

Abstract
Frataxin is a conserved mitochondrial protein, and deficiency underlies the neurodegenerative disease Friedreich’s ataxia. Frataxin interacts with the core machinery for Fe–S cluster assembly in mitochondria. Recently we reported that in frataxin-deleted yeast strains, a spontaneously occurring mutation in one of two genes encoding redundant Isu scaffold proteins, bypassed the mutant phenotypes. In the present study we created strains expressing a single scaffold protein, either Isu1 or the bypass mutant M107I Isu1. Our results show that in the frataxin-deletion strain expressing the bypass mutant Isu1, cell growth, Fe–S cluster protein activities, haem proteins and iron homoeostasis were restored to normal or close to normal. The bypass effects were not mediated by changes in Isu1 expression level. The persulfide-forming activity of the cysteine desulfurase was diminished in the frataxin deletion (Δyfh1 ISU1) and was improved by expression of the bypass Isu1 (Δyfh1 M107I ISU1). The addition of purified bypass M107I Isu1 protein to a Δyfh1 lysate conferred similar enhancement of cysteine desulfurase as did frataxin, suggesting that this effect contributed to the bypass mechanism. Fe–S cluster-forming activity in isolated mitochondria was stimulated by the bypass Isu1, albeit at a lower rate. The rescuing effects of the bypass Isu1 point to ways that the core defects in Friedreich’s ataxia mitochondria can be restored.
Keywords: cysteine desulfurase, eukaryote, frataxin, iron–sulfur, Isu1 scaffold, mitochondrion
INTRODUCTION
Frataxin is a conserved mitochondrial protein implicated in causation of Friedreich’s ataxia, an inherited neurodegenerative disease [1]. It is the deficiency of frataxin in certain tissues, such as the dorsal root ganglia and heart, that is responsible for the disease. The cellular disease phenotype is characterized by deficiencies of Fe–S cluster proteins and haem proteins [2]. Iron homoeostatic abnormalities also ensue, including accumulation of mitochondrial iron in the biologically unavailable form of ferric phosphate particles [3]. The frataxin protein is highly conserved, and the human protein complements the yeast deletion mutant, indicating an orthologous relationship [4]. In isolates of slow-growing yeast lacking the frataxin homologue (Yfh1), an outgrowth of clones with frataxin-bypassing activity was observed. A single point mutation in Isu1, the Fe–S scaffold protein, was identified as the mediator of the frataxin-bypassing effect [5].
This observation is interesting because of the implications for frataxin function, which is still poorly defined [6]. Fe–S clusters, essential cofactors for numerous proteins, are synthesized by a multisubunit machinery in mitochondria. Frataxin has been found to be a constituent of an Fe–S cluster assembly complex that also includes Nfs1 (NiFS-like)–Isd11 (iron–sulfur desulfurase-interacting protein) and Isu [7–9]. This assembly complex is needed to form Fe–S cluster intermediates on the Isu scaffold, which are subsequently transferred to target apoproteins by the mediation of chaperones, glutaredoxins and other components. The Nfs1–Isd11 complex provides the essential cysteine desulfurase activity for sulfur donation, binding the amino acid cysteine, generating a persulfide on Nfs1 and transferring the sulfur for synthesis of Fe–S cluster intermediates on the Isu scaffold [10]. The source of iron for the intermediate is unknown, and frataxin may play a role in the iron donation step [11]. Another function proposed for frataxin is as an allosteric activator of the cysteine desulfurase activity of the complex [12]. How exactly the mutant Isu bypasses frataxin is not known, but most probably it is able to fulfil some of frataxin’s functions in Fe–S cluster assembly in the absence of frataxin.
A curious feature of the frataxin-bypassing mutation of Isu1 is that it involves substitution of an amino acid present in all eukaryotic scaffold proteins with an amino acid present in many prokaryotic proteins [5]. The methionine residue at position 107 of Isu1 is present in all eukaryotes, including metazoans, flies, worms, plants and even primitive protists. In contrast, the isoleucine residue is found in prokaryotic scaffolds such as Escherichia coli, Shigella and Salmonella. Isu sequences and functions are highly conserved across evolution, and so this change is likely to be significant. The metamorphic structure extrapolated from the bacterial proteins consists of α-helices superimposed over a β-sheet platform, and the methionine to isoleucine mutation occurs on an exposed surface of a large C-terminal α-helix predicted to be close to interaction sites for Nfs1 [13], frataxin and the Hsp70 (heat-shock protein 70) chaperone Ssq1 (stress-seventy subfamily Q) [14]. In prokaryotes such as E. coli, the lack of frataxin induces a mild phenotype without altering iron homoeostasis or sensitivity to oxidants [15], as contrasted with eukaryotes where frataxin loss is severely deleterious or lethal [4]. Thus the methionine to isoleucine mutation, which improves mitochondrial function in the absence of frataxin, can be envisaged as creating a more prokaryotic-like Fe–S cluster machinery. The mechanistic details of how this occurs are not known.
The bypass mutation in Isu1 was identified in a strain of yeast deleted for YFH1. In yeast there are two functionally redundant genes encoding Isu1 and Isu2 [16]. Thus the bypass activity, conferred by the Isu1 substitution in the presence of an unaltered Isu2 implies some degree of genetic dominance. The M107I Isu1 protein might work as an alternative scaffold or might work in trans via the normal scaffold, and experiments up to now have not distinguished between these possibilities. In the present study we constructed strains in which a single Isu, wild-type or mutant, is expressed, either in the setting of normal frataxin (YFH1) or frataxin deletion (Δyfh1). Assays of these cells and mitochondria isolated from these cells have been performed to more fully characterize the features and mechanisms of frataxin bypass.
MATERIALS AND METHODS
Yeast strains, plasmids and growth conditions
Strains were constructed in the YPH499 background of Saccharomyces cerevisiae (Table 1). To construct the single copy Isu strains, the ISU2 gene from YPH499 was interrupted by insertion of pRS406-containing γ-oriented fragments of the gene. A GAL1 promoter swap was introduced upstream of ISU1, creating the strain 106-61, which was dependent on galactose for growth and Isu1 expression [17]. Plasmids were introduced containing wild-type Isu1 or the bypass Isu1 carrying a single amino acid substitution (M141I in the precursor protein or M107I in the mature protein, referred to as M>I). The CEN (centromere)-based YCplac22 plasmids contained 700 bp 5′ of the ORF and 200 bp 3′ of the ORF in a polylinker (EagI-Isu1 promoter-Nde1-Isu1 ORF-XhoI-stop-3′non-coding-Sac1). The resulting strains, 115-18 or 115-21 respectively, were viable and expressed only the plasmid-borne Isu1 or M>I when grown in media with raffinose or glucose as a carbon source. YFH1 was knocked out in these strains by transforming pRS405 with γ-oriented gene fragments and selecting for leucine prototrophy in an argon-flushed container. Three different colony sizes were noted: tiny colonies that were leucine auxotrophs on restreaking, intermediate sized colonies that were confirmed by PCR to be deleted for YFH1 and large colonies which resulted from incorrect integration. The confirmed Δyfh1 knockout strains exhibited moderately slow growth that was mitigated by the microaerophilic conditions in the argon-flushed chamber. The genotypes were confirmed by PCR amplification of Δyfh1 or YFH1 flanking regions. The resulting strains carried Δyfh1 and YCplac22-Isu1 (strain 115-26) or Δyfh1 and YCplac22-M107I (strain 115-28). For simplicity the four strains are referred to by a simple name or short genotype (Table 1). Strain 115-18 is called ‘WT’ or YFH1 ISU1. Strain 115-21 is called YFH1 M>I. Strain 115-26 is called ‘deletion’ or Δyfh1 ISU1. Strain 115-28 is called ‘bypass’ or Δyfh1 M>I. The ‘bypass’ terminology is meant to refer to the ability of the M107I mutant form of Isu1 to bypass the effects of frataxin deficiency (Table 1).
