

Abstract
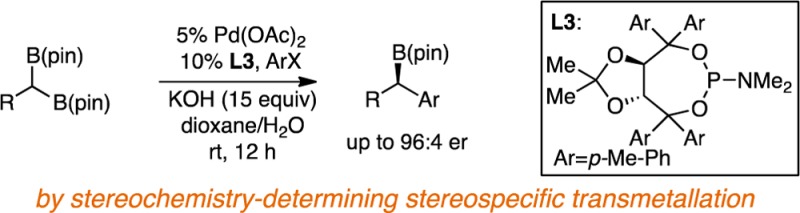
Catalytic enantiotopic-group-selective cross-couplings of achiral geminal bis(pinacolboronates) provide a route for the construction of nonracemic chiral organoboronates. In the presence of a chiral monodentate taddol-derived phosphoramidite ligand, these reactions occur with high levels of asymmetric induction. Mechanistic experiments with chiral 10B-enriched geminal bis(boronates) suggest that the reaction occurs by a stereochemistry-determining transmetalation that occurs with inversion of configuration at carbon.
The versatility of nonracemic chiral organoboronates has made them sought after intermediates in asymmetric synthesis.1 Recent catalytic enantioselective strategies for constructing such compounds include hydroboration,2 conjugate borylation,3 allylic borylation,4 conjugate addition and allylic substitution with borylated electrophiles,5 and cycloadditions with vinyl boronate derivatives.6 Also developed are conjugate reduction7 and hydrogenation of vinyl boronates,8 and diborylation of alkenes9 and alkynes.10 In this connection, our laboratory has pursued the enantioselective construction and transformation of vicinal diboronates with the notion that selective mono-cross-coupling reactions result in products wherein the remaining boronate can be used in subsequent strategically useful chemical transformations.11 It was considered that a similarly useful strategy for construction of organoboronates might arise by the mono-cross-coupling of geminal bis(boronates). These reactions have been extensively studied by Shibata and Endo who employed Pd(PtBu3)2 as an effective catalyst for a range of such reactions.12 While related reactions of chiral nonracemic geminal bis(boronates) have been developed by Hall13 and Yun14 (Scheme 1), we considered that readily accessed symmetric geminal bis(boronates), reacting in the presence of an appropriately designed chiral catalyst, might participate in an enantiotopic-group-selective Suzuki reaction and that such a strategy might provide a useful new route to nonracemic organoboronate derivatives that are not readily prepared by other methods.15 In this manuscript, we present such a process and show that it can be employed to prepare a range of chiral boronates in an enantioselective fashion; we also provide mechanistic insight about critical steps in the catalytic cycle.
Scheme 1.

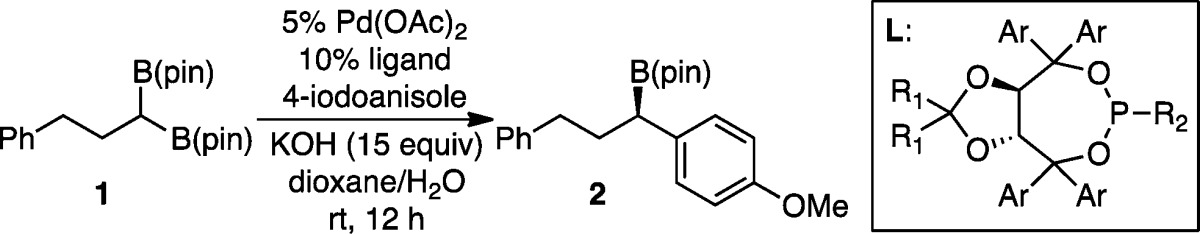
Our preliminary experiments surveyed the cross-coupling reaction between geminal bis(boronate) 1 and p-iodoanisole in the presence of Pd(OAc)2 and selected ligands (Table 1). Initial observations revealed a high background reaction rate in the absence of ligand (entry 1), as well as nonselective reaction in the presence of bidentate phosphines (entries 2–3) or with a preformed JosiphosPdCl2 complex. These observations suggested a catalytic reaction pathway requiring access to three-coordinate L1PdAr(X), ostensibly as a precursor to transmetalation with bis(boronate), and it was suspected that that complexes derived from bidentate ligands might prove sufficiently unreactive that nonselective background reactions could prevail. In line with this hypothesis, reactions with chiral monodentate ligands proved more selective with L3 being most effective. A survey of reaction conditions showed the following to be most critical: first, reactions of aryl iodides are significantly more selective than bromides (chlorides and triflates are unreactive); second, high concentrations of KOH are critical (reaction with 4.5 equiv of KOH results in 79:21 er); third, an excess of monodentate ligand is critical for highest selectivity (65:35 er was obtained with 1:1 L3/Pd), presumably as a result of competing background nonligated pathways at lower ligand loading. Lastly, it was found (vide infra) that aryl bromides can also be employed as electrophiles in asymmetric Suzuki reactions but require the addition of sodium iodide for high selectivity.
