity observed upon B-RAF knockdown.
Figure 8.
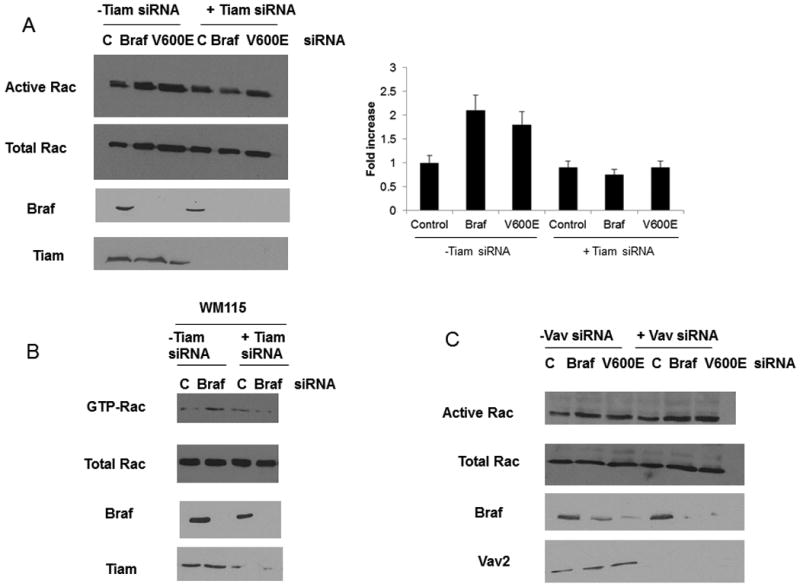
Rac activation observed upon BRAF knockdown requires Tiam1. WM793 (A) cells or WM115 cells (B) were transfected with control, Tiam1 (A, B) or Vav2 (C) siRNA in addition to BRAF or V600E siRNA. The cells were lysed and the activation of Rac was measured through a GST-PBD pulldown assay as described in the Methods. The levels of BRAF and Tiam1 (A,B) or Vav (C) were checked to confirm knockdown.
To confirm that Tiam1-dependent Rac1 activity was regulating the cadherin switch observed upon B-RAF ablation, both B-RAF and Tiam1 were knocked down with siRNA and the relative expression levels of E-cadherin and N-cadherin were assessed (Fig 9A and B). In cells subjected to a knockdown of B-RAF the levels of N-cadherin were observed to decrease while the levels of E-cadherin were observed to increase. However, when both Tiam1 and B-RAF were knocked down in the same cells the levels of N-cadherin remained elevated while the levels of E-cadherin remained undetectable. Tiam1 knockdown alone appeared to have no effect on the levels of N-cadherin and E-cadherin when compared to control cells. No effect on cadherin levels was observed when Vav and B-RAF were simultaneously knocked down in the WM793 cells (Fig 9C).
Figure 9.
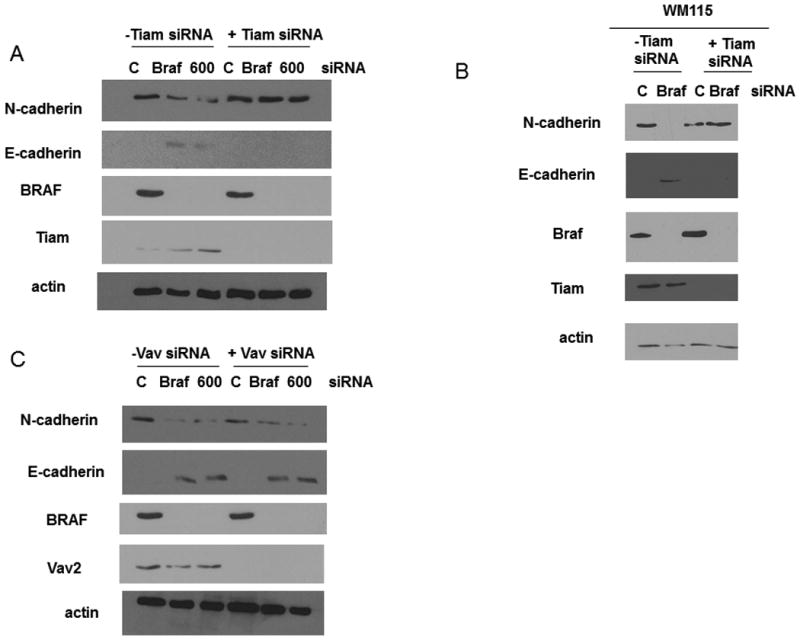
The cadherin switch observed upon BRAF knockdown requires Tiam1. WM793 cells (A) or WM115 cells (B) were transfected with control, BRAF, V600E and Tiam1 (A, B) or Vav2 (C) siRNA. N-cadherin and E-cadherin expression were analyzed by Western blotting. Actin was used as a loading control. The levels of BRAF and Tiam1 (A,B) or Vav (C) were checked to confirm knockdown.
