

Abstract
Cancer is a disease that results from the successive accumulation of genetic and epigenetic alterations. Despite intense study, many unanswered questions about the nature of the contribution of epigenetic changes to carcinogenesis remain. In this review, we describe principles of epigenetics as they relate to our current understanding of carcinogenesis. There are a number of in vivo models of specific pathways of carcinogenesis that are very useful for the characterization of epigenetic mechanisms that link environmental exposures or genetic susceptibility and cancer progression. Because epigenetic alterations are thought to be reversible, they offer great promise for treatment of cancer. The use of animal models to evaluate the effects of decitabine and zebularine has elucidated the mechanisms of action and indicated the potential for these types of treatment. Ultimately, the greatest challenge lies in the integration of laboratory and epidemiologic data to best prevent and treat this deadly disease.
Keywords: cancer, chromatin, epigenetics, histones, mechanisms, methylation
Introduction
The field of cancer epigenetics has thrived on discoveries from in vitro, in vivo, and human clinical and epidemiologic studies. Results from these complimentary approaches have challenged the classic view of cancer, which has traditionally been hypothesized as a disease that results from the successive accumulation of genetic alterations in oncogenes and tumor-suppressor genes, which leads to uncontrolled cell growth. It is now appreciated that epigenetic alterations contribute to carcinogenesis and a mechanistic link exists between environmental and dietary exposures and disease. Despite the rapidly developing breadth of knowledge in this field, many questions remain to understand the contribution of epigenetics to the carcinogenic process. Do environmental toxicants induce epigenetic changes to influence the initiation or promotion of cancer? Can epigenetic changes be initiators in the carcinogenic process, or are they a consequence of cellular transformation and genetic alterations? Most important, because epigenetic changes are largely thought to be reversible, can epigenetic therapy offer an avenue for cancer treatment? In this review, we will describe epigenetic changes in the context of carcinogenesis and offer examples of models of cancer progression and treatment that allow for the elucidation of the role epigenetics plays in cancer progression and treatment. First, we will introduce principles of epigenetic mechanisms in light of carcinogenesis. Then we will discuss how animal models contribute to our understanding of the contribution of epigenetics to understanding distinct pathways of carcinogenesis.
DNA Methylation
One of the most extensively studied epigenetic mechanisms is the methylation of the fifth carbon of a cytosine nucleotide to create 5-methylcytosine (5mC1). The methyl group of 5mC lies in the major groove of the double helix and can interfere with transcription factor binding to prevent gene expression. Additionally, there is a class of methylated DNA-binding proteins, specifically MECP2 and the MBD family of proteins, which bind to methylated cytosines and repress gene transcription by blocking transcription factors. Cytosine pairs with guanine by means of a phosphate group, and this dinucleotide (CpG) has been a major focus of epigenetic research because of its capacity to directly silence gene expression, particularly with respect to tumor-suppressor genes in carcinogenesis. CpG sites are unevenly distributed throughout the genome, concentrating in repetitive sequences such as tandem and interspersed repeats, distal gene regulatory regions, and CpG islands (Bird 2002; Ehrlich 2009; Ehrlich et al. 1982). DNA methylation is highly dysregulated in cancer. Aberrant patterns of methylation arise, leading to hypomethylation of distal regulatory regions and repetitive elements along with hypermethylation of CpG islands (Bird 2002; Ehrlich 2009). It has been known for some time that tumors from different sites display distinct CpG methylation profiles (Esteller et al. 2001) and exhibit distinct pathways of carcinogenesis within tumor sites (Sartor et al. 2011; Shen et al. 2007). However, how CpG methylation relates to epidemiologic and clinical characteristics is not yet fully understood.
Loss of DNA methylation was one of the first epigenetic changes described in human cancer. The first study of DNA methylation in human tumor tissue, using methylation-sensitive restriction enzyme digestion paired with Southern blotting, revealed that tumor tissues had a lower proportion of methylated cytosine than normal tissues (Feinberg and Vogelstein 1983). Shortly thereafter, whole genome enzymatic digests paired with high-performance liquid chromatography were used to show that overall 5mC content was inversely associated with tumor progression (Gama-Sosa et al. 1983). Since the publication of these seminal studies, almost every type of cancer has been shown to have an overall deficiency of 5mC compared with normal tissue, occurring specifically in intergenic repetitive regions, which increases genomic instability and promotes the progression of tumorigenesis.
Repetitive Elements
Repetitive elements make up about half of the genome and are normally heavily methylated. In cancer, hypomethylation of these genomic regions make up a large percentage of 5mC loss in cancers (Ehrlich 2009; Lander et al. 2001). Centromeric tandem repeats, adjacent-centromeric (juxtacentromeric) tandem repeats, and short- (Alu) and long-interspersed elements (LINE-1) are the most frequently studied repetitive elements in cancer that are found to be hypomethylated. Tandem repeats at and near the centromere play a role in keeping the DNA packaged into heterochromatin at the point of sister chromatid association, allowing for chromosome stability. Hypomethylation of these regions can lead to chromatin decondensation and chromosome rearrangements through unstable translocations, leading to widespread genomic instability (Eden et al. 2003). For example, in vitro experiments conducted to knock out DnmtI, a DNA methyltransferase (DNMT1), in murine embryonic stem cells showed an increase in chromosomal translocations (Chen et al. 1998). Additionally, loss of heterochromatin can affect the copy number of genes involved in tumorigenesis (Eden et al. 2003; Ehrlich 2009; Kokalj-Vokac et al. 1993). Hypomethylation of tandem repeats at or near centromeric regions contributes to tumorigenesis by unraveling the structure of the genome and amplifying genomic rearrangements (Kokalj-Vokac et al. 1993). However, chromosomal abnormalities are not the only process that occurs in tumor cells, and this is signified by other repetitive elements that are found to be hypomethylated in cancer.
