

Abstract
Osteoarthritis (OA) is the most common chronic disease of human joints. The basis of pathologic changes involves all the tissues forming the joint; already, at an early stage, it has the nature of inflammation with varying degrees of severity. An analysis of the complex relationships indicates that the processes taking place inside the joint are not merely a set that (seemingly) only includes catabolic effects. Apart from them, anti-inflammatory anabolic processes also occur continually. These phenomena are driven by various mediators, of which the key role is attributed to the interactions within the cytokine network. The most important group controlling the disease seems to be inflammatory cytokines, including IL-1β, TNFα, IL-6, IL-15, IL-17, and IL-18. The second group with antagonistic effect is formed by cytokines known as anti-inflammatory cytokines such as IL-4, IL-10, and IL-13. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of OA with respect to inter- and intracellular signaling pathways is still under investigation. This paper summarizes the current state of knowledge. The cytokine network in OA is put in the context of cells involved in this degenerative joint disease. The possibilities for further implementation of new therapeutic strategies in OA are also pointed.
1. Introduction
The most common chronic and currently regarded as potentially irreversible disease that affects the joints is osteoarthritis (OA) [1, 2]. The constantly growing number of causes for the development of the disease includes, for example, genetic predisposition, aging, obesity, trauma, and other systemic diseases (Figure 1). Although we are dealing with a diverse aetiology, which is most often the result of a number of overlapping factors, processes occur during the development of the pathomechanisms of disease at the tissue, cellular and ultrastructural level that gradually take a similar nature, resulting in the phenotypic image of OA. According to the latest medical knowledge, the participation of the immune system in the development and progression of OA is one of the key elements in the pathogenesis of the disease [3]. Currently, many independent authors are focused on identifying and describing the factors responsible for the development of inflammatory processes involved in OA [4]. An analysis of the ever-increasing number of reports directs attention to the special role of the cytokine network in the pathogenesis of OA. During the progression of OA, the production and operation of various cytokines can vary depending on the duration and severity of the disease [5]. The most important effect that cytokines have involves disturbing the catabolism and anabolism processes, particularly important in tissues that are often subject to high mechanical load, such as human joints [6]. As a result of disrupting the said balance, there is a progressive degeneration of articular cartilage performing a key role in the biomechanics of each joint and other components of the joint, which results in the development of a difficult-to-interrupt disease process that involves both inflammatory, degradation, and production processes, which together lead to a gradual loss of joint function and pain. Due to the effect of cytokines in the context of an inflammatory disease such as OA, they can be divided into inflammatory and anti-inflammatory [7]. It should be noted that the pathophysiological processes occurring in the joint affected by OA are largely mediated by inflammatory cytokines and other mediators intensifying catabolic effects. At the same time, one should not underestimate the role of anti-inflammatory cytokines that may modulate an inflammatory response and act protectively on joint tissue.
Figure 1.
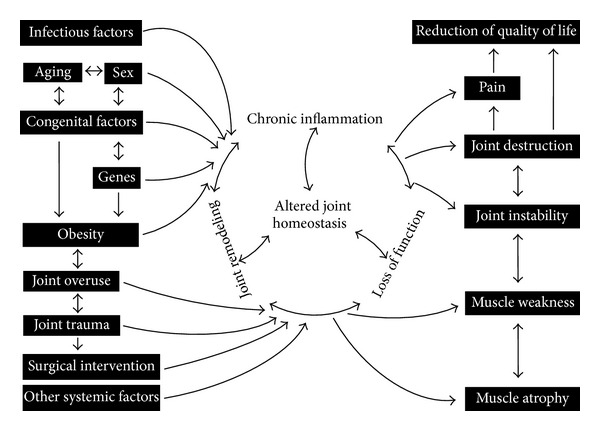
Schematic of a closed disease circle comprising the disease progression of osteoarthritis taking into account its causes and consequences.