Table 1.
List of yeast strains
| Strain number | Simple name | Short genotype | Genotype |
|---|---|---|---|
| 106-61 | Parent | – | MATa lys2-801(amber) ade2-101(ochre) trp1-Δ63 leu2-Δ1 pRS406-gamma isu2 (URA3) His3MX6-PGAL1-ISU1 |
| 115-18 | wt | YFH1 ISU1 | Parent with [YCplac22-Isu1] [pRS415] |
| 115-21 | – | YFH1 M>I | Parent with [YCplac22-M107I] [pRS415] |
| 115-26 | deletion | Δyfh1 ISU1 | Parent with [YCplac22-ISU1] [pRS405-Yfh1 gamma] |
| 115-28 | bypass | Δyfh1 M>I | Parent with [YCplac22-ISU1 M107I] [pRS405-Yfh1 gamma] |
| 116-14 | – | YFH1 tet-ISU1 | Parent with [pCM184-Isu1-His6 ][YEp351] |
| 116-15 | – | YFH1 tet-M>I | Parent with [pCM184-M107I-His6 ][YEp351] |
| 116-9 | – | Δyfh1 tet-ISU1 | Parent with [pCM184-Isu1-His6 ] Δyfh1::LEU2 |
| 116-11 | – | Δyfh1 tet-M>I | Parent with [pCM184-M107I-His6 ] Δyfh1::LEU2 |
| 116-16 | – | P100A | Parent with [pCM184-Isu1-P100A-His6 ] |
A second set of strains was constructed in which the tet07 promoter [18] replaced the native ISU1 promoter. Isu1-His6 or Isu1-M107I-His6 was subcloned into the pCM184 vector. The plasmids were inserted into the Gal-Isu1 Δisu2 strain 106-61, and YFH1 was knocked out as above. The set of strains 116-14, 116-15, 116-9 and 116-11 are referred to by their abbreviated genotypes YFH1 tet-ISU1, YFH1 tet-M>I, Δyfh1 tet-ISU1 and Δyfh1 tet-M>I respectively (Table 1). The plasmid pCM184-Isu1-His6 was modified by site-directed mutagenesis, changing amino acid 100 of the mature Isu1 from proline to alanine. The P100A mutant was inserted into the parent generating strain 116-16 (Table 1), and this strain was used as a control for detecting Isu1 stabilization as described previously [19]. For most experiments, cells were cultured in complete defined medium with raffinose as the carbon source. Air exposure was minimized by maintaining the cells in argon-flushed flasks. The cultures were brought out in air for the duration of the experiments, usually ranging between 15 and 48 h. Several independent clones of the Δyfh1 strains were tested to ensure that new genetic changes were not altering the phenotypes.
Enzyme assays and iron labelling
Mitochondrial lysates were assayed for activities of Fe–S cluster enzymes, succinate dehydrogenase and aconitase as described previously [20]. Iron labelling was performed by growing cells to steady state (four doublings) in the presence of radioactive 55Fe in a standard defined medium (100 nM 55FeCl3, 10 μM ferrous sulfate and 100 μM ascorbic acid). The cells were washed free of unincorporated iron and separated into cytoplasmic and mitochondrial fractions. The mitochondria were processed further by sonication in the presence of 0.1 % Triton X-100 in 50 mM Hepes/KOH and 150 mM NaCl. The supernatant (soluble) and pellet (insoluble) portions were separated by centrifugation (10 000 g for 15 min at 4 °C). All the fractions were analysed by scintillation counting and protein quantitation with BCA.
Purified proteins
pST39-Nfs1-His6/Isd11 was expressed and purified as described previously [21]. The ΔN1-51 mature form of Yfh1 was expressed and purified. The ΔN1-34 truncation of Isu1 was cloned into pET21b with a His6 tag, expressed in BL21(DE3) codon plus cells, and purified on Ni-NTA (Ni2+-nitrilotriacetate)–agarose. Additional washing steps in the presence of 0.5 M NaCl and 6 M urea while the protein was bound to the beads allowed a high level of purity to be obtained (greater than 95 %). The M107I substitution was introduced by QuikChange mutagenesis of the plasmid pET21b-Isu1-His6, and the mutant protein was similarly expressed and purified. Isd11 was expressed and purified as described previously [22]. Mitochondrial proteins were separated by SDS/PAGE (12 % gel), and immunoblotting was performed with specific rabbit antisera against various mitochondrial proteins. The secondary antibody was a goat anti-(rabbit IgG)–Alexa Fluor™ 680 (Life Technologies). Following scanning with the LiCor Odyssey infrared scanner, signal intensities were recorded.
Organelle assays
Persulfide formation on Nfs1 present in intact mitochondria was measured as described in [22]. Isolated and intact mitochondria were depleted for endogenous nucleotides by incubation for 10 min at 30 °C in buffer (20 mM Hepes/KOH, pH 7.5, and 0.6 M sorbitol). Labelling was carried out by the addition of 1 μl (10 μCi) of [35S]cysteine for 20 min at 30 °C. Mitochondria were recovered, proteins were separated by SDS/PAGE (12 % gel) and the persulfide was viewed by radioautography. Fe–S cluster formation was measured as described in [21]. Briefly, isolated mitochondria were labelled in the presence of added nucleotides (4 mM ATP, 1 mM GTP and 5 mM NADH) and iron (10 μM ferrous ascorbate). Mitochondria were recovered, and soluble proteins were released by freeze–thaw sonication and separated on a native gel. The signal from newly formed [Fe–35S]aconitase was visualized by radioautography. Quantification of radiolabelled protein bands was performed by densitometric analysis using NIH ImageJ software.
Statistics
All data were analysed using Student’s t test for unpaired data in a two-tailed analysis (except for the data in Figure 6C, which used a one-tailed analysis). P < 0.05 was considered statistically significant.
Figure 6. Overexpression of Isu1 and related phenotypes.

(A) Plate phenotypes. Strains with the indicated genotypes (native Isu1 promoter or tet07 promoter) were serially diluted and spotted on to YPAD agar plates. After 3 days of growth at 30 °C the plates were photographed. (B) Mitochondria were isolated from deletion (Δyfh1 ISU1) and wt (YFH1 ISU1) strains, and from the tet promoter set including Δyfh1 tet-ISU1, Δyfh1 tet-M>I, YFH1 ISU1 and YFH1 tet-M>I. Proteins were evaluated by immunoblotting using various antibodies. The images were developed using the LiCor system and infrared detector and the intensity signal was determined (shown in right-hand panel). Three independent mitochondrial preparations were analysed, and a representative immunoblot is shown. ∞, pixel saturation. Isu1 values are reported as ng/100 μg of mitochondrial protein. (C) Enzyme activities for succinate dehydrogenase (left-hand panel) and aconitase (right-hand panel) were measured in mitochondria. Three independent mitochondrial preparations were assayed. Results are means ± S.D. The mean succinate dehydrogenase activity was higher in the tet07 strain Δyfh1 tet-ISU1 compared with the native promoter strain Δyfh1 ISU1 although the difference did not reach statistical significance (P = 0.072). The mean aconitase activity was significantly higher in the tet07 strain compared with the native promoter strain (P = 0.018) as determined using a one-tailed Student’s t test.