Alu and LINE-1 elements are retrotransposons—that is, genetic elements that have the ability to amplify themselves by means of RNA intermediates. These elements together make up about 30% of the genome (Chen et al. 1998). There are more than 500,000 LINE-1 elements in the genome, although because of truncations, mutations, and deletions, only about 100 copies are functional. There are more than 1 million copies of Alu (Batzer and Deininger 2002). Both elements contain promoter sequences, which indicates their capacity for gene transcription if unregulated (Cordaux and Batzer 2009; Kazazian and Goodier 2002). In normal tissues, LINE-1 and Alu elements are silenced through DNA methylation; these elements are hypomethylated in cancer (Bird 2002; Thayer 1993). For example, it has been shown that hypomethylation of LINE-1 elements occurs in colorectal cancer early in tumorigenesis, disrupting normal patterns of gene expression (Suter et al. 2004). Hypomethylation of LINE-1 sequences has also been shown in urothelial and hepatocellular cancers (Jurgens et al. 1996; Takai et al. 2000). Alu elements, although less studied, have been shown to be hypomethylated with LINE-1 elements in prostate adenocarcinomas (Cho et al. 2007), pancreatic endocrine tumors, and carcinoid tumors (Choi et al. 2007). Hypomethylation of LINE-1 and Alu elements was found to be strongly linked to genomic instability early in non-small-cell lung cancer, playing a potential role in the formation of lung neoplasias (Daskalos et al. 2009).
Hypomethylation of these elements and their consequent activation has many implications for tumorigenesis. They can cause insertional mutagenesis and potentially disperse processed pseudogenes, which occur when spliced messenger RNA is reverse transcribed by L1 reverse transcriptase and reinserted into the genome. Transduction can occur when LINE-1 elements mobilize their 3’ and 5’ ends separately and carry them to new genomic locations. Rearrangements also take place when Alu and LINE-1 elements insert to potentially cause deletions or inversions in the genome (Kazazian 2004; Kazazian and Goodier 2002). This results in chromosomal abnormalities, aberrant gene expression, and overall genomic instability.
Other targets of hypomethylation are the CpG sites found in promoter regions that are outside CpG islands. These are found in promoter regions of normally repressed genes and are methylated in normal cells (Bird 2002; Ehrlich 2002, 2009). In cancer cells, these regions are found to be hypomethylated, affecting repression of normally silenced genes. For example, imprinted genes are normally monoallelically expressed; however, hypomethylation of CpG sites in promoter regions of these genes leads to their biallelic expression and is linked to carcinogenesis (Holm et al. 2005). Hypomethylation of promoter regions leads to activation of otherwise silenced genes, promoting aberrant gene expression, disruption of normal cellular processes, and overall genomic instability.
DNA Methyltransferases
Although cancer genomes are globally hypomethylated, some regions of the genome are found to be hypermethylated. The mechanism through which this occurs is DNMT overexpression. DNA methylation is regulated by DNMTs that act as the methyl donors to the cytosine residue. Although five members of the DNMT family have been discovered, only DNMT1, DNMT3a, and DNMT3b are known to contribute to the global pattern of cytosine methylation (Kulis and Esteller 2010; Okano et al. 1999). DNMT1 is classified as a maintenance protein and appears to be involved in methylation of CpG sites in newly synthesized daughter DNA strands to match the methylation pattern of the parental strand. It also directly binds histone deacetylases to promote heterochromatin formation and silence gene activity (Bird 2002; Kulis and Esteller 2010; Li et al. 1992). DNMT3a and DNMT3b are classified as de novo enzymes that are essential for establishment of mammalian development methylation patterns during embryogenesis and germ-cell development (Kulis and Esteller 2010).
DNMT overexpression seems to be a common characteristic of tumors, although only DNMT1 and DNMT3a/b are implicated in tumorigenesis (Issa et al. 1993). It has been proposed that these enzymes cooperate to initiate and maintain de novo methylation in cancer cells (Rhee et al. 2002). DNMT1 and DNMT3b have been shown to form a complex with oncogenic transcription factors to induce de novo methylation of CpG islands in promoter regions (Di Croce et al. 2002). Patients with DNMT3a mutations had significantly worse prognosis in acute myeloid leukemia (Ley et al. 2010). Therefore, DNMTs in cancer have a crucial role in the hypermethylation that is found on CpG islands and its subsequent downstream effects.
The genomic regions that are targeted for hypermethylation tend to be CpG islands. Contrary to individual CpG sites throughout the genome in intergenic regions, CpG islands are usually unmethylated in normal cells, regardless of gene expression (Jones and Laird 1999). However, there are very specific instances in which CpG islands are methylated in normal cells, such as in imprinted genes and X-chromosome inactivation.
CpG Islands and Gene Expression
CpG islands occupy approximately 60% of human gene promoters, most of which are constitutively expressed genes (Vu et al. 2000). A CpG island is generally defined as a 1000-kb stretch of DNA with GC content greater than 50%. The normal hypomethylated pattern of CpG islands is found to be consistent across various types of somatic tissues despite tissue-specific differences, illustrating that DNA methylation of these islands is not used as a regulatory mechanism of gene expression (Cotton et al. 2011). Therefore, when a CpG island becomes aberrantly methylated, it can have detrimental effects by stably silencing the associated gene (Cotton et al. 2011). The cancer cell genome is characterized by hypermethylation of CpG islands in promoter regions (Edwards and Ferguson-Smith 2007; Jones and Laird 1999; Meehan et al. 1992; Riggs and Pfeifer 1992). In contrast with hypomethylation of intergenic CpG sites in cancer that lead to genomic instability, hypermethylation of CpG islands promotes the progression of tumorigenesis by silencing tumor-suppressor genes. For example, PTEN, a protein that prevents rapid proliferation, is commonly hypermethylated in brain and thyroid cancers, whereas APC, a protein involved in cell-cycle regulation, cell–cell adhesion, and cell mobility, is inactivated by hypermethylation in many lung, breast, and colorectal cancers (Fan and Zhang 2009; Hatziapostolou and Iliopoulos 2011; Illingworth and Bird 2009). Suppression of p16, a cell-cycle regulator, occurs in essentially all common human cancers (Ligget and Sidransky 1998). Inactivating these tumor suppressors directly promotes tumorigenesis due to lack of control over cellular processes.
In addition to tumor-suppressor genes, hypermethylation of other classes of genes such as DNA repair genes and transcription factors can indirectly lead to tumorigenesis through silencing of further downstream targets or accumulation of genetic errors. For example, GATA-4 and GATA-5 are transcription factors silenced in colorectal and gastric cancers (Alvarez-Nunez et al. 2006). Inactivation of DNA repair genes, such as O-6-methylguanine-DNMT, is commonly found in primary neoplasias (Esteller et al. 2000). Therefore, hypermethylation of CpG islands in cancers can affect multiple pathways to promote carcinogenesis.