Although we are not able to prevent some of the causes of OA (genetic factors, aging, and trauma), we can try to alter the course of disease at the level of cellular communication through a better understanding of the sensitive dependences in the network of cytokines. This opens the possibility of using suitable targeted treatment with the use of antibodies and other recombinant biological factors that interrupt the self-perpetuating processes of the closed illness process [8]. This review aims to provide accurate and current knowledge on the role of inflammatory and anti-inflammatory cytokines in the pathogenesis of OA. It is worth noting that the role of anti-inflammatory cytokines in the context of OA has not been studied comprehensively so far.
2. Inflammatory Cytokines
The group of inflammatory cytokines is the most important group of compounds participating in the pathogenesis of OA. Their participation is by far the most widely and accurately documented in literature. They are responsible to the greatest extent for the loss of metabolic homeostasis of tissues forming joints by promoting catabolic and destructive processes. The key role they play in the pathogenesis of OA is a result of the effect these compounds have on the majority of cells that are in the joint and the impact via intracellular pathways of signal transduction on the production of cytokines as well as other inflammatory compounds and enzymes. Among the many representatives of this group, the greatest importance is attributed to IL-1β, TNFα, IL-6, IL-15, IL-17, and IL-18.
2.1. Interleukin-1 Beta (IL-1β)
Interleukin-1 beta (IL-1β) is considered one of the key cytokines involved in the pathogenesis of OA; it induces inflammatory reactions and catabolic effect independently as well as being combined with other mediators with respect to the articular cartilage and other elements of joints. It is one of the 11 representatives of the IL-1 family (IL-1F) [9] (Figure 2). It is originally formed as a cytosolic precursor protein (pro-IL-1β) consisting of 269 amino acid residues [9, 10]. The preparation of the active form of IL-1β with 153 amino acid residues is the result of intracellular proteolysis carried out by the enzyme Caspase 1 (IL-1β converting enzyme, ICE), followed by the release into the extracellular space [11]. Its synthesis in the joint is governed by chondrocytes, osteoblasts, cells forming the synovial membrane, and mononuclear cells that were previously present in the joint or infiltrated its structure during the inflammatory response [4, 12–16]. Patients with OA have an elevated level of IL-1β in both the synovial fluid, synovial membrane, cartilage, and the subchondral bone layer [12, 13, 15, 17]. The biological activation of cells by IL-1β is mediated by interaction with the membrane receptor, namely, the IL-1R1 (IL-1RI, CD121a), which can also bind IL-1α and IL-1Ra [18]. Another receptor capable of binding IL-1β is IL-1R2 (IL-1RII, CD121b), which after binding a ligand, such as the IL-1α, IL-1β, or IL-1Ra, forms an inactive ligand-receptor complex, showing no ability to communicate and activate the intracellular signal [19]. At this point it is worth mentioning the presence of the receptor antagonist IL-1Ra produced by certain cells of the joint, which (as noted above) can bond with IL-1R1 and IL-1R2, thereby blocking their connection with IL-1β, which may to some extent affect the decrease in the activity of IL-1β [20, 21]. The expression of the IL-1R1 receptor is increased in patients with OA on the surface of chondrocytes and fibroblast-like synoviocytes (FLS) compared to treatment groups [22, 23]. Binding the IL-1β to a receptor of the TLR family, such as IL-1R1, is followed by recruitment of additional IL-1R3 chain (IL-1RAcP), thereby forming a complex which, through its intracellular domain Toll-IL-1R (TIR), recruits the adapter protein MyD88 [24]. The entire previously described complex binds serine-threonine kinases of the IRAK group, which affect the TRAF6 protein, which induces further binding of TAK1, TAB1, and TAB2 [25]. TAK1 affects the phosphorylation of the IκB kinase complex (IKK), thereby activating the transcription