Table 1. Catalytic Enantioselective Suzuki Reactiona.
| entry | ligand | Ar | R1 | R2 | yield (%)a | erb |
|---|---|---|---|---|---|---|
| 1 | none | – | – | – | 98 | – |
| 2 | binap | – | – | – | 78 | 50:50 |
| 3 | JosiPhos | – | – | – | 55 | 50:50 |
| 4 | L1 | Ph | Me | Ph | 80 | 75:25 |
| 5 | L2 | Ph | H | NMe2 | 74 | 77:23 |
| 6 | L3 | p-tol | Me | NMe2 | >98 | 94:6 |
| 7 | L4 | 4-t-BuPh | Me | NMe2 | 92 | 92:8 |
Yield was determined by NMR in comparison to an internal standard.
Enantiomer ratio (er) determined by chiral SFC analysis.
The substrate scope of enantiotopic-group-selective cross-couplings involving geminal bis(boronates) was surveyed with a number of aryl halides and geminal bis(boronates) (Figure 1).16 The reactions with either p-iodoanisole (method A) or p-bromoanisole combined with sodium iodide (method B) provide comparable yield and selectivity in the production of 2, although it should be noted that with other substrates (product 4) there is a moderate difference between the two methods. Also of note, electron-neutral electrophiles (products 5–7), as well as more hindered ortho substituted electrophiles (products 10, 13), also couple with good enantioselection. While protodeboronation occurred when conjugating groups were present at the para position (CN, CO2Et; data not shown), this problem could be ameliorated for the case of ketone substrates by the use of ketal protection (product 11). Lastly, it was found that neither heterocycles (products 9, 14) nor an electron-withdrawing p-fluoro group (product 12) interfered in the process. In terms of the scope of geminal bis(boronates), the reaction applies to linear aliphatic substrates (products 16, 17), as well as more hindered substrates (product 18), although the larger substituent appears to retard the reaction rate.
Figure 1.

Substrate scope of the enantioselective Suzuki coupling.
The utility of the asymmetric cross-coupling reaction for the enantioselective construction of pharmaceutically relevant benzhydryl derivatives was examined by targeting the construction of (R)-tolterodine (Detrol LA), a therapeutic used for the treatment of urinary incontinence (Scheme 2).17 Construction of substrate 22 was accomplished by deprotonation and alkylation of 1,1-di(pincolatoboryl)methane (21).16a Subsequent Suzuki reaction provided 23 in 74% yield and 93:7 enantiomer ratio. Subsequent stereoretentive cross-coupling of 23 with 2-iodo-4-methylanisole employing conditions similar to those developed by Crudden18 occurred with some erosion of enantiomeric excess, but in very good yield. Lastly, deprotection of 23 furnished 24, a known precursor19 to the clinically used therapeutic.
Scheme 2. A Route for the Synthesis of (R)-Tolterodine with the Enantiotopic-Group-Selective Suzuki Reaction.