RESULTS
Single-copy Isu strains
S. cerevisiae has two redundant genes encoding the Fe–S scaffold proteins, ISU1 and ISU2, and a single gene encoding the yeast frataxin homologue YFH1. In several independent isolates of Δyfh1 yeast, an altered ISU1 was found to confer improved growth and recovery of many of the Δyfh1-related phenotypes. The bypass effect was traced to a point mutation in ISU1, changing amino acid 107 in the mature protein from methionine to isoleucine (M>I) [5]. We considered different scenarios for explaining the function of the bypass Isu1 protein. The bypass Isu1 mutant might work by interacting with the wild-type and unmutated Isu, encoded by ISU2, activating it during the course of Fe–S cluster intermediate formation or transfer. Alternatively, the bypass Isu1 might function as a scaffold on its own, being used in preference to the other unmutated Isu2. To distinguish between these possibilities we constructed congenic strains expressing a single Isu. The frataxin deletion strain (Δyfh1 ISU1) grew more slowly than the others under all conditions (Figure 1). The slow growth was exacerbated by use of non-fermentable substrates that require respiratory activity for their metabolism (Figure 1, right-hand panel). Individual clones of the frataxin deletion (Δyfh1 ISU1) appeared heterogeneous with larger and smaller colonies, posing a problem for phenotypic characterization. However, if the strains were maintained in an argon-flushed jar under low oxygen tension, growth was uniform, albeit slower than the wild-type, and new mutations did not appear. Therefore for construction and maintenance of these yeast strains, they were kept in the argon-flushed jar as much as possible, taking them out only for use in experiments. In terms of growth characteristics in standard medium, the bypass strain (Δyfh1 M>I) was indistinguishable from the wt (YFH1 ISU1) strains. The strain with the mutated Isu1 in the YFH1 context (YFH1 M>I) also grew normally (Figure 1).
Figure 1. Growth of single-copy Isu strains.

The four single-copy Isu strains (Δyfh1 ISU1, Δyfh1 M>I, YFH1 ISU1 and YFH1 M>I) were grown overnight in rich medium (YPAD), and 106 cells were subjected to serial dilutions and spotted on to agar plates containing 2 % glucose or 3 % ethanol as a carbon source. The plates were incubated at 30 °C for 3 days and photographed.
Enzyme activities
Activity of succinate dehydrogenase, a mitochondrial enzyme complex (complex II of the respiratory chain with haem, Fe2S2, Fe3S4 and Fe4S4 cofactors) [23] was measured. In the frataxin deletion (Δyfh1 ISU1), succinate dehydrogenase activity was approximately 24 %, significantly lower (P = 0.001) than in the wt (YFH1 ISU1) (Figure 2A). In the bypass mutant (Δyfh1 M>I), activity was restored and was no longer different from the wt (Figure 2A). For aconitase, an abundant Fe4S4 cluster protein of the mitochondrial tricarboxylic acid cycle, the activity was very low or undetectable in the frataxin-deletion strain (Δyfh1 ISU1), which was significantly lower (P = 0.012) than in the wt (YFH1 ISU1). By contrast, in the bypass mutant (Δyfh1 M>I), the activity was almost completely restored and was no longer different from the wt (Figure 2B). The activity gel assay for aconitase corroborated these findings, showing absent aconitase in the deletion and restoration in the bypass (Figure 2C).
Figure 2. Enzyme activities.

Mitochondria were prepared from three independent biological replicates from the four single-copy Isu strains. (A) Succinate dehydrogenase activity (means ± S.D.). Δyfh1 ISU1 was significantly different (P = 0.001) from YFH1 ISU1. YFH1 M>I was not significantly different (P = 0.422) from YFH1 ISU1. (B) Aconitase activity (means ± S.D.). Δyfh1 ISU1 was significantly different (P = 0.012) from YFH1 ISU1. YFH1 M>I was not significantly different (P = 0.368) from YFH1 ISU1. (C) The in-gel activity assay for aconitase activity on these mitochondria [39,40].
Isu1 and other protein levels in mitochondria
Aconitase protein was decreased in mitochondria from the frataxin-deletion strain (Δyfh1 ISU1), but recovered in the bypass (Δyfh1 M>I) (Figure 3). Equivalent levels were present in the frataxin-plus strains (YFH1 ISU1 and YFH1 M>I) (Figure 3). As the frataxin-deletion (Δyfh1 ISU1) strain had no detectable aconitase activity, the results are consistent with partial destabilization and increased turnover of the apoprotein. The levels of the individual components of the Fe–S cluster assembly complex consisting of Nfs1, Isd11, Isu1 and frataxin [7–9] were measured. Nfs1 levels were equivalent in all cases, showing no signs of regulatory variation. Isd11 also was unchanged. Yfh1 protein expression was consistent with the genotype, being absent in the deleted strains and present in the YFH1 strains as expected. Only Isu levels showed regulatory changes as has been reported previously [19]. Isu1 abundance was increased in the Δyfh1 ISU1 strain by 3–4-fold, but returned to normal in the bypass Δyfh1 M>I (Figure 3).
Figure 3. Protein levels in mitochondria.

Mitochondria isolated from single-copy Isu strains were analysed by SDS/PAGE. After immunoblotting with specific rabbit antibodies for aconitase (Aco1), Nfs1, yeast frataxin homologue (Yfh1), Isu1 and Isd11, the images were developed using the LiCor system and infrared detector and the intensity signal was determined (shown in lower panel). Three independent preparations of mitochondria were evaluated, and a representative immunoblot is shown. nd, not detected.
The amounts of several key proteins in mitochondria were measured by quantitative immunoblotting of wt (YFH1 ISU1) mitochondria. The quantities of Nfs1 and Isd11 present in mitochondria were similar (0.9 and 0.8 pmol per 100 μg of mitochondrial proteins respectively), consistent with the idea that they are tightly associated with each other [24] and form a cysteine desulfurase enzyme complex. Nfs1 and Isd11 in turn are associated with Isu and Yfh1 as part of an Fe–S cluster assembly complex. Isu1 was present in similar amounts as Nfs1–Isd11 (0.8 pmol per 100 μg of mitochondrial proteins) (Supplementary Figure S1 at http://www.biochemj.org/bj/459/bj4590071add.htm). Interestingly, Yfh1 was less abundant than the other proteins, being present at 0.5 pmol per 100 μg of mitochondrial proteins, significantly less than the amounts of Nfs1, Isd11 or Isu1 in wt strain mitochondria. The results suggest that Yfh1 may serve a stimulatory or catalytic role, coming on and off the Fe–S cluster assembly complexes as required. In the frataxin-deletion strain (Δyfh1 ISU1), Isu1 abundance was increased compared with the wt strain (Figure 3), suggesting that, in these mitochondria, it is present at molar excess over Nfs1 and Isd11. As such, the excess Isu might perform functions independently of the Fe–S cluster assembly complex. For example surplus Isu might store iron or Fe–S cluster intermediates in mitochondria.