factor NF-κB [25]. Moreover, p38MAPK and c-Jun N-terminal kinase (JNK) are also activated [24, 25]. Activation of these transcription factors very quickly results in the expression of hundreds of genes whose products include other cytokines, chemokines, adhesion molecules, inflammatory mediators, and enzymes [26]. The effect of IL-1β is manifested by its significant effect on the metabolism of cells and the extracellular matrix (ECM) [27]. In the course of the disease, the gradual loss of articular cartilage is of paramount importance. Many studies confirm that the effect of IL-1β blocks chondrocytes in the context of the synthesis of ECM components, interfering with the synthesis of the key structural proteins such as type-II collagen and aggrecan [28, 29]. In addition to the decrease in the synthesis of the building blocks, the IL-1β affects the operation of chondrocytes in the synthesis of enzymes from the group of metalloproteinases (MMPs), mainly interstitial collagenase (MMP-1), stromelysin-1 (MMP-3), and collagenase 3 (MMP-13), which have a destructive effect on cartilage components [30–32]. Besides the induction of enzymes of the MMPs family, IL-1β affects the chondrocytes' production of ADAMTS metalloproteinases, which are responsible for the proteolysis of aggrecan molecules [33]. A major role is attributed to ADAMTS-4, whose production is stimulated by both IL-1β and TNFα, while ADAMTS-5 has no correlation with the influence of cytokines and is produced constitutively [33, 34]. Chondrocytes subjected to the effect of IL-1β and TNFα also tend to age more rapidly and to induce apoptosis [35–37]. When analyzing the above information, one can observe the manifold effect of IL-1β on cartilage by inhibiting its restoration possibility, intensifying its deterioration by enzymes and a direct adverse effect on chondrocytes. In the cells of the joint, IL-1β is able to induce its own secretion in an autocrine manner to stimulate the synthesis of other cytokines such as, for example, TNFα, IL-6, IL-8, and CCL5 chemokine [38–42]. IL-1β has been shown to inhibit the signal pathway of the receptor-regulated SMADs (R-SMAD), crucial to the activation of transcription factors associated with TGF-β [43]. This is done by increasing the expression of the inhibitor protein SMAD7 and inhibition of synthesis of the TGF-β type II receptor in chondrocytes. In addition, effects are observed on the secretion of a number of other enzymes and mediators involved in the pathophysiology of OA. These compounds may include the iNOS generating NO, phospholipase A2 (PLA2), cyclooxygenase-2 (COX-2), prostaglandin E synthase 2 (PGE2 synthase) producing prostaglandin E2 (PGE2) [44–46]. During the course of the disease, IL-1β stimulates the production of reactive oxygen species (ROS), which generate the formation of, for example, peroxides and hydroxylated radicals, which directly damage the articular cartilage; the intensification of this process is also associated with decreased expression of oxidative enzymes, which is observed in the joint affected by the disease [47].
Figure 2.
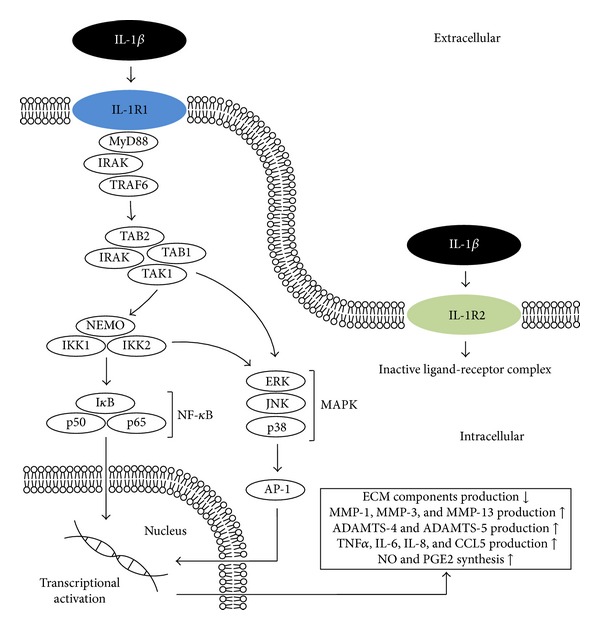
IL-1β associated intracellular signaling pathways and downstream cellular targets and effects. IL-1R1: interleukin-1 receptor, type 1; IL-1R2: interleukin-1 receptor, type 2; MyD88: myeloid differentiation primary response gene (88); IRAK: interleukin-1 receptor-associated kinase; TRAF6: TNF receptor-associated factor 6; TAK1: also known as mitogen-activated protein kinase kinase kinase 7 (MAP3K7); TAB1: also known as mitogen-activated protein kinase kinase kinase 7 interacting protein 1 (MAP3K7IP1); TAB2: also known as mitogen-activated protein kinase kinase kinase 7 interacting protein 2 (MAP3K7IP2); p50, p65: subunits of proteins forming NF-κB; IκB: (inhibitor of κB) an endogenous complex of proteins inhibiting the activation of NF-κB; IKK1,2/NEMO: NF-κB inhibitor kinase 1,2 (IκB kinase 1,2)/NF-κB kinase inhibitor (NF-κB essential modulator); ERK: extracellular-signal-regulated kinase; JNK: c-Jun N-terminal kinase; p38: p38 mitogen-activated protein kinases; MAPK: mitogen-activated protein kinases; AP-1: activator protein 1.