Promoter hypermethylation is often an early event in tumorigenesis. Several mechanisms have been proposed for targeting CpG islands for hypermethylation. One explanation is that the location of these islands in genomic regions that have potentially undergone massive epigenetic reprogramming leads to hypermethylation as a byproduct or for prevention of error (Bird 2002). Another explanation is that some gene promoters are targeted specifically by DNMTs complexed to oncogenic transcription factors (Okano et al. 1999). Finally, it has been proposed that hypermethylation is a result of histone marks created in a tumor-specific manner (Hatziapostolou and Iliopoulos 2011).
Although it may appear that hypomethylation and hypermethylation in cancer are opposing forces, the patterns usually coexist within the same tumor, although they occur in different genomic regions. Further, the epigenetic abnormalities that occur because of hypo- and hypermethylation can interact in various ways to produce distinct subtypes of cancer. However, these patterns are stable but not irreversible and remain flexible as the cellular environment changes, contributing to the complexity of the cancer cell epigenome.
The dysregulation of DNA methylation patterns observed in cancer does not occur independent of other epigenetic changes. Methylated DNA-binding proteins, which are attracted to methylated cytosine residues and contribute in gene silencing, have been shown to interact with a number of other partners involved in epigenetic regulation. In particular, methylated DNA-binding proteins have been shown to interact with proteins that are involved in controlling the interaction between DNA and histones, the proteins involved in DNA packaging.
Chromatin Remodeling in Cancer
The estimated 1.8 linear meters of DNA in the human cell are organized into a 3-dimensional structure and compacted within the cell nucleus by means of associations with histones, the major DNA packaging proteins. These DNA–histone complexes are the primary components of chromatin, which makes up the bulk of the material in the nucleus. The basic chromatin unit is the nucleosome, which consists of a protein octamer containing pairs of each of the four core histone proteins (H2A, H2B, H3, H4). Nucleosome structures are highly conserved and repetitive throughout the genome, forming a “beads-on-a-string” structure. Nucleosomes are organized by histone protein H1, a linker protein found outside the main histone octamer complex that binds to linker DNA at the entry and exit points of the nucleosome (Allan et al. 1980).
There are two common higher levels of nucleosome organization that are defined by the level of compaction of the nucleosome structures euchromatin and heterochromatin. Euchromatin is loosely packed and typically represents transcriptionally active genic regions due to the increased accessibility of the DNA in the nucleosome structure. Heterochromatin is densely packed, with intense cytological nuclear staining due to the high density of nuclear proteins. Heterochromatin is further classified into constitutive heterochromatin, or permanently silenced chromatin, and facultative heterochromatin, which is silenced chromatin that can become reactivated in response to appropriate genetic or environmental cues. Thus, throughout an organism's lifetime, chromatin conformation is a fluid, cell type–specific process, and it is prone to restructuring in response to environmental or physiologic signals. Altered or abnormal chromatin conformation has also now been recognized as an epigenetic hallmark of many cancers.
Chromatin conformation is controlled by chemical modifications, mainly covalent modifications, of the N-terminus tails of the histone proteins that form the core of the nucleosome. Histone modifications can affect the interaction between histone proteins and DNA as well as between adjacent histone proteins. Histone modification is a dynamic process, with enzymes catalyzing the addition of covalent modifications (“writers”), their removal (“erasers”), and recognition of marks previously laid down (“readers”) (Wang et al. 2007). Dysregulation of each of these classes of enzymes has been associated with a variety of cancer types. Here, we will detail the functional consequences of aberrant control of these enzymes during the carcinogenic process for histone methylation and acetylation, the two best-characterized histone modifications.
Histone Methylation
Histone methylation has been widely shown to regulate transcription; methylation at specific histone tail residues is associated with both transcriptional activation and repression. Histone methylation occurs at both arginine and lysine residues on the tails of histone proteins H3 and H4. A summary of enzymes that modify or read histone methylation marks that have been shown to be dysregulated in cancer is shown in Table 1. Lysine methylation is catalyzed by histone-lysine-N-methyltransferases, also known as K-methyltransferases, and involves the transfer of methyl groups from the cofactor S-adenosyl methionine. A key protein involved in control of stem cell maintenance and differentiation, EZH2 (enhancer of Zeste 2), is a K-methyltransferase that catalyzes the trimethylation of H3K27 (Cao et al. 2002). EZH2 is a member of the polycomb repressive complex 2, a protein complex that involves both a K-methyltransferase and “reader” proteins that recognize H3K27me3. The H3K27me3 mark is normally involved in silencing genes related to development and stem cell differentiation, including the Hox gene cluster (Lewis 1978). In many cancers, however, EZH2 is overexpressed both at the transcriptional and protein levels. EZH2 overexpression has been described as important in prostate cancer, where an increase in EZH2 protein staining in the cell nucleus was observed with a progression from benign to metastatic disease (Varambally et al. 2002). Further studies have identified overexpression of EZH2 as a key feature in breast cancer, lymphomas, and glioblastomas, among other cancers (Kleer et al. 2003; Suvà et al. 2009; van Kemenade et al. 2001). In cancer cells, H3K27me3 has also been shown to repress gene expression independent of gene-promoter DNA methylation (Kondo et al. 2008), whereas in normal cells, EZH2 has been shown to control DNA methylation by interacting with DNMTs (Vire et al. 2006). Additionally, dysregulation of other members of the polycomb repressive complex, including proteins that interact with polycomb repressive complex 2 proteins following the transfer of the H3K27me3 mark by EZH2, have also been recently described. In contrast with the silencing histone modification H3K27me3, histone methylation can also be a mark associated with transcriptional activation. The mixed lineage leukemia (MLL) is a K-methyltransferase that catalyzes the methylation of H3K4. MLL acts in opposition to polycomb repressive complex proteins, activating genes involved in development and differentiation (Milne et al. 2002). MLL genetic events, particularly gene fusions and overamplification, have been shown to be an important characteristic of leukemia. An experimental mouse model with an MLL–AF9 gene fusion introduced by homologous recombination led to the development of acute leukemia in all chimeric mice (Corral et al. 1996). A study of acute lymphoblastic leukemia patients with MLL translocations found a unique gene expression profile when compared with patients with conventional B-precursor acute lymphoblastic leukemia (Armstrong et al. 2002). Specifically, patients with MLL translocations were found to have multilineage gene expression, aberrantly overexpressing genes associated with early-stage hematopoiesis.
Table 1.