Mutant B-RAF has been shown to modulate invasiveness in melanoma cells (7). We found that knockdown of B-RAF alone caused a significant decrease (p<0.05, Student's t test) in the invasive capacity of the melanoma cells. To investigate whether Rac was required for the decrease in invasion observed upon B-RAF knockdown cells were subjected to double knockdown of B-RAF and Rac. We found that under these conditions the invasive capacity of the double knockdown cells was not significantly different than the control cells (Fig 10A). As we had previously demonstrated in Fig 8 that Tiam1 activity was responsible for the increase in Rac1 activation observed upon B-RAF knockdown, we wanted to determine of Tiam1 activity was required for the decrease in the invasive phenotype observed after B-RAF knockdown. We tested this idea by using siRNA directed against both B-RAF and Tiam1. We again found that knockdown of B-RAF alone caused a significant decrease (p<0.05, Student's t test) in the invasive capacity of the melanoma cells. However, when we knocked down both Tiam1 and B-RAF we observed that the invasive capacity of the melanoma cells was not significantly different than that of the control cells (Fig 10B). When we knocked down both B-RAF and Vav2, another Rac1 GEF, we found that in both cases the melanoma cells continued to show significantly decreased (p<0.05, Student's t test) invasion equivalent to the B-RAF knockdown alone (Fig 10C). Collectively, these results suggest that both Rac and Tiam1 activity are required for the decrease in invasion seen upon B-Raf knockdown. One concern, however, was that the change in invasiveness could actually represent a change in the proliferative rate of the cells upon B-RAF knockdown. To test this possibility, we performed a proliferation assay of control, BRAF and V600E siRNA treated cells. We analyzed proliferation using an electrical impedance assay to quantify cell number (28). The RTCA (Real-Time Cell Analysis) is an automated real-time method to monitor impedance generated by cells grown on microelectrode sensors. WM793 cells were transfected with control, BRAF or V600E siRNA, trypsinized and replated onto microelectrode dishes and analyzed for 72 hours. A representative impedance trace (represented as Cell Index) is shown in Fig 10D. Knockdown of BRAF, with either the BRAF or V600E siRNA, showed no significant difference in Cell Index (impedance) when compared to the control cells. This result finds no significant difference in the proliferation rate of BRAF knockdown cells when compared to control cells. Taken together, thes e results suggest that mutant B-RAF regulates the invasive capacity of melanoma cells in part through the regulation of Tiam1-dependent Rac1 activity.
Figure 10.
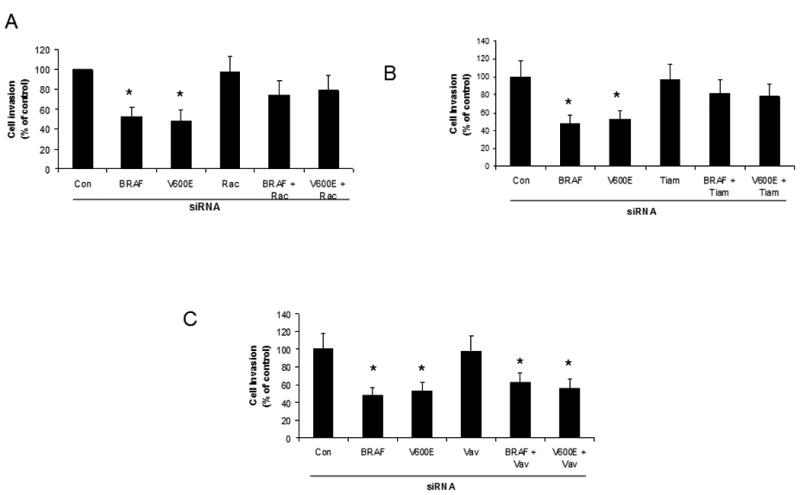
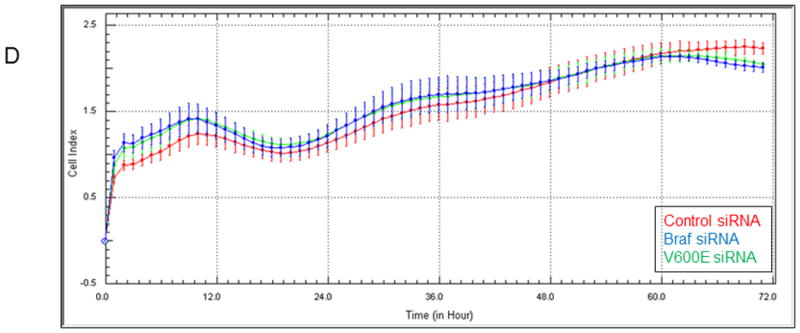
BRAF knockdown regulates the invasive capacity of melanoma cells through Tiam1 and Rac. WM793 cells were transfected with control, BRAF, V600E and Rac (A), Tiam1 (B) or Vav2 (C) siRNA. A 24 hour Matrigel cell invasion assay was performed after knockdown. Asterisks denote statistical significance comparing knockdown cells with controls. D) WM793 cells were transfected with control, BRAF or V600E siRNA and proliferation was measured through an RTCA assay. * indicates statistical significance when compared to control treatment (p<0.05, Student's t-test).
Discussion