To provide insight into the processes that underlie the enantiotopic-group-selective Suzuki reaction, the cycle in Scheme 3 was considered. On the basis of findings that Suzuki reactions in the presence of base and water may occur through the intermediacy of Pd(hydroxide) intermediates,20 it was considered that, subsequent to oxidative addition, substitution of halide for hydroxide might occur. Next, transmetalation might furnish α-borylorganopalladium complex 25; reductive elimination would then furnish the product. Other possibilities notwithstanding, in this scenario the stereoselectivity-determining step might be transmetalation, or if the intermediate 25 is subject to isomerization, reductive elimination might control the product outcome.
Scheme 3.

One possible mechanism for equilibration of intermediate 25 involves reversible deprotonation.21 This possibility was excluded by examination of the reaction run in the presence of D2O: less than 5% deuterium was incorporated in the product (eq 1, Scheme 4). While other mechanisms might epimerize 25 (i.e., reversible β-hydrogen elimination), the experiments in eqs 2 and 3 strongly suggest that the transmetalation is stereospecific and this step likely is stereochemistry-determining. In these experiments, enantiomerically enriched, isotopically chiral (S)-10B-26, prepared from 10B-boric acid, was subjected to the asymmetric cross-coupling with either enantiomer of chiral ligand L3. For the reaction with (R,R)-L3, mass spectral analysis of the product shows an isotopic composition consistent with one expected for a reaction where the 10B labeled boronate participates selectively, while the reaction with the (S,S) enantiomer of L3 gives an isotope pattern expected for replacement of the natural abundance B(pin) group.22 With the reasonable assumption that reductive elimination occurs with retention of configuration at carbon, the product isotopic composition observed in both experiments is most consistent with that calculated for transmetalation occurring with inversion of configuration at carbon.
Scheme 4.

Additional information about the nature of the species involved in the cross-coupling of geminal bis(boronates) was revealed by the experiment in eq 4. When a 1:1 mixture of doubly labeled boronates was subjected to KOH in H2O/dioxane, complete scrambling of the boronates was observed. Similarly, when the mixture is subjected to catalytic cross-coupling, both products bear deuterated and nondeuterated pinacol boronates (data not shown). While this experiment does not reveal the mechanism of pinacol exchange, it does suggest that bis(boronates) or their mono- or bis(hydrolysis) products (or the derived “ate” complexes) are candidate reactants in the transmetalation. Thus the stereoselective transmetalation might occur either by a desymmetrization if both boronates are equivalent or by a dynamic kinetic resolution if the geminal boron atoms are not substituted equivalently.
Stereoinversion has been observed in other transmetalations and is most often associated with an open transition state that does not involve preassociation between Pd and the reacting organometallic reagent.23 This feature was unanticipated in the present reaction and will be the subject of forthcoming studies. Further studies on expanding the scope of this reaction to the construction of other chiral boron derivatives and on further elucidation of catalytic mechanisms is in progress.
Acknowledgments
The NIH (GM-59471) is acknowledged for financial support; AllyChem (B2(pin)2) and BASF (pinacolborane) are acknowledged for donations of reagents.