2.2. Tumor Necrosis Factor Alpha (TNFα)
Tumor necrosis factor alpha (TNFα), together with IL-1β, is considered a key inflammatory cytokine involved in the pathophysiological processes occurring in the course of OA (Figure 3). It is one of the 19 ligands within the tumour necrosis factors superfamily (TNF superfamily) [48]. It is originally formed as a homotrimeric transmembrane protein type II (mTNFα), which can be secreted into the environment in the next stage involving the TACE/ADAM17 metalloproteinase, creating a free form of TNFα (sTNFα) [49–51]. TNFα is secreted by the same cells in the joint that synthesize IL-1β, and its increased concentration is also observed in the same elements, such as synovial fluid, synovial membrane, cartilage, and subchondral bone layer, where increased levels of IL-1β are also detected [12, 13, 15, 17]. The cytokine has the ability to bind to the two isotypes of membrane receptors located on the surface of almost every nucleated cell TNF-R1 (p55, CD120a, and TNFRSF1a) and TNF-R2 (p75, CD120b, and TNFRSF1b) [52, 53]. The TNF-R1 receptor can be efficiently activated by the soluble and membrane forms, while TNF-R2 mainly binds the membrane form [53]. So far the participation of TNF-R1 seems to have a greater impact on the local loss of articular cartilage than TNF-R2; this, however, does not change the fact that both receptors are involved in signal transduction after being activated by TNFα in the processes occurring in OA [54, 55]. The expression of TNF-R1 is also increased within the FLS cells [56, 57]. As mentioned above, both receptors differ in their affinity to the TNFα and also in their amino acid composition, degree of glycosylation, and structure, primarily in their intracellular part, which within the TNF-R1 includes the so-called death domain (DD), which does not occur in the TNF-R2 receptor [51, 58]. In view of the significant lack of a homology in the intracellular portion, both receptors have the ability to transmit different signals. TNF-R1 is able to recruit two different TNF-R1 signalling complexes (TNF-RSC) [59, 60]. Complex I is mainly involved in the activation of pathways whose end products stimulate an inflammatory response, especially the production and secretion of cytokines and the production of proteins that prevent apoptosis, whereas the main function of complex II involves signal transduction leading to cell disintegration [60, 61]. The association of TNFα with TNF-R1 causes interaction between the TRADD adapter protein with the DD domain and gradual binding of other adapter proteins, such as TRAF2, c-IAP1, c-IAP2, and RIP1 [62–64]. The creation of the complex is followed by ubiquitination of the RIP1 protein, which also binds TAK1, TAB1, and TAB2, resulting in the phosphorylation of the IKK complex and ultimately, the activation of one of the most important transcription pathways in the course of OA—NF-κB [65, 66]. Furthermore, during the formation and effect of complex I, the other important signalling pathway is also activated by JNK kinase, as well as others such as extracellular-regulated kinase (ERK) pathway and p38MAPK [60, 67, 68]. The formation of complex II is accompanied by endocytosis of the activated receptor, changing its conformation and