The ability of cells to invade and eventually progress to a metastatic disease is an important hallmark in the development of melanoma. Acquiring the metastatic phenotype in melanoma changes the disease from one with a high survival rate (approximately 80%) to one with a low survival rate (less than 20%). Although the importance of the invasive phenotype is well-known, the molecular mechanisms that control this event are still poorly understood. Here we demonstrate that mutant B-RAF expression in melanoma cells regulates the invasive capacity of those cells through modulation of Rac1 activity and cadherin expression.
We initially focused on mutant B-RAF upregulation of N-cadherin expression and downregulation of E-cadherin expression. In the normal skin, melanocyte growth is regulated by the surrounding keratinocytes (29). Keratinocyte control is often lost during oncogenesis after the downregulation of E-cadherin, which is typically accompanied by an increase in N-cadherin (30). This event is known as the cadherin switch and it enables cell-cell interactions between adjacent melanoma cells as well as melanoma cell interactions with both fibroblasts and endothelial cells (12), (13), (14)). In addition, this cadherin switch promotes both the survival and migration of melanoma cells (14). The mutant B-RAF induced cadherin switch provides us with one mechanism underlying increased melanoma invasion. Our data demonstrate that the cadherin switch can be regulated by mutant B-RAF signaling which inhibits Rac1 activity. When this inhibition on Rac1 activity is lifted, either through B-RAF knockdown or pharmacological inhibition of B-RAF or B-RAF-dependent downstream MEK/ERK signaling, we observe a reversal of the cadherin switch, where N-cadherin is down regulated and E-cadherin is upregulated. This reversal in cadherin composition is concomitant with a decrease in the invasive capacity of the melanoma cells. Several transcription factors have linked mutant BRAF signaling in melanoma with the cadherin switch and with changes in invasiveness. Perhaps the most well-studied is the transcription factor Snail, which is known to be highly upregulated in melanoma and to inhibit the transcription of E-cadherin (31). Recent work has demonstrated that BRAF signaling through Erk leads to activation of Snail and the subsequent inhibition of the tumor suppressor, CYLD. This inhibition of CYLD allows for the transcription factor BCL3 to act on the N-cadherin promoter resulting in increased expression of N-Cadherin (32). Another interesting study has demonstrated the BRAF signaling leads to the activation of the transcription factor BRN2 which subsequently leads to an increase in invasion (33) (34). These previous studies provide interesting avenues of exploration for future work with the goal of determining what transcriptional events may link BRAF signaling to Rac signaling and the subsequent cadherin switch.
The small GTPases of the Rho family including RhoA, Rac1 and Cdc42 regulate the establishment and maintenance of adherens junctions in epithelial cells (15), (35), (16), (36). Rac1 activity in particular is considered one of the key signaling components controlling the cadherins at sites of cell-cell contact (16), (17), (37). We observed that in melanoma cells mutant B-RAF signaling attenuates Rac1 activity and results in a more invasive phenotype. This is in agreement with earlier studies demonstrating that increased Rac1 activity inhibits the migration and invasion of certain epithelial cells (17). However, this observation is also in contrast to published reports that demonstrate enhanced activation of Rac1 induces invasion of certain cell types, including murine T-lymphoma cells (38). It seems that Rac1 signaling is more complicated than simply high activity leading to high (or low) levels of invasion. Instead it is likely that the functions of Rac1 on invasion are at least in part cell-type and effector dependent.