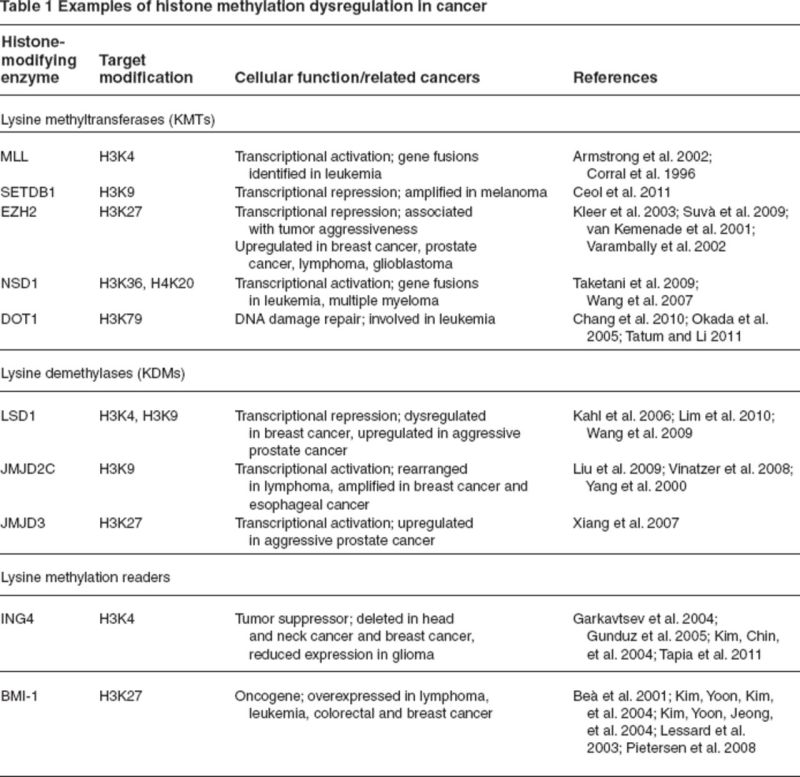
|
Histone methylation marks are removed by a variety of enzymes, with marks at specific histone tail residues interacting with distinct histone lysine demethylases, or K-demethylases. JMJD2C is a K-demethylase that catalyzes the removal of methylation marks from H3K9, a mark typically associated with gene repression (Snowden et al. 2002). Amplification of JMJD2C has been observed in a variety of cancers, including breast and esophageal cancer (Liu et al. 2009; Yang et al. 2000). Lysine specific demethylase 1, a K-demethylase that targets H3K9 and H3K4 methylation, has recently shown to be overexpressed in estrogen receptor–negative breast cancer (Lim et al. 2010), mesenchymal tumors (Schildhaus et al. 2011), and bladder cancers (Hayami et al. 2011). Although more research is necessary to fully understand the functional consequences of dysregulation of histone methylation, it is clear that K-demethylases and K-methyltransferases are important in the carcinogenic process and represent novel targets for therapy.
Histone Acetylation
Unlike histone methylation, which can be associated with transcriptional activation or repression based on the specific residue methylated, histone acetylation is strongly associated with transcriptional activation. Histone acetylation occurs on lysine residues and is thought to enhance transcription by charge neutralization of the positively charged histones, decreasing their interaction with the negatively charged DNA phosphate backbone. Maintenance of histone acetylation marks and the dynamic state of chromatin conformation are controlled by histone acetyltransferases (HATs), also known as K-acetyltransferases, and histone deacetylases (HDACs1). HATs catalyze the addition of acetyl groups to histone lysines using acetyl coenzyme A as a cofactor and induce an open or permissive chromatin state, whereas HDACs remove acetyl groups and induce a closed or repressive state (Roth et al. 2001). The normal in vivo role of HATs and HDACs is often obfuscated in cancer, leading to an abnormal chromatin phenotype.
There are three distinct families of HATs: The Gcn5 family, the p300/CBP family, and the MYST family (Lee and Workman 2007). HATs from each of these families have been shown to play a role in carcinogenesis, from either inappropriate activation or repression of target gene activity. The Wnt signaling pathway, previously shown to be commonly dysregulated in cancers, particularly those with a stem cell phenotype, has been shown to be augmented by the HAT Gcn5 in breast cancer (Chen et al. 2010). CBP (cyclic AMP response element-binding [CREB] protein) and p300, have been shown to be capable of acetylation of all four core histones as well as a number of other nonhistone proteins, including p53, Rb, E2F, and myb (Iyer et al. 2004). Loss of heterozygosity at either p300 or CBP has been detected in a large proportion of cancer cell lines examined, with 51% of cell lines experiencing loss at p300 and 35% experiencing loss at CBP (Tillinghast et al. 2003). These findings suggest that both p300 and CBP are important tumor-suppressor genes that may be lost through loss of heterozygosity in a number of different cancers. MYST family HATs have been identified as important in hematopoesis and, as such, also identified as dysregulated in acute myeloid leukemia (Yang and Ullah 2007). In the M4/M5 subset of leukemia cases, a stable and recurrent translocation t(8;16)(p11;p13) causes a fusion between MOZ, a MYST family acetyltransferase, and CBP, leading to aberrant chromatin acetylation (Borrow et al. 1996). Similarly, MOZ is found fused to p300 following a t(8;22)(p11;q13) translocation observed in a subset of acute monocytic leukemia cases (Chaffanet et al. 2000).
HDACs are enzymes that catalyze the removal of histone acetyl marks and are involved in transcriptional repression. HDACs, like HATs, also have nonhistone proteins as potential substrates and are involved in the deacetylation of a number of proteins identified as important in carcinogenesis, including p53, YY1, and STAT3 (Glozak et al. 2005). The 18 human proteins identified with HDAC activity suggest that there is likely some redundancy in function between HDACs as well as the potential for different histone tail residues or other nonhistone proteins as targets.
Studies of multistage models of carcinogenesis have identified histone deactylation as an early step in the process (Fraga et al. 2005). Specifically, early loss of monoactylation of histone H4K16 was observed in a mouse model of multistage skin carcinogenesis. Additionally, a number of cancer cell lines, as well as primary lymphomas and colorectal adenomas, were also found to be hypoacetylated compared with normal cells, suggesting that histone deacetylation is a widespread event in cancer. HDACs are often overexpressed in many different tumor types, including breast (Krusche et al. 2005), prostate (Weichert, Röske, Niesporek, et al. 2008), and colorectal cancer (Weichert, Röske, Gekeler, et al. 2008). A study of the function of HDAC3, a class 1 HDAC, in cancer cells, found that long term knockdown by means of RNA interference led to inhibition of #57442;-catenin's translocation to the nucleus (Godman et al. 2008). In addition to disrupting Wnt signaling, HDAC3 inhibition also increased expression of the vitamin D receptor, rendering those cells more sensitive to the effects of vitamin D. The common pattern of HDAC deregulation in cancer cells has provided a novel target for chemotherapeutic intervention—the HDAC inhibitor. HDAC inhibitors, both natural and synthetic, have been widely used in the treatment of a number of diseases, including psychiatric diseases and cancer. There are two HDAC inhibitors currently approved by the US Food and Drug Administration for the treatment of cutaneous T-cell lymphoma—suberoylanilide hydroxaminc acid (vorinostat) and romidepsin. Additionally, there are a number of other HDAC inhibitors under investigation in early- and late-stage clinical trials, which may provide novel epigenetic therapies for cancer treatment.
