

Abstract
Recessive mutations in the ANO5 gene, encoding anoctamin 5, cause proximal limb girdle muscular dystrophy (LGMD2L), Miyoshi-type distal myopathy (MM3) and asymptomatic hyper- CKemia.
We report a woman with exertion-induced myalgia and weakness in the hip girdle manifesting at the age of 40. Creatine kinase (CK) was increased 20-fold. Histologically the dominating feature was necrotizing myopathy, but long-term immunosuppressive therapy did not change CK level or myopathic symptoms. Molecular genetic investigation led to the finding of the homozygous ANO5 c.191dupA mutation. This is a report of a muscular dystrophy due to ANO5 mutation presenting histologically as necrotizing myopathy. For this reason our finding extends the histological spectrum of myopathies due to ANO5 mutations as well as the possible differential diagnoses for necrotizing myopathy.
Key words: Anoctamin 5, limb girdle muscular dystrophy 2L, necrotizing myopathy
Case report
Recessive mutations in the ANO5 gene (ANO5, MIM 6086629) are associated with limb girdle muscular dystrophy (LGMD) 2L; known to be the third most common LGMD in Northern and Central Europe (1-3) but also with a distal non-dysferlin Miyoshi type dystrophy (MM3) or with asymptomatic hyperCKemia (4, 5). We present here a patient homozygous for the ANO5 mutation c.191dupA with necrotizing myopathy as the dominating histological feature.
A 40-year-old athletic Caucasian woman started to complain about exertion-induced weakness and myalgia, especially in thighs and buttocks. At the time she had been weight training and mountain biking several times a week. Creatine kinase (CK) was 20-fold increased. A muscle biopsy from the gastrocnemius muscle presented as necrotizing myopathy (Fig. 1). Due to MHC upregulation myositis therapy with prednisolone and methotrexate (MTX) was initiated which diminished myalgia but the CK remained constantly raised (10- to 20-fold, maximum 35-fold) over several years. Investigations for myotoxic medication, potential malignancies and antibodies against signal recognition particle were negative. Magnetic resonance imaging (MRI) revealed asymmetric fatty atrophy of both thighs with accentuation of the posterior compartment (Fig. 2). First presentation of the patient in our clinic was five years after disease onset. She then complained of ongoing exercise intolerance with myalgia of the upper legs and hip girdle, difficulties in climbing stairs and rising from sitting or squatting position. Examination revealed mildly asymmetric weakness of the hip flexors and foot plantarflexors with restricted monopedal jumping and tiptoe walking. The CK was elevated 15-fold. Some fibrillations in the gastrocnemius muscle were obvious. The permanent elevation of CK despite immunosuppression, and the clinical presentation with prevalent proximal leg weakness as well as the charcteristic MRI-findings prompted us to search for causative mutations in ANO5. Amplification and sequencing of exon 5 of the ANO5 gene revealed the homozygous mutation c.191dupA (4) a founder mutation frequently identified in northern Europe patients with ANO5 myopathy (2-4, 6, 7). Therefore we diagnosed a necrotizing myopathy characterized by necrotic fibres and the absence of inflammatory and dystrophic signs, accompanied by an otherwise typical clinical presentation of ANO5 myopathy with LGMD phenotype.
Figure 1.
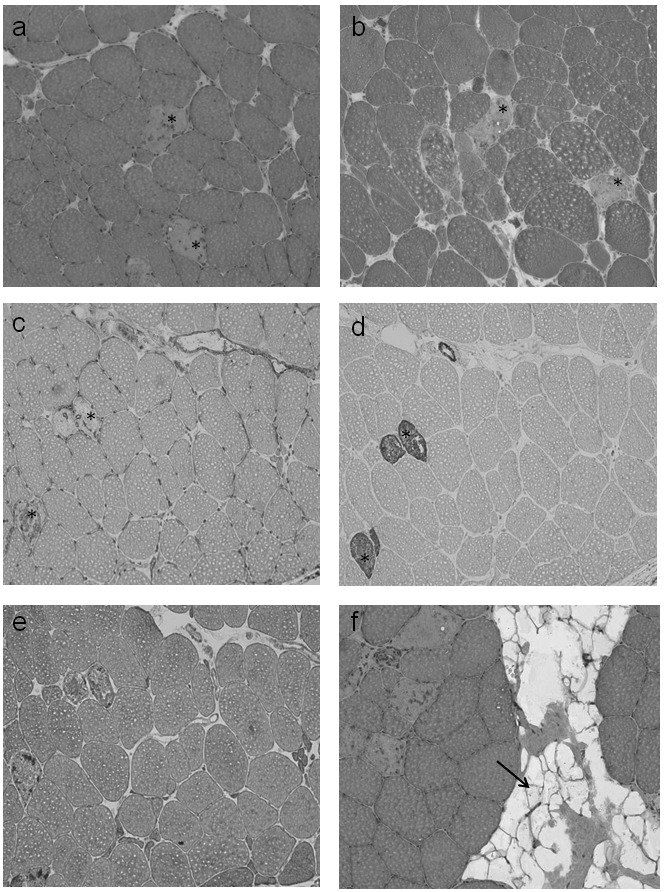
(a-d). Muscle biopsy from the gastrocnemic muscle. (a) H&E, (b) Gomori Trichrome. Occurence of 8% disseminated necrotic muscle fibers *, positive for (c) major histocompatibility complex (MHC I) and (d) complement (C5b9). Some smaller muscle fibres (25 μm) contained internalized caveolin and (e) dysferlin, indicating regenerating fibres. No inflammatory infiltrates, fibrosis, fatty degeneration, or further myopathic changes were detected. (f) H&E. Some lipocytes (arrow) were visible in the perifascicular connective tissue. Magnification x 10.