Supporting Information Available
Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hall D. G.Boronic Acids, 2nd ed.; Wiley-VCH: Weinheim, 2011. [Google Scholar]
- For a review of Rh-catalyzed asymmetric hydroboration, see:; a Carroll A.-M.; O’Sullivan T. P.; Guiry P. J. Adv. Synth. Catal. 2005, 347, 609. [Google Scholar]; For recent examples, see:; b Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3160. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062. [DOI] [PubMed] [Google Scholar]; d Smith S. M.; Takacs J. M. J. Am. Chem. Soc. 2010, 132, 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Corberán R.; Mszar N. W.; Hoveyda A. H. Angew. Chem., Int. Ed. 2011, 50, 7079. [DOI] [PubMed] [Google Scholar]; f Smith S. M.; Hoang G. L.; Pal R.; Khaled M. O. B.; Pelter L. S.; Zeng X. C.; Takacs J. M. Chem. Commun. 2012, 48, 12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review of catalytic enantioselective conjugate borylation, see:Calow A. D. J.; Whiting A. Org. Biomol. Chem. 2012, 10, 5485. [DOI] [PubMed] [Google Scholar]
- a Ito H.; Ito S.; Sasaki Y.; Matsuura K.; Sawamura M. J. Am. Chem. Soc. 2007, 129, 14856. [DOI] [PubMed] [Google Scholar]; b Guzman-Martinez A.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10634. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ito H.; Kunii S.; Sawamura M. Nat. Chem. 2010, 2, 972. [DOI] [PubMed] [Google Scholar]; d Park J. K.; Lackey H. H.; Ondrusek B. A.; McQuade D. T. J. Am. Chem. Soc. 2011, 133, 2410. [DOI] [PubMed] [Google Scholar]
- a Lee J. C. H.; Hall D. G. J. Am. Chem. Soc. 2010, 132, 5544. [DOI] [PubMed] [Google Scholar]; b Carosi L.; Hall D. G. Angew. Chem., Int. Ed. 2007, 46, 5913. [DOI] [PubMed] [Google Scholar]
- Gao X.; Hall D. G. J. Am. Chem. Soc. 2003, 125, 9308. [DOI] [PubMed] [Google Scholar]
- Ding J.; Lee J. C. H.; Hall D. G. Org. Lett. 2012, 14, 4462. [DOI] [PubMed] [Google Scholar]
- a Morgan J. B.; Morken J. P. J. Am. Chem. Soc. 2004, 126, 15338. [DOI] [PubMed] [Google Scholar]; b Moran W. J.; Morken J. P. Org. Lett. 2006, 8, 2413. [DOI] [PubMed] [Google Scholar]; c Paptchikhine A.; Cheruku P.; Engman M.; Andersson P. G. Chem. Commun. 2009, 5996. [DOI] [PubMed] [Google Scholar]; d Ganić A.; Pfaltz A. Chem.—Eur. J. 2012, 18, 6724. [DOI] [PubMed] [Google Scholar]; e Gazić Smilović I.; Casas-Arcé E.; Roseblade S. J.; Nettekoven U.; Zanotti-Gerosa A.; Kovačevič M.; Časar Z. Angew. Chem., Int. Ed. 2012, 51, 1014. [DOI] [PubMed] [Google Scholar]
- a Morgan J. B.; Miller S. P.; Morken J. P. J. Am. Chem. Soc. 2003, 125, 8702. [DOI] [PubMed] [Google Scholar]; b Trudeau S.; Morgan J. B.; Shrestha M.; Morken J. P. J. Org. Chem. 2005, 70, 9538. [DOI] [PubMed] [Google Scholar]; c Pelz N. F.; Woodward A. R.; Burks H. E.; Sieber J. D.; Morken J. P. J. Am. Chem. Soc. 2004, 126, 16328. [DOI] [PubMed] [Google Scholar]; d Burks H. E.; Kliman L. T.; Morken J. P. J. Am. Chem. Soc. 2009, 131, 9134. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kliman L. T.; Mlynarski S. N.; Morken J. P. J. Am. Chem. Soc. 2010, 132, 13949. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Coombs J. R.; Haeffner F.; Kliman L. T.; Morken J. P. J. Am. Chem. Soc. 2013, 135, 11222. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Toribatake K.; Nishiyama H. Angew. Chem., Int. Ed. 2013, 52, 11011. [DOI] [PubMed] [Google Scholar]