Animal Models of Carcinogenesis
Findings from in vivo models of carcinogenesis can be used to predict how the most susceptible humans in the population may respond to genetic lesions or exposure to environmental carcinogens. Additionally, these studies can identify epigenetic biomarkers and provide insight into the specific mechanisms of tumor progression.
There are a number of in vivo models of carcinogenesis that allow for the characterization of epigenetic mechanisms that link environmental exposures or genetic susceptibility and cancer progression. These models typically involve the induction of tissue-specific cancer through toxicant exposure or transgenic manipulation. A carefully designed animal model can specifically characterize molecular pathways of carcinogenesis, providing evidence for a sequential series of epigenetic and genetic effects as a malignancy progresses from carcinoma in situ to metastatic disease. Often these models are particularly useful for elucidating the contribution of epigenetic dysregulation of specific pathways in carcinogenesis in a temporal fashion. Lung cancer is an example of a cancer where epidemiologic studies have identified relevant exposures, but the early events in carcinogenesis are not well characterized (Betancourt et al. 2010; Jenkins et al. 2009). Exposure to 3-methylcholanthrene and diethylnitrosamine has been known for at least two decades to induce lung tumors in animal models (Henry et al. 1981; Schuller et al. 1988). These models have proven useful to understand the basic processes that underlie neoplastic lung adenocarcinoma initiation and progression. More recently, researchers have extended the use of these lung carcinogenesis models to understand the specific epigenetic mechanisms involved in lung cancer progression, including increases in promoter methylation of the cell-cycle regulator genes p27 and p57 (Liu et al. 2010). Epidemiologic studies have consistently identified inflammation as an important initiator and promoter of lung carcinogenesis, but human studies are limited in studying the sequential molecular events that characterize this pathway. Blanco and colleagues (2007) used a silica exposure–based inflammatory in vivo model of lung carcinogenesis to study the role of inflammation in lung carcinogenesis. They identified multiple epigenetic alterations, particularly methylation of the cell-cycle control proteins p16, APC, and Cdh13, during tumor progression that characterize lung cancers arising from this pathway. Similarly, chronic inflammation is hypothesized to drive somatic mutation and neoplastic transformation in prostate cancer. A study of prostate carcinogenesis using transgenic adenocarcinoma of mouse prostate (TRAMP) mice identified that expression of the oxidative stress–sensing enzyme Nrf2 is suppressed by DNA methylation and chromatin silencing in prostate cancer (Yu et al. 2010). There are also a number of in vivo models of carcinogenesis that focus on the progression of hematological malignancies, such as leukemias and lymphomas. A mouse model of acute lymphoblastic leukemia, Il15 transgenic FVB/NJ mice, was used to identify epigenetic alterations specific to leukemia progression, finding that the putative tumor-suppressor gene Idb4 was epigenetically silenced in both mouse and human leukemias but not in solid tumors (Yu et al. 2005). Epigenetic alterations, particularly the DNA methylation silencing of tumor-suppressor genes Pten and p53, after overexpression of the oncogene MYC were found using a bitransgenic mouse model of T cell lymphoma (Opavsky et al. 2007).
The elucidation of epigenetic mechanisms through animal models has given rise to therapeutic interventions in the treatment of carcinogenesis. Because methylation of tumor-suppressor genes is a common characteristic of tumorigenesis, demethylating agents such as 5-azacytidine, decitabine, and zebularine have been studied in various animal models. Mice induced to develop oral cavity carcinogenesis were treated with 5-azacytidine and exhibited reduced lesions compared with untreated mice (Tang et al. 2009). Zebularine administered to BALB/c nu/nu mice with human bladder carcinoma xenografts significantly reduced tumor size through demethylation of the p16 promoter (Cheng et al. 2003). Aside from methylation, histone acetylation also occurs early in carcinogenesis, and as such, HDAC inhibitors have been considered as treatment options. Bachmann and colleagues (2010) used the acute lymphoblastic leukemia xenograft SCID mouse model to find that vorinostat, an HDAC inhibitor, reinstated gene expression of BIM, a tumor suppressor, which is silenced in lymphoid malignancies. Valproic acid, another HDAC inhibitor, was found to induce histone hyperacetylation and inhibit angiogenesis, resulting in prolonged survival of orthotopic xenograft mouse models of medulloblastoma (Zhang et al. 2011). Although therapeutic effects have been ascertained from administering demethylating agents and HDAC inhibitors alone, combination treatments of epigenetic therapies with either chemotherapeutic drugs or each other have been found to be most effective in stimulating synergistic antitumor activity (Fang et al. 2010; Gagnon et al. 2003; Gollob et al. 2006; Lemaire et al. 2009; Plumb et al. 2000; Venturelli et al. 2007). Drug-resistant ovarian and colon tumor xenograft mouse models treated with decitabine increased hMLH1 expression through promoter demethylation. Although this did not affect tumor growth, the treatment sensitized the xenografts to cisplatin and other chemotherapeutic drugs, increasing their efficacy (Plumb et al. 2000). Xenograft hepatoma models were used to examine the effects of azacytidine and an HDAC inhibitor when administered alone or in combination. It was found that the combination therapy generated significant antitumor effects compared with each agent administered alone (Venturelli et al. 2007). The potential of combination therapy with epigenetic agents has been established through the use of in vivo models, and many demethylating agents and HDAC inhibitors are currently in or have completed clinical trials for human use (Amatori et al. 2010; Fang et al. 2010; Gollob et al. 2006).