The MAPK family is activated in virtually all melanomas (39). The most common mutations are in B-RAF, present in ∼60% of melanoma (3), and N-Ras, present in ∼15% of melanomas (40). Both B-RAF and N-Ras mutations lead to enhanced MEK/ERK signaling; however, the mechanism of MEK/ERK activation differs with these mutations. Cells with mutant N-Ras use C-RAF, as opposed to B-RAF, to signal to MEK/ERK (41) and this results in the propagation of different downstream signals (41). Indeed, we found that our mutant B-RAF cells signal through MEK to initiate a Rac-dependent cadherin switch. However, no such Rac-dependency in cadherin expression is observed in the mutant N-Ras cells. The role of the MEK/ERK pathway is regulating Rac1 activity is also diverse. In gastrointestinal epithelia EGF induces MEK/ERK activation that, in turn, induces Rac1 activation (42). This is in contrast to our results that demonstrate B-RAF dependent MEK/ERK activation inhibits Rac1 activity in melanoma cells. The combined data reinforce the idea that the MEK/ERK pathway balances Rac1 activation, although context-dependent differences in the mechanisms of crosstalk are present.
Tiam1, an upstream activator of Rac1 has been implicated in the regulation of adherens junction components, including cadherins (17), (43). When we examined Tiam1 activity in melanoma cells we found that, similar to Rac1, Tiam1 activity was low in cells expressing mutant B-RAF. Inhibition of B-RAF activity through RNAi resulted in enhanced Tiam1 activity which, in turn, led to the activation of Rac1. We demonstrate that the activity of Tiam1 is required for the cadherin switch and the decrease in invasion observed upon B-RAF knockdown. The effects of Tiam1 on cadherin expression are consistent with earlier reports showing that Tiam1 promotes the accumulation of E-cadherin and other adherens junction components while simultaneously decreasing cellular invasion (17), (44). Taken together with our data these observations support the idea that Tiam1 plays a role in regulating the potential malignancy of various cancers, including melanoma. It has been shown that expression of Tiam1 in the MV3 metastatic melanoma cells line inhibits the invasion and migration of those cells (45). It has also been observed that Ras-induced skin tumors progress more frequently to malignancy in Tiam1 -/- mice than in wild-type (45).
Additional studies have linked Rho GTPases and their effectors to B-RAF dependent effects in melanoma invasion. Aplin's group has demonstrated (46) (47) that the Rho antagonist Rnd3 regulates Rho activity and invasion through mutant BRAF-dependent mechanisms. Their results clearly show that B-RAF dependent MEK/ERK signaling regulates Rnd3 activity and our results show that B-RAF dependent MEK/ERK signaling regulates Tiam1 activity. In both cases, however, the exact mechanism of regulation remains unknown. Important future studies should be directed at determining the mechanisms through which ERK is able to regulate multiple GTPase effector pathways.
In conclusion, we provide evidence that mutant B-RAF regulates a cadherin switch to modulate the invasive capacity of melanoma cells (Figure 11). These effects are mediated through regulation of a Tiam1/Rac1/cadherin dependent pathway. In addition, Tiam1 acts downstream of B-RAF and modulates the crosstalk between the MEK/ERK pathway and the Rac1/cadherin pathway. These results emphasize the need to further identify the role of Tiam1/Rac1 in regulating melanoma invasiveness.
Figure 11.