recruitment of FADD and pro-Caspase 8, which ends with cell death [59, 69]. In turn, binding mTNFα to the TNF-R2 receptor will result in the binding and mutual interaction of proteins, which includes TRAF2, which is crucial in the signal transduction and others such as TRAF3, c-IAP1, and c-IAP2. The effect of interaction of the complex is the activation of the JNK kinase and the transcription factor NF-κB [70–73]. It has been proven that polymorphism in the gene (M196R) encoding the receptor protein TNF-R2 may predetermine the development of OA by increasing the number of receptor proteins on the surface of chondrocytes, which leads to disturbance of their functions due to excessive activation by mTNFα [74]. Moreover, not only polymorphism in the receptor but also in the TNFα ligand may favour the occurrence of OA in relation to the specific populations studied [75, 76]. It is worth to mention the presence of an additional ligand capable of incorporating both TNF-R1 and TNF-R2 with still increasing importance in the pathogenesis of OA, which is progranulin (PGRN) also known as granulin epithelin precursor (GEP), PC-cell derived growth factor (PCDGF), proepithelin (PEPI), or acrogranin [77–80]. PGRN is a growth factor having anti-inflammatory and immunomodulatory properties [81]. It was shown that PGRN level is significantly elevated in patients suffering from cartilage arthropathies including OA [82]. The PGRN binding ability to the dedicated TNFα receptors, as well as its elevated levels during the course of the disease, makes it a natural antagonist of TNF that interfere with the signaling pathway TNFα/TNF-R1 and TNFα/TNF-R2 [83]. This implies that an imbalance of TNFα/PGRN can both accelerate and inhibit the development of OA. In addition, the presence of PGRN autoantibodies was observed in some joint diseases with an autoimmune component [84–86]. The effect of TNFα in most cases coincides with the action of IL-1β, and in the case of many phenomena occurring in the course of OA there is a marked synergism between the two cytokines [87]. This effect is the result of activation of the same group of intracellular signalling pathways, which in turn triggers similar effects that increase the inflammation and catabolism in joint tissues [26, 27]. TNFα affects blocking the chondrocytes' synthesis of proteoglycan components, proteins binding proteoglycans, and type II collagen [88, 89]. Activated chondrocytes also produce MMP-1, MMP-3, MMP-13, and ADAMTS-4 [33, 90, 91]. As described earlier, there is an induction of chondrocyte death and a disorder in the migration of chondrogenic progenitor cells (CPCs), which strips the cartilage of any possibility of regeneration [35–37, 92]. A clear impact of TNFα and IL-1β on reducing the efficiency of the respiratory chain was also observed, and hence the decrease in ATP produced within the mitochondria located in chondrocytes; additionally, there is a decrease in the potential of the mitochondrial membrane [93]. TNFα is responsible for increased synthesis of, for example, IL-6, IL-8, RANTES, and VEGF [39–41, 94]. Moreover, as described above, IL-1β and TNFα induce the production of iNOS, COX-2, and PGE2 synthase, thereby increasing the amounts of their products [44, 46, 95, 96].
Figure 3.