Conclusions
The greatest challenge to cancer researchers is the integration of human and animal data to realize the translational potential of findings. Although epidemiologic studies have identified dietary and environmental factors as associated with risk of cancer, animal models can identify the mechanisms and temporal relationship between these factors and carcinogenesis. Translational research that integrates results from human and animal studies will provide insight into the temporal nature of epigenetic dysregulation during tumor initiation and progression and into how environmental and dietary exposures influence the epigenetic phenotype of a tumor. Additionally, these studies will determine which cancer subtypes are susceptible to chemotherapy that influences epigenetic state, including DNA demethylating agents, HDAC inhibitors, or a number of the other promising epigenetic therapies currently in preclinical and clinical trials. Molecular characterization of tumor progression, including genetic and epigenetic profiles, is a key step in the development of individualized treatment modalities and personalized cancer therapy.
Acknowledgments
Support for this work was provided by the National Cancer Institute (R01CA158286). Support for S. Virani was provided by the National Institute on Aging Institutional Training Grant, Interdisciplinary Studies in Public Health and Aging (T32 AG027708). Support for J.A. Colacino was provided by institutional training grants from the National Institute of Environmental Health Sciences (T32 ES007062) and the National Human Genome Research Institute (T32 HG00040).
Biography
Shama Virani, BS, is a graduate student; Justin A. Colacino, MPH, is a graduate student; Jung H. Kim, PhD, is a postdoctoral fellow; and Laura S. Rozek, PhD, is an assistant professor, all at the Department of Environmental Health Sciences, University of Michigan School of Public Health, Ann Arbor.
Footnotes
Abbreviations that appear ≥3x throughout this article: 5mC, 5-methylcytonsine; DNMT, DNA methyltransferase; HDAC, histone deacetylase.
References
- Allan J, Hartman PG, Crane-Robinson C, Aviles FX. 1980. The structure of histone H1 and its location in chromatin. Nature 288:675–679 [DOI] [PubMed] [Google Scholar]
- Alvarez-Nunez F, Bussaglia E, Mauricio D, Ybarra H, Vilar M, Lerma E, de Leiva A, Matias-Guiu X. 2006. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 16:17–23 [DOI] [PubMed] [Google Scholar]
- Amatori S, Bagaloni I, Donati B, Fanelli M. 2010. DNA demethylating antineoplastic strategies: A comparative point of view. Genes Cancer 1:197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. 2002. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nature Genetics 30:41–47 [DOI] [PubMed] [Google Scholar]
- Bachmann PS, Piazza RG, Janes MN, Wong NC, Davies C, Mogavero A, Bhadri VA, Szymanska B, Geninson G, Magistroni V, Cazzaniga G, Biondi A, Miranda-Saavedra D, Gottgens B, Saffery R, Craig JM, Marshall GM, Gambacorti-Passerini C, Pimanda JE, Lock RB. 2010. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia and its reversal by histone deacetylase inhibition. Blood 116:3013–3022 [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3(5):370–379 [DOI] [PubMed] [Google Scholar]
- Beà S Tort F, Pinyol M, Puig X, Hernández L, Hernández S, Fernández PL, van Lohuizen M, Colomer D, Campo E. 2001. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res 61:2409–2412 [PubMed] [Google Scholar]
- Betancourt AM, Eltoum IA, Desmond RA, Russo J, Lamartiniere CA. 2010. In utero exposure to bisphenol A shifts the window of susceptibility for mammary carcinogenesis in the rat. Environ Health Perspect 118:1614–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21 [DOI] [PubMed] [Google Scholar]
- Blanco DS, Vicent S, Fraga MF, Fernandez-Garcia I, Freire J, Lujambio A, Esteller M, Ortiz-de-Solorzano C, Pio R, Lecanda F, Montuenga LM. 2007. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia 9:840–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow JU, Stanton VP, Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM, Horsman D, Mitelman F, Volinia S, Watmore AE, Housman DE. 1996. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet 14:33–41 [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043 [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F. 2011. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471:513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffanet M, Gressin L, Preduhomme C, Soenen Cornu V, Birnbaum D, Pébusque MJ. 2000. MOZ is fused to p300 in an acute monocytic leukemia with t (8; 22). Genes Chromosomes Cancer 28:138–144 [DOI] [PubMed] [Google Scholar]
- Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, Hemenway CS, Zhang W. 2010. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res 70:10234–10242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Luo Z, Yuan Y, Huang X, Cai W, Li C, Wei T, Zhang L, Yang M, Liu Q, Ye G, Dai X, Li B. 2010. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol Cell Biol 30:5621–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. 1998. DNA hypomethylation leads to elevated mutation rates. Nature 395:89–93 [DOI] [PubMed] [Google Scholar]
- Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. 2003. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 95:399–409 [DOI] [PubMed] [Google Scholar]
- Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, Kim D, Kang GH. 2007. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 211:269–277 [DOI] [PubMed] [Google Scholar]
- Choi IS, Estecio MR, Nagano Y, Kim do H, JA White, Yao JC, Issa JP, Rashid A. 2007. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol 20:802–810 [DOI] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie AN, King G, Rabbitts TH. 1996. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 85:853–861 [DOI] [PubMed] [Google Scholar]
- Cotton AM, Lam L, Affleck JG, Wilson IM, Penaherrera MS, McFadden DE, Kobor MS, Lam WL, Robinson WP, Brown CJ. 2011. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum Genet 130: 187–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalos A, Nikolaidis G, Xinarianos G, Savvari P, Cassidy A, Zakopoulou R, Kotsinas A, Gorgoulis V, Field JK, Liloglou T. 2009. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer 124:81–87 [DOI] [PubMed] [Google Scholar]
- Di Crose L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S, Pelicci PG. 2002. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295: 1079–1082 [DOI] [PubMed] [Google Scholar]
- Eden A, Gaudet F, Waghmare A, Jaenisch R. 2003. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455. [DOI] [PubMed] [Google Scholar]
- Edwards CA, Ferguson-Smith AC. 2007. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol 19:281–289 [DOI] [PubMed] [Google Scholar]
- Ehrlich M. 2002. DNA methylation in cancer: Too much but also too little. Oncogene 21:5400–5413 [DOI] [PubMed] [Google Scholar]
- Ehrlich M. 2009. DNA hypomethylation in cancer cells. Epigenomics 1:239–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. 1982. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10:2709–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, Herman JG. 2001. A gene hypermethylation profile of human cancer. Cancer Res 61:3225–3229 [PubMed] [Google Scholar]
- Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG. 2000. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res 60:2368–2371 [PubMed] [Google Scholar]