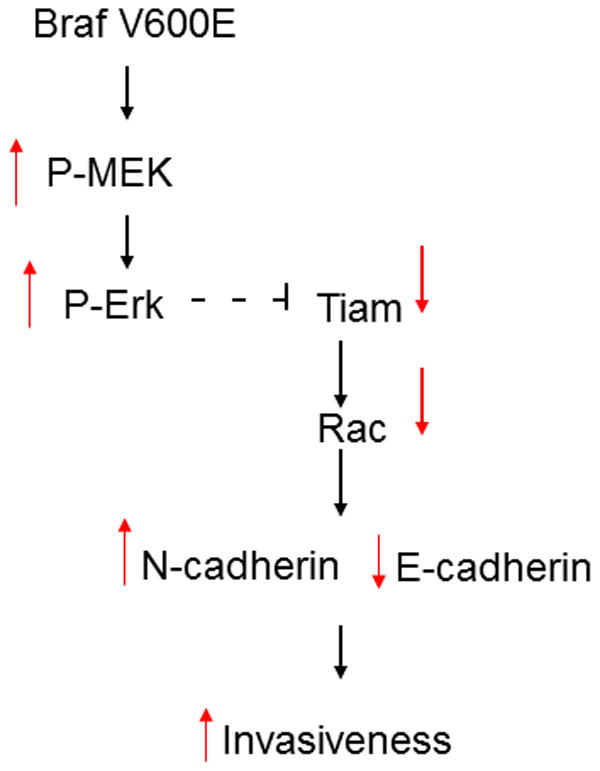
Model of mutant BRAF Rac-dependent cadherin switch. In melanoma cells expressing BRAFV600E the activity of MEK and ERK is high, leading to low Tiam1 activity followed by low Rac activity. Low Rac activity results in high expression of N-cadherin and low expression of E-cadherin and enhanced invasiveness. (Black arrows outline the pathway. Red arrows show the relative activity level or expression of the indicated molecule).
Methods
Cell Culture
Human WM793 and WM115 melanoma cells were kindly provided by Dr. Andrew Aplin (Thomas Jefferson University, Philadelphia, PA). WM793 and WM115 melanoma cells were cultured in MCDB 153 (Sigma-Aldrich, St. Louis, MO) containing 20% Leibovitz L-15 medium (Sigma-Aldrich), 2% fetal bovine serum (Sigma-Aldrich), 0.2% (wt/vol) sodium bicarbonate (Sigma-Aldrich) and 5 ug/ml insulin (Sigma-Aldrich) at 37C with 5% CO2. Human VMM39 melanoma cells were kindly provided by Dr. Jim Bear (University of North Carolina Chapel Hill, Chapel Hill, NC). HaCat keratinocytes were kindly provided by Paul Higgins (Albany Medical College, Albany, NY). VMM39 melanoma cells, HaCat and MRC5 cells were grown in DMEM containing 10% FBS. Normal Human Epidermal Melanocytes (NHEM) were purchased from ATCC and grown in Melanocyte Medium plus Bullet Kit (ATCC).
Antibodies and Inhibitors
The following antibodies were used: B-RAF and Tiam1 were from Santa Cruz Biotechnology (Santa Cruz, CA); E-cadherin, phospho-ERK1/2, N-cadherin, Vav2 and ERK were from Cell Signaling Technology (Danvers, MA); Rac1 was from BD Biosciences; actin was from Sigma-Aldrich. In some experiments, cells were treated with the MEK inhibitor U0126 (Cell Signaling Technology), the BRAF inhibitor PLX4720 (Selleck Chemicals) or the Rac inhibitor NSC (Calbiochem).
siRNA Transfections
The following siRNAs were used B-RAF, Tiam1, Vav and Rac (Dharmacon) as well as B-RAFV600E described in Calipel et al 2003. Melanoma cells were transfected for 4h with 25nM siRNA using OligofectAMINE (Invitrogen) according to manufacturer's instructions. After transfection cells were cultured in complete medium for an additional 72h in complete medium, and then they were processed for further analysis.
Immunofluorescence
WM793 cells were fixed with 3.7% formaldehyde in phosphate-buffered saline and permeabilized in 0.5% Triton X-100. E-cadherin was visualized with anti-E-cadherin antibody (Cell Signaling) followed by an anti-mouse Alexa 594-conjugated secondary antibody. Images were recorded with a Zeiss LSM510 confocal microscope.
Rac1 Activity Assays
Cells were treated as indicated in figure legends. After treatments cells were kept on ice, washed with ice-cold PBS and assayed for Rac activation with glutathione S-transferase-p21-activating-kinase, as described by Sandler et al. The beads were washed four times with lysis buffer and after the final wash beads were resuspended in sample buffer. Samples were then analyzed by SDS-PAGE.
Melanocyte Transfections
NHEM were transfected with PMCEF BRAF V600E (kindly provided by Richard Marais, Institute of Cancer Research, London). Transfections were carried out using X-tremeGENE HP (Roche) according to manufacturer's instructions. 24 hours after transfection cells were harvested.