Recovery of haem proteins, iron homoeostasis and iron solubility
Ccp1 (cytochrome c peroxidase) and Cyc1 (cytochrome c) are haem proteins. These proteins reside in the intermembrane space and co-ordinate b and c type haems respectively. In the single-copy Isu strains, Ccp1 protein was decreased to approximately 50 % in the absence of Yfh1 (Δyfh1 ISU1), but completely recovered in the bypass strain (Δyfh1 M>I) (Figure 4). Cyc1, which strongly depends on haem for both its transcription and protein stability [25], was not detected at all in the frataxin-deletion strain (Δyfh1 ISU1), but was nearly normal in the bypass strain (Δyfh1 M>I) (Figure 4). The results suggest that haem was deficient in the frataxin-deletion yeast as reported in [26], whereas it was recovered in the bypass, similar to frataxin-plus yeast.
Figure 4. Haem protein levels.

Mitochondria isolated from single-copy Isu strains were analysed by SDS/PAGE. The proteins were transferred on to nitrocellulose membranes and probed with rabbit polyclonal antibodies against Ccp1 and Cyc1. The blots were also probed for Yfh1 and Nfs1 to confirm the genotype and to serve as controls The images were developed using the LiCor system and infrared detector and the intensity signal was determined (shown in lower panel). Three independent preparations of mitochondria were evaluated and a representative immunoblot is shown. nd, not detected.
Iron homoeostasis was evaluated by growing cells to steady state in the presence of 55Fe radionuclide. Cells were fractionated into cytoplasm and mitochondria, and iron in the fractions was measured by scintillation counting. The expected iron homoeostatic phenotype [26,27] was observed for the frataxin-deletion strain (Δyfh1 ISU1), as reflected by increased total cellular iron (P = 0.010) (Figure 5A) and mitochondrial iron (P = 0.013) (Figure 5C) compared with the wt strain. In the bypass strain (Δyfh1 M>I), the abnormal iron uptake and distribution were reverted, and these values were not significantly different from the wt strain (YFH1 ISU1) (Figures 5A–5C). A feature of the iron homoeostatic abnormality associated with lack of frataxin is the formation of ferric phosphate nanoparticles in mitochondria [26,28]. Thus the accumulated mitochondrial iron in the frataxin mutant strain (Δyfh1 ISU1) was predominantly insoluble, as determined by a biochemical test involving detergent solubilization and centrifugation of isolated mitochondria. Significantly more iron was present in the insoluble fraction in the frataxin mutant (P = 0.002) compared with the wt strain (Figure 5D). By contrast, the bypass mutant behaved like the wt strain; iron did not accumulate in mitochondria (Figure 5C) and iron in the insoluble fraction was not increased significantly compared with wt strain (Figure 5D). The M>I substitution in the YFH1 context did not lead to any discernible changes in cellular iron content, distribution or solubility (Figure 5).
Figure 5. Iron homoeostasis.

The single-copy Isu strains were grown in the presence of 55Fe radionuclide tracer in defined medium supplemented with 10 μM ferrous ascorbate. Three independent labelling and fractionation experiments were performed. Results are means ± S.D. Whole cells, cytoplasmic and mitochondrial fractions were subjected to scintillation counting for 55Fe. (A) Iron per cell was determined by cell counting, and the amount in Δyfh1 ISU1 was significantly greater than the amount in YFH1 ISU1 (P = 0.010), whereas the amount in YFH1 M>I was not significantly different from the amount in YFH1 ISU1. (B) Cytoplasmic iron was determined, and the amount in Δyfh1 ISU1 was significantly greater than the amount in YFH1 ISU1 (P = 0.005), whereas the amount in YFH1 M>I was not significantly different from the amount in YFH1 ISU1. (C) Mitochondrial iron was determined and the amount in Δyfh1 was significantly greater than the amount in YFH1 ISU1 (P = 0.013), whereas the amount in YFH1 M>I was not significantly different from the amount in YFH1 ISU1. (D) For solubility determinations, mitochondria were sonicated in buffer containing 0.1 % Triton X-100. The supernatant and pellet fractions were separated by centrifugation and subjected to scintillation counting to ascertain the iron content of each fraction. The insoluble fraction was significantly greater in the Δyfh1 ISU1 than in the wt YFH1 ISU1 (P = 0.002), whereas the insoluble fraction in the YFH1 M>I was not significantly different from the amount in YFH1 ISU1.
Forced overexpression of Isu1
Isu abundance was increased in the frataxin-deletion strain (Figure 3) and in other settings with deficient Fe–S cluster synthesis [13]. We wondered if this increase in Isu could perform some function, perhaps acting to improve viability or Fe–S cluster synthesis. To test this notion, a set of single-copy Isu strains was constructed in which Isu1 was placed under control of the tet07 promoter. Driven by the tet07 promoter, expression of Isu1 was increased by more than 5-fold compared with the native promoter (Figure 6B, Isu1 in lanes 1–4 compared with lane 6). The phenotypes of the Isu-overexpressing strains were examined further. The frataxin-deleted strain (Δyfh1 tet-ISU1) was slower growing than the others. As before, the overexpressed bypass Isu1 strain (Δyfh1 tet-M>I) completely restored normal growth and was indistinguishable from the frataxin-plus strains (YFH1 tet-ISU1 and YFH1 tet-M>I) in terms of growth and colony size on YPAD agar plates (Figure 6A). There was no discernible effect of the strong overexpression of Isu1 in enhancing (or inhibiting) growth under standard conditions, compared with the strains with much lower Isu1 expression from the native promoter (Figure 6A). However, Cyc1 protein, which was undetectable in the frataxin-deletion strain (Δyfh1 ISU1), was increased to 47 % of the wt strain levels as a consequence of overexpression of Isu1 in the Δyfh1 tet-ISU1 strain (Figure 6B, lane 1 compared with lane 6). Succinate dehydrogenase was increased from 16 % to 31 % by overexpression of Isu1 in the Δyfh1 context (Figure 6C, left-hand panel), although this difference did not reach statistical significance (P = 0.072). Aconitase activity, which was barely detected, was significantly higher in the Isu1-overexpressing strain (P = 0.018), increasing to 14 % of the wt strain level (Figure 6C, right-hand panel). In conclusion, overexpression of Isu1 resulted in uneven phenotypic effects, with no enhancement of growth, but with significant improvement in some biochemical traits of Δyfh1 yeast. An effect of increased Isu level in enhancing fitness and responses to some forms of environmental stress has been described previously [19]. Conversely, decreases in Isu levels, as occured in Δisu1 strains, are synthetically lethal when combined with Δyfh1 [29]. By contrast, the M>I substituted Isu1 produced a more complete correction of the Δyfh1 mutant phenotypes, and its bypass activity was independent of the expression level (Figure 6).