- Fan S, Zhang X. 2009. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem Biophys Res Commun 383:421–425 [DOI] [PubMed] [Google Scholar]
- Fang F, Balch C, Schilder J, Breen T, Zhang S, Shen C, Li L, Kulesavage C, Snyder AJ, Nephew KP, Matei DE. 2010. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent platinum-resistant, epithelial ovarian cancer. Cancer 116:4043–4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132:6–13 [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400 [DOI] [PubMed] [Google Scholar]
- Gagnon J, Shaker S, Primeau M, Hurtubise A, Momparler RL. 2003. Interaction of 5-aza-2’-deoxycytidine and depsipeptide on antineoplastic activity and activation of 14-3-3sigma, E-cadherin and tissue inhibitor of metalloproteinase 3 expression in human breast carcinoma cells. Anticancer Drugs 14:193–202 [DOI] [PubMed] [Google Scholar]
- Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. 1983. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11:6883–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH, Jain RK. 2004. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 428:328–332 [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. 2005. Acetylation and deacetylation of non-histone proteins. Gene 363:15–23 [DOI] [PubMed] [Google Scholar]
- Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang H, Shin DG, Rosenberg DW, Giardina C. 2008. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol Ther 7:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollob JA, Sciambi J, Peterson BL, Richmond T, Thoreson M, Moran K, Dressman HK, Jelinek J, Issa JP. 2006. Phase I trial of sequential low-dose 5-aza-2’-deoxycytidine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res 12:4619–4627 [DOI] [PubMed] [Google Scholar]
- Gunduz M, Nagatsuka H, Demircan K, Gunduz E, Cengiz B, Ouchida M, Tsujigiwa H, Yamachika E, Fukushima K, Beder L. 2005. Frequent deletion and down-regulation of ING4, a candidate tumor suppressor gene at 12p13, in head and neck squamous cell carcinomas. Gene 356:109–117 [DOI] [PubMed] [Google Scholar]
- Hatziapostolou M, Iliopoulos D. 2011. Epigenetic aberrations during oncogenesis. Cell Mol Life Sci 68:1681–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. 2011. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 128:574–586 [DOI] [PubMed] [Google Scholar]
- Henry C, Billups L, Avery M, Rude T, Dansie D, Lopez A, Sass B, Whitmire C, Kouri R. 1981. Lung cancer model system using 3-methylcholanthrene in inbred strains of mice. Cancer Res 41:5027. [PubMed] [Google Scholar]
- Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM, 3rd, Jaenisch R. 2005. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 8:275–285 [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP. 2009. CpG islands—“A rough guide.” FEBS Lett 583:1713–1720 [DOI] [PubMed] [Google Scholar]
- Issa JP, Vertino PM, Wu J, Sazawal S, Celano P, Nelkin BD, Hamilton SR, Baylin SB. 1993. Increased cytosine DNA-methyltransferase activity during colon cancer progression. J Natl Cancer Inst 85:1235–1240 [DOI] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. 2004. p300/CBP and cancer. Oncogene 23:4225–4231 [DOI] [PubMed] [Google Scholar]
- Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. 2009. Oral exposure to bisphenol a increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect 117:910–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Laird PW. 1999. Cancer epigenetics comes of age. Nat Genet 21:163–167 [DOI] [PubMed] [Google Scholar]
- Jurgens B, Schmitz-Drager BJ, Schulz WA. 1996. Hypomethylation of L1 LINE sequences prevailing in human urothelial carcinoma. Cancer Res 56:5698–5703 [PubMed] [Google Scholar]
- Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E. 2006. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res 66:11341–11347 [DOI] [PubMed] [Google Scholar]
- Kazazian HH., Jr 2004. Mobile elements: Drivers of genome evolution. Science 303:1626–1632 [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Goodier JL. 2002. LINE drive.Retrotransposition and genome instability. Cell 110:277–280 [DOI] [PubMed] [Google Scholar]
- Kim JH, Yoon SY, Jeong SH, Kim SY, Moon SK, Joo JH, Lee Y, Choe IS, Kim JW. 2004. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast 13:383–388 [DOI] [PubMed] [Google Scholar]
- Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK, Choe IS, Choe YK, Kim JW. 2004. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett 203:217–224 [DOI] [PubMed] [Google Scholar]
- Kim S, Chin K, Gray JW, Bishop JM. 2004. A screen for genes that suppress loss of contact inhibition: identification of ING4 as a candidate tumor suppressor gene in human cancer. Proc Natl Acad Sci U S A 101:16251–16256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CC, Cao Q, Varambally S, Shen R, Ota I, SA Tomlins, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 100:11606–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokalj-Vokac N, Almeida A, Viegas-Pequignot E, Jeanpierre M, Malfoy B, Dutrillaux B. 1993. Specific induction of uncoiling and recombination by azacytidine in classical satellite-containing constitutive heterochromatin. Cytogenet Cell Genet 63:11–15 [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, Gold DL, Sekido Y, Huang TH, Issa JP. 2008. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40:741–750 [DOI] [PubMed] [Google Scholar]
- Krusche CA, Wülfing P, Kersting C, Vloet A, Böcker W, Kiesel L, Beier HM, Alfer J. 2005. Histone deacetylase-1 and-3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res Treat 90:15–23 [DOI] [PubMed] [Google Scholar]
- Kulis M, Esteller M. 2010. DNA methylation and cancer. Adv Genet 70:27–56 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fultron RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Eberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itho T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel MA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu K, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins MS, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: One size doesn’t fit all. Nat Rev Mol Cell Biol 8:284–295 [DOI] [PubMed] [Google Scholar]
- Lemaire M, Momparler LF, Raynal NJ, Bernstein ML, Momparler RL. 2009. Inhibition of cytidine deaminase by zebularine enhances the antineoplastic action of 5-aza-2’-deoxycytidine. Cancer Chemother Pharmacol 63:411–416 [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423:255–260 [DOI] [PubMed] [Google Scholar]
- Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276:565–570 [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. 2010. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363:2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926 [DOI] [PubMed] [Google Scholar]
- Liggett WH, Sidransky D. 1998. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol 16:1197–1206 [DOI] [PubMed] [Google Scholar]
- Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. 2010. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31512–520 [DOI] [PubMed] [Google Scholar]
- Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang ZQ. 2009. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28:4491–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WB, Liu JY, Ao L, Zhou ZY, Zhou YH, Cui ZH, Coa J. 2010. Epigenetic silencing of cell cycle regulatory genes during 3-methylcholanthrene and diethylnitrosamine-induced multistep rat lung cancer. Mol Carcinog 49:556–565 [DOI] [PubMed] [Google Scholar]