Nucleotide free Rac1 Pulldown Assays
Affinity precipitation of exchange factors with the nucleotide-free Rac1 mutant (G15A) has been described in detail in previous work from our laboratory (Garcia-Mata et al., 2006). Briefly, cells were lysed in 20 mM HEPES (pH 7.6), 150 mM NaCl, 1% Triton X-100, 5 mM MgCl2, 200 μM orthovanadate plus protease inhibitors. Equalized and clarified lysates were incubated with 20 μg of purified Rac1(15A) bound to glutathione-sepharose beads for 60 minutes at 4°C. Samples were then washed in lysis buffer and processed for SDS-PAGE.
Subcellular Fractionation
Cells were washed and incubated with ice-cold hypotonic lysis buffer (10mM Hepes pH 7.3, 1.5mM MgCl2 , 5mM KCl, 1mM DTT and protease inhibitors) for 10 minutes. Cells were scraped and homogenized with 20 strokes of Dounce homogeneizer. Homogenates were centrifuged at 700g for 3 minutes to pellet nuclei and intact cells. The supernatants were then centrifuged at 40,000g for 30 minutes at 4°C. The pellets (membrane fraction) were then resuspended in Sample Buffer.
Invasion Assays
Polycarbonate filters (8um pore size; Corning, Lowell, MA) of traswell cell culture chambers were coated with 50ul of growth-factor reduced Matrigel (BD Biosciences). Knockdown melanoma cells were added to the upper chamber in serum-free medium. The lower chamber was filled with normal melanoma growth medium, thereby establishing a soluble gradient of chemoattractants that promoted cellular invasion. The cells were allowed to invade for 24h at 37C, at which time the Matrigel and cells associated with the upper chamber were removed with cotton swabs. The undersides of the filters were then trypsinized to collect cells that had invaded through the Matrigel and cells were counted with a hemocytometer.
Immunoblotting
Treatment doses and times are provided in the figure legends. Cells were rinsed with PBS and lysed in sample buffer. Lysates were electrophoresed into SDS-PAGE gels. Proteins were transferred to nitrocellulose (Schleicher and Schuell Bioscience) and processed for Western blot analysis using the antibodies described in the figure legends. Bound antibodies were detected by enhanced chemiluminescence.
TER
Co-cultures of WM793 cells and MRC5 cells or WM793 cells and HaCat cells were plated at confluence onto Transwell filters (0.4 uM pore, 12mm diameter, Corning) and cultured for 3 days with medium replacement daily. TER was measured using an Endohm-12 voltohmeter (World Precision) according to manufacturer's instructions.
Real Time Cell Analysis (RTCA) of Proliferation
The xCELLigence Real-Time Cell Analyzer (RTCA) system (Acea Biosciences/Roche Applied Science 05-469-759-001) was used to measure electrical impedance, as a readout of proliferation. This method uses electrical impedance signals to monitor the status of cells grown directly on micro-electrode coated surfaces. Changes in impedance reflect changes cell number. WM793 cells Transfected with the indicated siRNA were counted with a hemocytometer, then plated (in quadruplicate) directly onto a microelectrode-surface within the wells of an E-Plate 16 (Roche Applied Science, 05-469-813-001). Impedance readings were taken automatically every hour for another 72 hrs and plotted as Cell Index ± SEM.
Acknowledgments
This work was supported by NIH grants GM029860 and HL080166 to KB. E M-B is supported by American Cancer Society PF-09_119-01-CSM.