Figure 2.
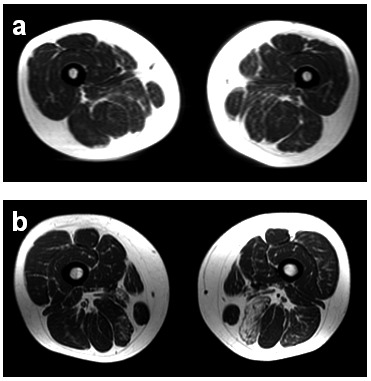
Magnetic resonance imaging (MRI, T1 fat suppressed) of the upper legs of the patient at 42 years old, with very mild asymmetric fatty replacement in the posterior compartment, accentuated in the left semimembranous muscle (a). Further progression at follow-up after one year (b).
Necrotizing myopathy has been associated with a variety of neoplasms and with autoimmune processes, e.g. as in the anti-signal recognition particle (anti-SRP) syndrome. However, we did not find any evidence of such etiological causes in our patient. Interestingly most recently Claeys at al. presented a patient with immune mediated necrotizing myopathy with antibodies against 3-hydoxy-3-methylglutaryl-coenzyme-Areductase without previous statin exposure in which 2 pathogenetic mutations of ANO5 (c.191dupA, exon 5; c.1627dupA, exon 15) were identified (8). Since we did not test for other antibodies, we cannot completely rule out the possibility of myositis or immune mediated necrotizing myopathy, although it is very unlikely because immunosuppressive therapy had no beneficial effect in our patient. There was also no hint of other underlying causes for myocyte necrosis; e.g. rhabdomyolysis in metabolic myopathies, necrosis as a sequelae of inflammation, toxin and drug-induced causes.
Few necrotic fibers can also occur as part of the dystrophic pattern in ANO5 myopathy (2, 9, 10), but in contrast to necotizing myopathy they are invariably associated with fatty and fibrotic remodelling.
Deficiency in Anoctamin 5, a putative calcium-activated chloride channel in skeletal muscle, is associated with multifocal loss of the costameres and gaps in the sarcolemmal membrane. Therefore a defective membrane repair might result in a higher vulnerability of muscle fibres, causing ongoing hyperCKemia and necrosis even in early (histological) stages of ANO 5 myopathy.
ANO5 myopathy can present as necrotizing myopathy extending the histological spectrum of myopathies due to ANO5 mutations as well as the possible differential diagnoses for necrotizing myopathy.
Acknowledgements
The authors thank Prof. Rolf Schröder, Institute of Neuropathology, University Erlangen for histological analysis of the muscle biopsy, Thekla Wangemann for performing the PCR, and Dr. Kathryn Birch for copy editing the manuscript.
References
- 1.Witting N, Duno M, Petri H, et al. Anoctamin 5 muscular dystrophy in Denmark: prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. J Neurol. 2013;260:2084–2093. doi: 10.1007/s00415-013-6934-y. [DOI] [PubMed] [Google Scholar]
- 2.Sarkozy A, Hicks D, Hudson J, et al. ANO5 Gene Analysis in a Large Cohort of Patients with Anoctaminopathy: Confirmation of Male Prevalence and High Occurrence of the Common Exon 5 Gene Mutation. Hum Mutat. 2013;34:1111–1118. doi: 10.1002/humu.22342. [DOI] [PubMed] [Google Scholar]
- 3.Hicks D, Sarkozy A, Muelas N, et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain. 2011;134:171–182. doi: 10.1093/brain/awq294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deschauer M, Joshi Pr, Glaser D, et al. [Muscular dystrophy due to mutations in anoctamin 5: clinical and molecular genetic findings]. Nervenarzt. 2011;82:1596–1603. doi: 10.1007/s00115-011-3325-4. [DOI] [PubMed] [Google Scholar]
- 5.Bouquet F, Cossee M, Behin A, et al. Miyoshi-like distal myopathy with mutations in anoctamin 5 gene. Rev Neurol (Paris) 2012;168:135–141. doi: 10.1016/j.neurol.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Bolduc V, Marlow G, Boycott Km, et al. Recessive mutations in the putative calcium- activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86:213–221. doi: 10.1016/j.ajhg.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahjneh I, Jaiswal J, Lamminen A, et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscul Disord. 2010;20:791–795. doi: 10.1016/j.nmd.2010.07.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claeys KG, Gorodinskaya O, Handt S, et al. Diagnostic challenge and therapeutic dilemma in necrotizing myopathy. Neurology. 2013;81:932–935. doi: 10.1212/WNL.0b013e3182a35285. [DOI] [PubMed] [Google Scholar]
- 9.Magri F, Bo Rd, D'angelo Mg, et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord. 2012;22:934–943. doi: 10.1016/j.nmd.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penisson-Besnier I, Saint-Andre Jp, Hicks D, et al. Myopathy caused by anoctamin 5 mutations and necrotizing vasculitis. J Neurol. 2012;259:1988–1990. doi: 10.1007/s00415-012-6502-x. [DOI] [PubMed] [Google Scholar]