Mechanism of Isu regulation
The Isu level was increased in the Δyfh1-deletion strain (Δyfh1 ISU1) and it returned to normal in the bypass strain (Δyfh1 M>I). To assess Isu1 protein stability, pulse–chase experiments were performed. The strains with overexpressed Isu1 under the tet07 promoter were grown to log-phase, pulsed with [35S]methionine and chased with unlabelled methionine. The Isu1 protein was recovered with a C-terminal His6 tag, and protein turnover was evaluated. The results showed that Isu1 was rapidly turned over, with the signal declining to less than 20 % of the starting signal within 30 min of chase (Supplementary Figure S2B at http://www.biochemj.org/bj/459/bj4590071add.htm). The rapid turnover was observed for Isu1 or M>I in the YFH1 strains, and similarly rapid turnover was observed in the Δyfh1 context (Supplementary Figure S2A). Numerous other labelled proteins were recovered on the Ni-NTA–agarose and visualized on the gel (Supplementary Figure S2), probably due to non-specific binding, and most of these proteins were stable during the time frame of the experiment. A single amino acid substitution was introduced into the plasmid pCM184-Isu1-His6 (P100A), and protein stability of this mutant protein was tested. As reported previously [19], the P100A Isu1 protein was completely stabilized, showing almost no decline in the labelled cohort after 180 min. Steady-state levels of Isu1 in the tet07 strains were almost equal (Figure 6B) except in the case of the stabilized P100A mutant for which the level was increased (results not shown). The ISU1 promoter has binding sites for Aft1/2 (activator of ferrous transport 1/2) [13,16], the transcriptional regulators for iron. Thus the increased Isu1 expression in the Δyfh1 setting could be due, at least in part, to perturbed iron homoeostasis and increased Aft1/2-dependent transcription. The ISU1 transcripts were directly evaluated by real-time PCR. The deletion had a ΔCT value of 4.2, which was significantly higher (P = 0.020) than the wt strain (ΔCT = 3.3), using TAF1 (TATA-binding protein-associated factor 1) as an endogenous standard. The difference corresponded to a 1.8-fold difference in the RNA levels. There was no difference between the ΔCT of the bypass strain and the wt strain (Supplementary Figure S3 at http://www.biochemj.org/bj/459/bj4590071add.htm). Thus changes in Isu1 protein level in the deletion (Δyfh1 ISU1) were attributable principally to mRNA level regulation and were corrected in the bypass mutant (Δyfh1 M>I).
Nfs1 persulfide formation
An initial step in formation of the Fe–S cluster assembly intermediate on the Isu scaffold is sulfur donation. This requires the activity of the Nfs1 cysteine desulfurase in mitochondria [30]. As part of its reaction cycle, Nfs1 binds the amino acid cysteine at its substrate-binding site. Then, in a step that depends on a conformational change induced by the accessory protein Isd11, the Nfs1 active-site cysteine forms a persulfide with the -SH group of the substrate cysteine [22]. The persulfide can be labelled with [35S]cysteine substrate, allowing detection by gel electrophoresis and providing a quantitative measure of a single round of cysteine desulfurase activity in mitochondria [21].
Mitochondria were isolated from the four single-copy Isu strains, and these were briefly (10 min) incubated at 30 °C to deplete endogenous nucleotides including ATP, GTP and NAD(P)H. The nucleotide-depleted intact mitochondria, although able to form persulfide on Nfs1, cannot transfer the persulfide because of a block in the nucleotide-dependent steps [21]. These intact mitochondria were labelled with [35S]cysteine, recovered and analysed by non-reducing SDS/PAGE. The Nfs1 persulfide signal in the frataxin-deletion mitochondria (Δyfh1 ISU1) was found to be diminished to 28 %of the wt strain level (Figure 7A, lane1). In the bypass mitochondria (Δyfh1 M>I) the persulfide signal was recovered to 91 % of the wt strain level (Figure 7A, lane 2). The persulfide was detected at a level comparable with the wt stain in the mutant Isu1 in the YFH1 context (YFH1 M>I) (Figure 7A, lane 4). MA14, carrying a hypomorphic allele of NFS1, showed no detectable persulfide in mitochondria, and the purified Nfs1–Isd11 showed a strong persulfide signal, migrating slightly slower than the mitochondrial signal due to the His6 tag on the purified Nfs1 [21]. The changes in mitochondrial persulfide were not due to changes in Nfs1 protein levels, as the Nfs1 protein levels were equivalent in these mitochondria (Figure 3).
Figure 7. Persulfide-forming activity in mitochondria.

Mitochondria were isolated from the single-copy Isu strains (native Isu1 promoter strains). (A) Nfs1 persulfide in intact mitochondria. Intact mitochondria were depleted of endogenous nucleotides by incubation for 10 min at 30 °C. After labelling with [35S]cysteine, mitochondria were recovered, proteins were separated by non-reducing SDS/PAGE and Nfs1 persulfide (Nfs1-S-35SH) was visualized by radioautography. Lane 1, Δyfh1 ISU1; lane 2, Δyfh1 M>I; lane 3, YFH1 ISU1; lane 4, YFH1 M>I; lane 5, MA14 mitochondria; lane 6, purified Nfs1–Isd11 complex. The Nfs1 persulfide is indicated by an arrow. Four independent experiments were performed and a representative autoradiogram is shown. (B) Nfs1 persulfide in mitochondrial lysate. Lysate from the deletion (Δyfh1 ISU1) mitochondria was supplemented with different amounts of purified Yfh1 protein (lanes 2–4) or purified mutant M>I Isu1 protein (lanes 5–7). The lysates were incubated with [35S]cysteine, and radiolabelled persulfide was detected by separation on non-reducing SDS gel and radioautography. Lysate from MA14, a hypomorphic Nfs1 mutant, was included as a negative control (lane 8), and purified Nfs1–Isd11 protein complex was included as a positive control (lane 9). The experiment was repeated three times and a representative autoradiogram is shown.
Persulfide on Nfs1, reflecting cysteine desulfurase activity, can also be assayed in mitochondrial lysates. A lysate was generated from frataxin-deletion mitochondria (Δyfh1 ISU1), and this exhibited low, but detectable, Nfs1 persulfide (Figure 7B, lane 1). Mature frataxin, expressed and purified from bacteria, was added back to the Δyfh1 mitochondrial lysate. A stimulatory effect was observed, and the stimulation increased with increasing amounts of added frataxin (Figure 7B, lanes 2–4). Similarly, mature M>I Isu1 protein was expressed and purified from bacteria. Addition of this substituted bypass Isu1 also produced a stimulatory effect on Nfs1 persulfide-forming activity in the lysate (Figure 7B, lanes 5–7). The degree of stimulation was similar to the frataxin effect, with the maximum occurring at approximately 2 μg of added protein. Thus either frataxin or the M>I bypass Isu was able to stimulate cysteine desulfurase activity in the mitochondrial lysate from the frataxin-deletion (Δyfh1 ISU1) strain. In summary, the mitochondrial Nfs1 persulfide level, reflecting a single round of cysteine desulfurase activity, was decreased in mitochondria lacking Yfh1 (Δyfh1 ISU1 compared with YFH1 ISU1), consistent with a role of Yfh1 as a positive regulator of cysteine desulfurase [12]. The M>I substituted Isu1 (Δyfh1 M>I) restored persulfide-forming activity when expressed as the sole Isu1 in mitochondria lacking frataxin (Figure 7A), or when added to mitochondrial lysate lacking frataxin, but already carrying endogenous Isu1 (Figure 7B). Frataxin or M>I bypass Isu1 exhibited similar stimulatory effects on the cysteine desulfurase in a Δyfh1 mitochondrial lysate, suggesting that they exert their effects by a similar mechanism.