Footnotes
Conflict of Interest: The authors declare no conflict of interest
Reference List
- 1.Sauter ER, Herlyn M. Molecular biology of human melanoma development and progression. Mol Carcinog. 1998 Nov;23(3):132–43. doi: 10.1002/(sici)1098-2744(199811)23:3<132::aid-mc2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 2.Chin L. The genetics of malignant melanoma: lessons from mouse and man. Nat Rev Cancer. 2003 Aug;3(8):559–70. doi: 10.1038/nrc1145. [DOI] [PubMed] [Google Scholar]
- 3.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002 Jun 27;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003 May;34(1):108–12. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 5.Dong J, Phelps RG, Qiao R, Yao S, Benard O, Ronai Z, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003 Jul 15;63(14):3883–5. [PubMed] [Google Scholar]
- 6.Sumimoto H, Miyagishi M, Miyoshi H, Yamagata S, Shimizu A, Taira K, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004 Aug 12;23(36):6031–9. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 7.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008 Feb 26;105(8):3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vleminckx K, Kemler R. Cadherins and tissue formation: integrating adhesion and signaling. Bioessays. 1999 Mar;21(3):211–20. doi: 10.1002/(SICI)1521-1878(199903)21:3<211::AID-BIES5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 9.Hsu MY, Wheelock MJ, Johnson KR, Herlyn M. Shifts in cadherin profiles between human normal melanocytes and melanomas. J Investig Dermatol Symp Proc. 1996 Apr;1(2):188–94. [PubMed] [Google Scholar]
- 10.Silye R, Karayiannakis AJ, Syrigos KN, Poole S, van NS, Batchelor W, et al. E-cadherin/catenin complex in benign and malignant melanocytic lesions. J Pathol. 1998 Dec;186(4):350–5. doi: 10.1002/(SICI)1096-9896(199812)186:4<350::AID-PATH181>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 11.Tang A, Eller MS, Hara M, Yaar M, Hirohashi S, Gilchrest BA. E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J Cell Sci. 1994 Apr;107(Pt 4):983–92. doi: 10.1242/jcs.107.4.983. [DOI] [PubMed] [Google Scholar]
- 12.Hsu M, Andl T, Li G, Meinkoth JL, Herlyn M. Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J Cell Sci. 2000 May;113(Pt 9):1535–42. doi: 10.1242/jcs.113.9.1535. [DOI] [PubMed] [Google Scholar]
- 13.Sandig M, Voura EB, Kalnins VI, Siu CH. Role of cadherins in the transendothelial migration of melanoma cells in culture. Cell Motil Cytoskeleton. 1997;38(4):351–64. doi: 10.1002/(SICI)1097-0169(1997)38:4<351::AID-CM5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 14.Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001 May 1;61(9):3819–25. [PubMed] [Google Scholar]
- 15.Braga VM, Machesky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol. 1997 Jun 16;137(6):1421–31. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell-cell adhesion by rac and rho small G proteins in MDCK cells. J Cell Biol. 1997 Nov 17;139(4):1047–59. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hordijk PL, ten Klooster JP, van der Kammen RA, Michiels F, Oomen LC, Collard JG. Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science. 1997 Nov 21;278(5342):1464–6. doi: 10.1126/science.278.5342.1464. [DOI] [PubMed] [Google Scholar]
- 18.Habets GG, Scholtes EH, Zuydgeest D, van der Kammen RA, Stam JC, Berns A, et al. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994 May 20;77(4):537–49. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 19.Michiels F, Collard JG. Rho-like GTPases: their role in cell adhesion and invasion. Biochem Soc Symp. 1999;65:125–46. [PubMed] [Google Scholar]
- 20.Engers R, Springer E, Michiels F, Collard JG, Gabbert HE. Rac affects invasion of human renal cell carcinomas by up-regulating tissue inhibitor of metalloproteinases (TIMP)-1 and TIMP-2 expression. J Biol Chem. 2001 Nov 9;276(45):41889–97. doi: 10.1074/jbc.M105049200. [DOI] [PubMed] [Google Scholar]
- 21.Calipel A, Lefevre G, Pouponnot C, Mouriaux F, Eychene A, Mascarelli F. Mutation of B-Raf in human choroidal melanoma cells mediates cell proliferation and transformation through the MEK/ERK pathway. J Biol Chem. 2003 Oct 24;278(43):42409–18. doi: 10.1074/jbc.M308709200. [DOI] [PubMed] [Google Scholar]
- 22.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010 Sep 30;467(7315):596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004 May 18;101(20):7618–23. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akbar H, Cancelas J, Williams DA, Zheng J, Zheng Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. Methods Enzymol. 2006;406:554–65. doi: 10.1016/S0076-6879(06)06043-5. [DOI] [PubMed] [Google Scholar]
- 25.Albino AP, Nanus DM, Mentle IR, Cordon-Cardo C, McNutt NS, Bressler J, et al. Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene. 1989 Nov;4(11):1363–74. [PubMed] [Google Scholar]