- Meehan R, Lewis J, Cross S, Nan X, Jeppesen P, Bird A. 1992. Transcriptional repression by methylation of CpG. J Cell Sci Suppl 16:9–14 [DOI] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. 2002. MLL targets SET domain methyltransferase activity to Hox gene promoters. Molecular Cell 10:1107–1117 [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield V, Su L, Xu G, Zhang Y. 2005. hDOT1L links histone methylation to leukemogenesis. Cell 121:167. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257 [DOI] [PubMed] [Google Scholar]
- Opavsky R, Wang SH, Trikha P, Raval A, Huang Y, Wu YZ, Rodriguez B, Keller B, Liyanarachchi S, Wei G, Davuluri RV, Weinstein M, Felsher D, Ostrowski M, Leone G, Plass C. 2007. CpG island methylation in a mouse model of lymphoma is driven by the genetic configuration of tumor cells. PLoS Genet 3:1757–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietersen AM, Horlings HM, Hauptmann M, Langerod A, Ajouaou A, Cornelissen-Steijger P, Wessels LF, Jonkers J, van de Vijver MJ, van Lohuizen M. 2008. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res 10:R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. 2000. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res 60: 6039–6044 [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. 2002. RDNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416:552–556 [DOI] [PubMed] [Google Scholar]
- Riggs AD, Pfeifer GP. 1992. X-chromosome inactivation and cell memory. Trends Genet 8:169–174 [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. 2001. Histone acetyltransferases. Annu Rev Biochem 70:81–120 [DOI] [PubMed] [Google Scholar]
- Sartor MA, Dolinoy DC, Jones TR, Colacino JA, Prince MD, Carey TE, Rozek LS. 2011. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 6:777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildhaus HU, Riegel R, Hartmann W, Steiner S, Wardelmann E, Merkelbach-Bruse S, Tanaka S, Sonobe H, Schule R, Buettner R, Kirfel J. 2011. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors synovial sarcomas rhabdomyosarcomas, desmoplastic small round cell tumors and malignant peripheral nerve sheath tumors. Hum Pathol 42:1667–75 [DOI] [PubMed] [Google Scholar]
- Schuller HM, Becker KL, Witschi HP. 1988. An animal model for neuroendocrine lung cancer. Carcinogenesis 9:293–296 [DOI] [PubMed] [Google Scholar]
- Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. 2007. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A 104:18654–18659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden AW, Gregory PD, Case CC, Pabo CO. 2002. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol 12:2159–2166 [DOI] [PubMed] [Google Scholar]
- Suter CM, Martin DI, Ward RL. 2004. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis 19:95–101 [DOI] [PubMed] [Google Scholar]
- Suvà ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino D, Cironi L. 2009. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 69:9211. [DOI] [PubMed] [Google Scholar]
- Takai D, Yagi Y, Habib N, Sugimura T, Ushijima T. 2000. Hypomethylation of LINE1 retrotransposon in human hepatocellular carcinomas but not in surrounding liver cirrhosis. Jpn J Clin Oncol 30:306–309 [DOI] [PubMed] [Google Scholar]
- Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. 2009. NUP98-NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet 190:108–112 [DOI] [PubMed] [Google Scholar]
- Tang XH, Albert M, Scognamiglio T, Gudas LJ. 2009. A DNA methyltransferase inhibitor and all-trans retinoic acid reduce oral cavity carcinogenesis induced by the carcinogen 4-nitroquinoline 1-oxide. Cancer Prev Res (Phila) 2:1100–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia C, Zlobec I, Schneider S, Kilic E, Güth U, Bubendorf L, Kim S. 2011. Deletion of the inhibitor of growth 4 (ING4) tumor suppressor gene is prevalent in human epidermal growth factor 2 (HER2)-positive breast cancer. Hum Pathol 42:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatum D, Li S. 2011. Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J Biol Chem 286:17530–17535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer RE, Singer MF, Fanning TG. 1993. Undermethylation of specific LINE-1 sequences in human cells producing a LINE-1-encoded protein. Gene 133:273–277 [DOI] [PubMed] [Google Scholar]
- Tillinghast GW, Partee J, Albert P, Kelley JM, Burtow KH, Kelly K. 2003. Analysis of genetic stability at the EP300 and CREBBP loci in a panel of cancer cell lines. Genes Chromosomes Cancer 37:121–131 [DOI] [PubMed] [Google Scholar]
- van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. 2001. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood 97:3896–3901 [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629 [DOI] [PubMed] [Google Scholar]
- Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, Wehrmann M, Gregor M, Lauer UM, Bitzer M. 2007. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer 109:2132–2141 [DOI] [PubMed] [Google Scholar]
- Vinatzer U, Gollinger M, Mullauer L, Raderer M, Chott A, Streubel B. 2008. Mucosa-associated lymphoid tissue lymphoma: novel translocations including rearrangements of ODZ2, JMJD2C, and CNN3. Clin Cancer Res 14:6426–6431 [DOI] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. 2006. The polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874 [DOI] [PubMed] [Google Scholar]
- Vu TH, Li T, Nguyen D, Nguyen BT, Yao XM, Hu JF, Hoffman AR. 2000. Symmetric and asymmetric DNA methylation in the human IGF2-H19 imprinted region. Genomics 64:132–143 [DOI] [PubMed] [Google Scholar]
- Wang GG, Allis CD, Chi P. 2007. Chromatin remodeling and cancer part I: Covalent histone modifications. Trends Mol Med 13:363–372 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L. 2009. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138:660–672 [DOI] [PubMed] [Google Scholar]
- Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche F, Niesporek S, Denkert C, Dietel M. 2008. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Brit J Cancer 98:604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert W, Röske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. 2008. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res 14:1669. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. 2007. JMJD3 is a histone H3K27 demethylase. Cell Research 17:850–857 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ullah M. 2007. MOZ and MORF two large MYSTic HATs in normal and cancer stem cells. Oncogene 26:5408–5419 [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Imoto Y, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. 2000. Identification of a novel gene GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res 60:4735–4739 [PubMed] [Google Scholar]
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, Raval A, Liu TH, Ding W, Mao C, Liu S, Smith LT, Lee S, Rasssenti L, Marcucci G, Byrd J, Caligiuri MA, Plass C. 2005. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet 37:265–274 [DOI] [PubMed] [Google Scholar]
- Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, Foster BA, Kan YW, Kong AN. 2010. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One 5:e8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang G, Wang L, Song C, Wang X, Kang J. 2011. Valproic acid inhibits prostate cancer cell migration by up-regulating E-cadherin expression. Pharmazie 66:614–618 [PubMed] [Google Scholar]