Fe–S cluster assembly in isolated mitochondria
Isolated and intact mitochondria possess a complete machinery for synthesis of new Fe–S clusters. When supplied with [35S]cysteine, iron and nucleotides, the machinery is activated, and newly formed [Fe–35S] clusters are made and inserted into apoaconitase, providing a measure of the efficiency of the entire biosynthetic pathway [21]. In wt (YFH1 ISU1) mitochondria, new aconitase Fe–S cluster synthesis was apparent even at 15 min, increasing further at the 30 and 60 min time points (Figure 8A, lanes 1–3). In mitochondria with frataxin present and the substituted Isu1 (YFH1 M>I), the synthesis of new clusters was still quite active, although slightly less than that in the wt strain (Figure 8A, lanes 4–6). By contrast, in the frataxin-deletion strain (Δyfh1 ISU1), Fe–S cluster synthesis activity was severely impaired (Figure 8A, lanes 7–9). In the bypass mutant mitochondria (Δyfh1 M>I), biosynthetic activity was restored to some extent, but the amount was small and the rate of formation remained low (Figure 8A, lanes 10–12), reaching only 15 % of the wild-type amount after 60 min. These same bypass (Δyfh1 M>I) mitochondria have normal levels of aconitase protein (Figure 3) and almost normal aconitase activity (Figure 2). The data suggest that turnover of these slowly formed Fe–S clusters in living cells is slow enough to allow for eventual recovery of normal amounts of aconitase holoprotein.
Figure 8. Fe–S cluster forming activity in intact mitochondria.

(A) Isolated mitochondria from strains with the indicated genotypes were incubated with [35S]cysteine in the presence of nucleotides (4 mM ATP, 1 mM GTP and 5 mM NADH) and ferrous ascorbate (10 μM) at 30 °C for the indicated times. Mitochondria were recovered, lysed and soluble proteins were analysed by native gel electrophoresis. Radiolabelled aconitase (Aco1 [Fe-35S]) was visualized by radioautography. The time course experiment was repeated three times and a representative autoradiogram is shown. (B) Isolated mitochondria were labelled at 30 °C for 30 min in the presence of [35S]cysteine, nucleotides and different concentrations of ferrous ascorbate (0, 10 and 50 μM). Radiolabelled aconitase was visualized as in (A). The iron titration experiment was repeated three times and a representative autoradiogram is shown.
Iron dependence of Fe–S cluster synthesis was evaluated by adding different amounts of ferrous ascorbate to the isolated mitochondria during the labelling assay (Figure 8B). Iron dependence of new Fe–S cluster formation was observed for the wt strain (Figure 8B, lanes 1–3). Little new Fe–S clusters were seen in the deletion mitochondria (Δyfh1 ISU1) (Figure 8B, lanes 7–9). The addition of up to 50 μM iron enhanced Fe–S cluster assembly in the bypass mutant mitochondria (Δyfh1 M>I), but the improvement was modest and the amount of synthesis after 60 min was still quite low compared with the wild-type (Figure 8B, lanes 10–12). Thus the kinetic defect in Fe–S cluster assembly in the M>I mitochondria was not overcome by the addition of iron in vitro, suggesting that the function of frataxin could not be replaced by exogenously supplied iron.
Peroxide sensitivity
Frataxin deficiency has been associated with increased sensitivity to oxidative stress [31]. With this in mind, the strains were subjected to oxidative stress by spotting on to agar plates supplemented with H2O2. The frataxin-deletion strain (Δyfh1 ISU1) was sensitive as expected, exhibiting no colony formation at higher H2O2 concentrations (Figure 9, row 1 at 4 mM H2O2). The wt frataxin-plus strain was resistant (Figure 9, row 3). However, examination of the bypass strain (Δyfh1 M>I) revealed a unique phenotype (Figure 9, row 2). Growth was identical to the wt at low peroxide concentrations (Figure 9, 0.25 mM and 2 mM H2O2), but at higher H2O2 concentrations it exhibited decreased growth indicative of sensitivity to H2O2 (Figure 9, row 2 at 4 mM H2O2). Overexpression of wild-type or mutant Isu1 did little to alter the H2O2 sensitivity phenotypes (Figure 9, rows 5–8). The increased H2O2 sensitivity of the bypass mutant (Δyfh1 M>I) compared with the wt strain (YFH1 ISU1) is not likely to be explained by iron accumulation and the resultant free radical damage, because iron homoeostasis was corrected in that strain. Instead, a possible explanation is that H2O2 exposure induced a more rapid turnover of Fe–S clusters or their precursors. The low rate of synthesis of Fe–S clusters in the bypass strain then made it unable to keep up with the increased demand created by the oxidative stress conditions.
Figure 9. H2 O2 sensitivity.
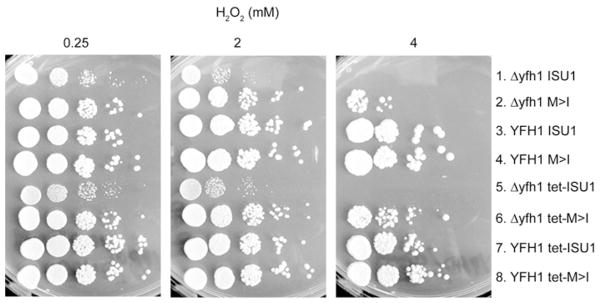
Yeast strains (Isu1 driven by the native promoter or tet07 promoter) were serially diluted and spotted on to YPAD agar plates containing H2 O2 at different concentrations (0.25 mM, 2 mM or 4 mM). The growth of the bypass strain (Δyfh1 M>I) was indistinguishable from the wt strain (YFH1 ISU1) at low H2 O2, but severely impaired at high H2 O2 concentration.
DISCUSSION
Mitochondria synthesize Fe–S clusters in a multi-step process, involving formation of Fe–S cluster intermediates on a scaffold and transfer to substrate apoproteins [10]. Frataxin is part of an Fe–S cluster assembly protein complex in mitochondria, interacting with the cysteine desulfurase, Nfs1–Isd11, and the scaffold, Isu [7–9]. Frataxin is required for efficient formation of the Fe–S cluster intermediate on the scaffold and for Fe–S cluster synthesis in cells [7]. An amino acid substitution in one of two redundant genes encoding Isu, the M>I change of amino acid 107 of Isu1, was found to promote recovery of almost all of the mutant phenotypes, including growth, Fe–S cluster protein activities, haem and iron homoeostasis [5]. In the present study we found that the recovery could be effected by single-copy Isu1 with the M>I substitution. The implication is that the bypass Isu1 was able to function as the sole Isu in the cell, mediating the scaffold function and also conferring the frataxin-bypass effect. An interaction with a wild-type copy of Isu1 or Isu2 was not required.