- 26.Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–37. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- 27.Stam JC, Sander EE, Michiels F, van Leeuwen FN, Kain HE, van der Kammen RA, et al. Targeting of Tiam1 to the plasma membrane requires the cooperative function of the N-terminal pleckstrin homology domain and an adjacent protein interaction domain. J Biol Chem. 1997 Nov 7;272(45):28447–54. doi: 10.1074/jbc.272.45.28447. [DOI] [PubMed] [Google Scholar]
- 28.Zhang JD, Koerner C, Bechtel S, Bender C, Keklikoglou I, Schmidt C, et al. Time-resolved human kinome RNAi screen identifies a network regulating mitotic-events as early regulators of cell proliferation. PLoS One. 2011;6(7):e22176. doi: 10.1371/journal.pone.0022176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G, Satyamoorthy K, Herlyn M. Dynamics of cell interactions and communications during melanoma development. Crit Rev Oral Biol Med. 2002;13(1):62–70. doi: 10.1177/154411130201300107. [DOI] [PubMed] [Google Scholar]
- 30.Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998 Aug;153(2):333–9. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poser I, Dominguez D, de Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J Biol Chem. 2001 Jul 6;276(27):24661–6. doi: 10.1074/jbc.M011224200. [DOI] [PubMed] [Google Scholar]
- 32.Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T, et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med. 2009 Jan 16;206(1):221–32. doi: 10.1084/jem.20082044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arozarena I, Sanchez-Laorden B, Packer L, Hidalgo-Carcedo C, Hayward R, Viros A, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011 Jan 18;19(1):45–57. doi: 10.1016/j.ccr.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Goodall J, Carreira S, Denat L, Kobi D, Davidson I, Nuciforo P, et al. Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res. 2008 Oct 1;68(19):7788–94. doi: 10.1158/0008-5472.CAN-08-1053. [DOI] [PubMed] [Google Scholar]
- 35.Kuroda S, Fukata M, Fujii K, Nakamura T, Izawa I, Kaibuchi K. Regulation of cell-cell adhesion of MDCK cells by Cdc42 and Rac1 small GTPases. Biochem Biophys Res Commun. 1997 Nov 17;240(2):430–5. doi: 10.1006/bbrc.1997.7675. [DOI] [PubMed] [Google Scholar]
- 36.Zhong C, Kinch MS, Burridge K. Rho-stimulated contractility contributes to the fibroblastic phenotype of Ras-transformed epithelial cells. Mol Biol Cell. 1997 Nov;8(11):2329–44. doi: 10.1091/mbc.8.11.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jou TS, Schneeberger EE, Nelson WJ. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol. 1998 Jul 13;142(1):101–15. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stam JC, Michiels F, van der Kammen RA, Moolenaar WH, Collard JG. Invasion of T-lymphoma cells: cooperation between Rho family GTPases and lysophospholipid receptor signaling. EMBO J. 1998 Jul 15;17(14):4066–74. doi: 10.1093/emboj/17.14.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003 May 1;104(5):527–32. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 40.Alsina J, Gorsk DH, Germino FJ, Shih W, Lu SE, Zhang ZG, et al. Detection of mutations in the mitogen-activated protein kinase pathway in human melanoma. Clin Cancer Res. 2003 Dec 15;9(17):6419–25. [PubMed] [Google Scholar]
- 41.Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006 Oct 1;66(19):9483–91. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- 42.Ray RM, Vaidya RJ, Johnson LR. MEK/ERK regulates adherens junctions and migration through Rac1. Cell Motil Cytoskeleton. 2007 Mar;64(3):143–56. doi: 10.1002/cm.20172. [DOI] [PubMed] [Google Scholar]
- 43.Zondag GC, Evers EE, ten Klooster JP, Janssen L, van der Kammen RA, Collard JG. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000 May 15;149(4):775–82. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sander EE, van DS, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, et al. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998 Nov 30;143(5):1385–98. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uhlenbrock K, Eberth A, Herbrand U, Daryab N, Stege P, Meier F, et al. The RacGEF Tiam1 inhibits migration and invasion of metastatic melanoma via a novel adhesive mechanism. J Cell Sci. 2004 Sep 15;117(Pt 20):4863–71. doi: 10.1242/jcs.01367. [DOI] [PubMed] [Google Scholar]
- 46.Klein RM, Spofford LS, Abel EV, Ortiz A, Aplin AE. B-RAF regulation of Rnd3 participates in actin cytoskeletal and focal adhesion organization. Mol Biol Cell. 2008 Feb;19(2):498–508. doi: 10.1091/mbc.E07-09-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein RM, Aplin AE. Rnd3 regulation of the actin cytoskeleton promotes melanoma migration and invasive outgrowth in three dimensions. Cancer Res. 2009 Mar 15;69(6):2224–33. doi: 10.1158/0008-5472.CAN-08-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]