- a Lee Y.; Jang H.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 18234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynarski S. N.; Schuster C. H.; Morken J. P. Nature 2014, 505, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Endo K.; Ohkubo T.; Hirokami M.; Shibata T. J. Am. Chem. Soc. 2010, 132, 11033. [DOI] [PubMed] [Google Scholar]; b Endo K.; Ohkubo T.; Shibata T. Org. Lett. 2011, 13, 3368. [DOI] [PubMed] [Google Scholar]; c Endo K.; Ohkubo T.; Ishioka T.; Shibata T. J. Org. Chem. 2012, 77, 4826. [DOI] [PubMed] [Google Scholar]
- a Lee J. C. H.; McDonald R.; Hall D. G. Nat. Chem. 2011, 3, 894. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jeon H.; Yun J. Angew. Chem., Int. Ed. 2013, 52, 3989. [DOI] [PubMed] [Google Scholar]
- For an intramolecular enantiotopic-group-selective Suzuki reaction with a 1,7-bis(borane), see:; a Cho S. Y.; Shibasaki M. Tetrahedron: Asymmetry 1998, 9, 3751. [Google Scholar]; For a review of enantioselective desymmetrization, see:; b Willis M. C. J. Chem. Soc., Perkin Trans. 1 1999, 1765. [Google Scholar]
- For the preparation of bis(boronates), see:; a Matteson D. S.; Moody R. J. Organometallics 1982, 1, 20. [Google Scholar]; b Ito H.; Kubota K. Org. Lett. 2012, 14, 890. [DOI] [PubMed] [Google Scholar]
- a Nilvebrant L.; Andersson K.-E.; Gillberg P.-G.; Stahl M.; Sparf B. Eur. J. Pharmacol. 1997, 327, 195. [DOI] [PubMed] [Google Scholar]; For some asymmetric syntheses, see:; b Andersson P. G.; Schink H. E.; Österlund K. J. Org. Chem. 1998, 63, 8067. [Google Scholar]; c Hedberg C.; Andersson P. G. Adv. Synth. Catal. 2005, 347, 662. [Google Scholar]; d Ulgheri F.; Marchetti M.; Piccolo O. J. Org. Chem. 2007, 72, 6056. [DOI] [PubMed] [Google Scholar]; e Botteghi C.; Corrias T.; Marchetti M.; Paganelli S.; Piccolo O. Org. Process Res. Dev. 2002, 6, 379. [Google Scholar]; f Chen G.; Tokunaga N.; Hayashi T. Org. Lett. 2005, 7112285. [DOI] [PubMed] [Google Scholar]; g Roesner S.; Aggarwal V. K. Can. J. Chem. 2012, 90, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imao D.; Glasspoole B. W.; Laberge V. S.; Crudden C. M. J. Am. Chem. Soc. 2009, 131, 5024. [DOI] [PubMed] [Google Scholar]
- Jonsson N. A.; Sparf B. A.; Mikiver L.; Moses P.; Nilvebrant L.; Glas G. U.S. Patent 5,382,600 A1,1995.
- a Amatore C.; Jutand A.; Le Duc G. Chem.—Eur. J. 2011, 17, 2492. [DOI] [PubMed] [Google Scholar]; b Carrow B.; Hartwig J. J. Am. Chem. Soc. 2011, 133, 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Suzaki Y.; Osakada K. Organometallics 2006, 25, 3251. [Google Scholar]; d Braga A. A.; Morgon N. H.; Ujaque G.; Maseras F. J. Am. Chem. Soc. 2005, 127, 9298. [DOI] [PubMed] [Google Scholar]
- For deprotonation of geminal dimetallics, see: Reference (16a).
- Calculated isotopic composition accounts for the enantiomeric purity of the substrate, the enantioselectivity of the reaction, and the isotope composition of naturally occurring boron and naturally occurring carbon. See Supporting Information for details.
- a Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191. [DOI] [PubMed] [Google Scholar]; b Sandrock D. L.; Jean-Gérard L.; Chen C. Y.; Dreher S. D.; Molander G. A. J. Am. Chem. Soc. 2010, 17108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Labadie J. W.; Stille J. K. J. Am. Chem. Soc. 1983, 105, 6129. [Google Scholar]; d Kells K. W.; Chong J. M. J. Am. Chem. Soc. 2004, 126, 15666. [DOI] [PubMed] [Google Scholar]; e Hatanaka Y.; Hiyama T. J. Am. Chem. Soc. 1990, 112, 7793. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.