The process of Fe–S cluster assembly in mitochondria requires iron and sulfur. For the sulfur component, the assembly process involves the release of sulfur from the amino acid cysteine, formation of a persulfide on the cysteine desulfurase Nfs1 and donation of the sulfur to the Isu scaffold [32]. In eukaryotic cells, the Nfs1 cysteine desulfurase activity is apparently strongly regulated, with the enzyme remaining ‘off’ until the crucial activating steps occur [12,22]. In experiments with intact mitochondria and mitochondrial lysates, the persulfide signal generated by a single cycle of cysteine desulfurase was found to be decreased in frataxin-deletion mitochondria (Δyfh1 ISU1) compared with the wt strain (YFH1 ISU1). The activity was restored in vitro by the addition of frataxin to the mitochondrial lysate. Interestingly, the mutant form of Isu1, M>I, performed very much in the same way as frataxin. The bypass mitochondria expressing the single-copy Isu1 M>I mutant in the absence of frataxin were more active for cysteine desulfurase, and the addition of purified M>I mutant Isu1 protein stimulated persulfide formation in the deletion mitochondrial lysate. These data strongly suggest that the frataxin-bypassing effect of the M>I Isu1 is mediated, at least in part, by a stimulatory effect on cysteine desulfurase. In recent work with purified proteins, the stimulation was traced back to an effect on enhancing substrate binding to Nfs1 [33]. Either frataxin or M>I Isu1 increased the binding of the amino acid cysteine to Nfs1 in an early step of the reaction cycle, and the persulfide formation was subsequently stimulated by Isd11 interaction with Nfs1 in a later step of the reaction cycle [33]. How this might occur in terms of structural changes in the proteins is unknown, as no crystal structures of eukaryotic cysteine desulfurases or scaffolds have been solved. One can imagine that the M107I substitution might alter the interaction of Isu1 with Nfs1 in such a way to expose Nfs1 substrate-binding sites and enhance substrate binding. Thus frataxin or M>I could act in similar fashion as an allosteric effector of the cysteine desulfurase [12,33].
There is an evolutionary story here too. The Isu protein is highly conserved between prokaryotes and eukaryotes, consistent with the evolutionary origin of mitochondria from purple bacteria [14]. The M>I change occurs in a conserved amino acid. At residue 107 the methionine is present in all eukaryotes including plants, animals and protists, whereas the isoleucine is present in many prokaryotes including E. coli. Other branched chain amino acids such as leucine and valine are present in a similar position in Isu homologues from other prokaryotes [5]. The M>I change then can be considered to confer a prokaryotic feature to Isu and to the Fe–S cluster assembly apparatus. In this regard it is interesting that the E. coli Fe–S cluster assembly apparatus does not depend on frataxin as much as the eukaryotic apparatus [34]. The E. coli knockout has very mild phenotypes and does not accumulate iron or show severe Fe–S cluster deficiencies [15]. Also the cysteine desulfurase does not depend on CyaY, the prokaryotic frataxin, for activation in the way that the eukaryotic Nfs1 depends on Yfh1, the yeast frataxin homologue [35]. Rendering the mitochondrial Fe–S cluster assembly apparatus more prokaryote-like might also make it more frataxin independent.
Isolated and metabolically active mitochondria were examined in an assay for new Fe–S cluster synthesis on apo-aconitase [36]. There was strong Fe–S cluster biosynthetic activity in the wt strain (YFH1 ISU1) and negligible activity in the frataxin-deletion strain (Δyfh1 ISU1), consistent with the in vivo phenotypes. Although mitochondria with the bypass (Δyfh1 M>I) recovered activity, Fe–S clusters were made slowly such that the newly synthesized [Fe–35S] aconitase clusters were only approximately 15 % of the wild-type level after 60 min (Figure 8A). With this information, i.e. that the bypass mitochondria possess normal aconitase activity, but a very slow rate of new aconitase cluster synthesis, we can envision what occurs in living cells. The slow synthesis of Fe–S clusters in the absence of frataxin does not restore function instantaneously, but, over time, the mutant ‘catches up’ eventually accumulating enough Fe–S clusters to reconstitute holoenzymes in normal amounts. Cellular Fe–S clusters are slowly restored, and these include the regulatory Fe–S clusters that control iron homoeostasis [37,38]. The implications of this scenario are first, that the normally functioning Fe–S cluster assembly machinery has tremendous spare capacity, and secondly, that the turnover rate of assembled Fe–S clusters within target enzymes is low. As far as we know neither of these parameters, spare Fe–S cluster biosynthetic capacity or Fe–S cluster turnover, has been measured directly. The exposure of cells to H2O2, a strong oxidant, by damaging Fe–S clusters and Fe–S cluster intermediates might decrease the spare capacity and increase Fe–S cluster turnover. Thus, because the bypass mutant (Δyfh1 M>I) lacked spare biosynthetic capacity, it was able to grow normally under non-stress conditions, but only very slowly under H2O2 stress conditions. The deficit of spare biosynthetic capacity in the bypass mutant M>I might be accounted for by other frataxin functions that were not corrected as well as the cysteine desulfurase. The bypass mutant might effectively replace frataxin in terms of stimulating cysteine desulfurase, but it might fail to replace some other key function of frataxin, for example iron donation for Fe–S cluster assembly [11].
In summary, a detailed analysis has been presented of the frataxin-bypass phenotype conferred by the M>I mutant Isu1, expressed as the sole cellular Fe–S scaffold protein. Most Δyfh1 phenotypes were corrected in the bypass strain (Δyfh1 M>I). Stimulation of the cysteine desulfurase contributed to the bypass effect, although a kinetic defect in Fe–S cluster assembly in mitochondria remained. The results point to the multifaceted roles of Yfh1 in controlling mitochondrial Fe–S cluster assembly, and also suggest therapeutic targets for bypassing frataxin in Friedreich’s ataxia and for improving Fe–S cluster assembly generally.
Supplementary Material
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [grant numbers R37DK053953 (from the National Institute of Diabetes and Digestive and Kidney Diseases to A.D.) and RO1AG030504 (from the National Institute on Aging to D.P.)].
We thank Elise Lyver for assistance.
Abbreviations
- Aft1/2
activator of ferrous transport 1/2
- Ccp1
cytochrome c peroxidase
- Cyc1
cytochrome c
- Isd11
iron–sulfur desulfurase-interacting protein
- Nfs1
NiFS-like 1
- Ni-NTA
Ni2+-nitrilotriacetate
Footnotes
AUTHOR CONTRIBUTION
Heeyong Yoon and Simon Knight constructed the strains and performed biochemical experiments. Alok Pandey and Jayashree Pain performed the persulfide and Fe–S cluster loading experiments. Yan Zhang performed the pulse–chase experiments. Debkumar Pain and Andrew Dancis designed experiments and wrote the paper.
References
- 1.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 2.Rouault TA. Biogenesis of iron–sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech. 2012;5:155–164. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitnall M, Suryo Rahmanto Y, Huang ML, Saletta F, Lok HC, Gutierrez L, Lazaro FJ, Fleming AJ, St Pierre TG, Mikhael MR, et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc Natl Acad Sci USA. 2012;109:20590–20595. doi: 10.1073/pnas.1215349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet. 1997;16:352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 5.Yoon H, Golla R, Lesuisse E, Pain J, Donald JE, Lyver ER, Pain D, Dancis A. Mutation in the Fe–S scaffold protein Isu bypasses frataxin deletion. Biochem J. 2012;441:473–480. doi: 10.1042/BJ20111637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pastore A, Puccio H. Frataxin: a protein in search for a function. J Neurochem. 2013;126:43–52. doi: 10.1111/jnc.12220. [DOI] [PubMed] [Google Scholar]
- 7.Gerber J, Muhlenhoff U, Lill R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003;4:906–911. doi: 10.1038/sj.embor.embor918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmucker S, Martelli A, Colin F, Page A, Wattenhofer-Donze M, Reutenauer L, Puccio H. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron–sulfur assembly complex. PloS One. 2011;6:e16199. doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, Craig EA. Binding of yeast frataxin to the scaffold for Fe–S cluster biogenesis, Isu. J Biol Chem. 2008;283:12674–12679. doi: 10.1074/jbc.M800399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, Uzarska MA, Webert H, Wilbrecht C, Muhlenhoff U. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. 2012;1823:1491–1508. doi: 10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Cook JD, Kondapalli KC, Rawat S, Childs WC, Murugesan Y, Dancis A, Stemmler TL. Molecular details of the yeast frataxin-Isu1 interaction during mitochondrial Fe–S cluster assembly. Biochemistry. 2010;49:8756–8765. doi: 10.1021/bi1008613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 13.Andrew AJ, Song JY, Schilke B, Craig EA. Posttranslational regulation of the scaffold for Fe–S cluster biogenesis, Isu. Mol Biol Cell. 2008;19:5259–5266. doi: 10.1091/mbc.E08-06-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markley JL, Kim JH, Dai Z, Bothe JR, Cai K, Frederick RO, Tonelli M. Metamorphic protein IscU alternates conformations in the course of its role as the scaffold protein for iron–sulfur cluster biosynthesis and delivery. FEBS Lett. 2013;587:1172–1179. doi: 10.1016/j.febslet.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li DS, Ohshima K, Jiralerspong S, Bojanowski MW, Pandolfo M. Knock-out of the cyaY gene in Escherichia coli does not affect cellular iron content and sensitivity to oxidants. FEBS Lett. 1999;456:13–16. doi: 10.1016/s0014-5793(99)00896-0. [DOI] [PubMed] [Google Scholar]
- 16.Garland SA, Hoff K, Vickery LE, Culotta VC. Saccharomyces cerevisiae ISU1 and ISU2: members of a well-conserved gene family for iron–sulfur cluster assembly. J Mol Biol. 1999;294:897–907. doi: 10.1006/jmbi.1999.3294. [DOI] [PubMed] [Google Scholar]
- 17.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 18.Gari E, Piedrafita L, Aldea M, Herrero E. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.Song JY, Marszalek J, Craig EA. Cysteine desulfurase Nfs1 and Pim1 protease control levels of Isu, the Fe–S cluster biogenesis scaffold. Proc Natl Acad Sci USA. 2012;109:10370–10375. doi: 10.1073/pnas.1206945109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Kogan M, Knight SA, Pain D, Dancis A. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J Biol Chem. 1999;274:33025–33034. doi: 10.1074/jbc.274.46.33025. [DOI] [PubMed] [Google Scholar]
- 21.Pandey A, Yoon H, Lyver ER, Dancis A, Pain D. Identification of a Nfs1p-bound persulfide intermediate in Fe–S cluster synthesis by intact mitochondria. Mitochondrion. 2012;12:539–549. doi: 10.1016/j.mito.2012.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandey A, Golla R, Yoon H, Dancis A, Pain D. Persulfide formation on mitochondrial cysteine desulfurase: enzyme activation by a eukaryote-specific interacting protein and Fe–S cluster synthesis. Biochem J. 2012;448:171–187. doi: 10.1042/BJ20120951. [DOI] [PubMed] [Google Scholar]
- 23.Smith PM, Fox JL, Winge DR. Biogenesis of the cytochrome bc1 complex and role of assembly factors. Biochim Biophys Acta. 2012;1817:276–286. doi: 10.1016/j.bbabio.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiedemann N, Urzica E, Guiard B, Muller H, Lohaus C, Meyer HE, Ryan MT, Meisinger C, Muhlenhoff U, Lill R, Pfanner N. Essential role of Isd11 in mitochondrial iron–sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006;25:184–195. doi: 10.1038/sj.emboj.7600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherman F. The importance of mutation, then and now: studies with yeast cytochrome c. Mutat Res. 2005;589:1–16. doi: 10.1016/j.mrrev.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Lesuisse E, Santos R, Matzanke BF, Knight SA, Camadro JM, Dancis A. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1) Hum Mol Genet. 2003;12:879–889. doi: 10.1093/hmg/ddg096. [DOI] [PubMed] [Google Scholar]
- 27.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 28.Miao R, Martinho M, Morales JG, Kim H, Ellis EA, Lill R, Hendrich MP, Munck E, Lindahl PA. EPR and Mossbauer spectroscopy of intact mitochondria isolated from Yah1p-depleted Saccharomyces cerevisiae. Biochemistry. 2008;47:9888–9899. doi: 10.1021/bi801047q. [DOI] [PubMed] [Google Scholar]
- 29.Ramazzotti A, Vanmansart V, Foury F. Mitochondrial functional interactions between frataxin and Isu1p, the iron-sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 2004;557:215–220. doi: 10.1016/s0014-5793(03)01498-4. [DOI] [PubMed] [Google Scholar]
- 30.Muhlenhoff U, Gerber J, Richhardt N, Lill R. Components involved in assembly and dislocation of iron–sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulteau AL, Dancis A, Gareil M, Montagne JJ, Camadro JM, Lesuisse E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic Biol Med. 2007;42:1561–1570. doi: 10.1016/j.freeradbiomed.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 32.Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron–sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 33.Pandey A, Gordon DM, Pain J, Stemmler TL, Dancis A, Pain D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites and a mutant Fe–S cluster scaffold protein with frataxin-bypassing ability acts similarly. J Biol Chem. 2013;288:36773–36786. doi: 10.1074/jbc.M113.525857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roche B, Aussel L, Ezraty B, Mandin P, Py B, Barras F. Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim Biophys Acta. 2013;1827:455–469. doi: 10.1016/j.bbabio.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Bridwell-Rabb J, Iannuzzi C, Pastore A, Barondeau DP. Effector role reversal during evolution: the case of frataxin in Fe–S cluster biosynthesis. Biochemistry. 2012;51:2506–2514. doi: 10.1021/bi201628j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amutha B, Gordon DM, Dancis A, Pain D. Nucleotide-dependent iron–sulfur cluster biogenesis of endogenous and imported apoproteins in isolated intact mitochondria. Methods Enzymol. 2009;456:247–266. doi: 10.1016/S0076-6879(08)04414-5. [DOI] [PubMed] [Google Scholar]
- 37.Kumanovics A, Chen OS, Li L, Bagley D, Adkins EM, Lin H, Dingra NN, Outten CE, Keller G, Winge D, et al. Identification of FRA1 and FRA2 as genes involved in regulating the yeast iron regulon in response to decreased mitochondrial iron–sulfur cluster synthesis. J Biol Chem. 2008;283:10276–10286. doi: 10.1074/jbc.M801160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ojeda L, Keller G, Muhlenhoff U, Rutherford JC, Lill R, Winge DR. Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J Biol Chem. 2006;281:17661–17669. doi: 10.1074/jbc.M602165200. [DOI] [PubMed] [Google Scholar]
- 39.Amutha B, Gordon DM, Gu Y, Lyver ER, Dancis A, Pain D. GTP is required for iron–sulfur cluster biogenesis in mitochondria. J Biol Chem. 2008;283:1362–1371. doi: 10.1074/jbc.M706808200. [DOI] [PubMed] [Google Scholar]
- 40.Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron–sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.