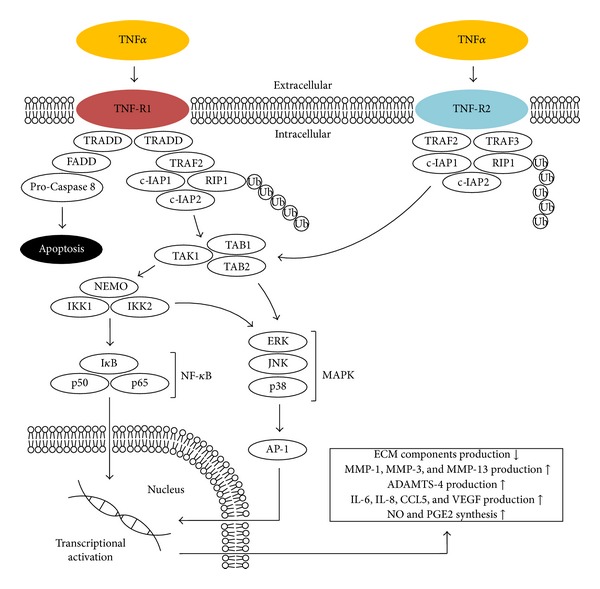
TNFα associated intracellular signaling pathways and downstream cellular targets and effects. TNF-R1: tumor necrosis factor receptor superfamily member 1; TNF-R2: tumor necrosis factor receptor superfamily member 2; TRADD: tumor necrosis factor receptor type 1 associated death domain protein; FADD: Fas-associated protein with death domain; TRAF2: TNF receptor-associated factor 6; c-IAP1: also known as Baculoviral IAP repeat-containing protein 2 (BIRC2); c-IAP2: also known as Baculoviral IAP repeat-containing protein 3 (BIRC3); RIP1: receptor-interacting protein kinase 1; Ub: ubiquitin; TRAF3: TNF receptor-associated factor 3; TAK1: also known as mitogen-activated protein kinase kinase kinase 7 (MAP3K7); TAB1: also known as mitogen-activated protein kinase kinase kinase 7 interacting protein 1 (MAP3K7IP1); TAB2: also known as mitogen-activated protein kinase kinase kinase 7 interacting protein 2 (MAP3K7IP2); p50, p65: subunits of proteins forming NF-κB; IκB: (inhibitor of κB) an endogenous complex of proteins inhibiting the activation of NF-κB; IKK1,2/NEMO: NF-κB inhibitor kinase 1,2 (IκB kinase 1,2)/NF-κB kinase inhibitor (NF-κB essential modulator); ERK: extracellular-signal-regulated kinase; JNK: c-Jun N-terminal kinase; p38: p38 mitogen-activated protein kinases; MAPK: mitogen-activated protein kinases; AP-1: activator protein 1.
2.3. Interleukin-6 (IL-6)
Interleukin-6 (IL-6) is a compound characterized by omnidirectional interactions in the processes occurring in the human body. It is considered a cytokine that strongly activates the immune system and enhances inflammatory response, although considering some of its effects, it may be classified as anti-inflammatory interaction. IL-6 is a glycoprotein consisting of 184 amino acid residues, which in the process of posttranslational processing ultimately becomes an interconnected structure of four α-helices [97]. The production of IL-6 in the tissues of the affected joint is usually in response to IL-1β and TNFα and is mainly implemented by chondrocytes, osteoblasts, FLS, macrophages, and adipocytes [39, 98–102]. The increased concentration of IL-6 is present in both the synovial fluid and serum and is positively correlated with the intensity of lesions in X-ray imaging [103–107]. The effect of IL-6 can be observed through a unique receptor system. There are two subtypes of the IL-6R receptor (gp80, CD126), namely, the membrane form of mIL-6R and the soluble sIL-6R, which may be formed by cutting or by the ADAM17/TACE by metalloproteinase or by means of alternative splicing [108]. Transferring the signal into the cell requires the association of additional gp130 protein (CD130) [109, 110]. The gp130 protein is also present in the membrane form mgp130 and in soluble form sgp130 [111, 112]. The association of the IL-6 and IL-6R with sgp130 is associated with inhibiting the signalling pathway of IL-6 [111, 112]. Recruitment of the ligand-receptor complex to mgp130 causes homodimerization and consequently the formation of hexamer that allows effective signal transduction to the cell [113]. JAK kinase mediates the phosphorylation of tyrosine residues that are an integral part of the gp130, which in further stages results in the activation of STAT3, phosphorylation of MAPK, and activation of the PI3 K/AKT pathway [114, 115]. While analyzing the role of IL-6 in OA, one should first note that the polymorphism of gene (−174G/C) encoding IL-6 may predetermine the development of OA [116, 117]. The effect of IL-6 on joint cartilage is not different from other cytokines and, in synergy with them, causes a decrease in the production of type II collagen and increases the production of enzymes from the MMPs group [118–120]. It was also found that these effects can be enhanced under the influence of injury [121]. IL-6 is considered to be the key cytokine, which causes changes in the subchondral bone layer [122, 123]. Its effect is largely based on promoting the formation of osteoclasts and thus bone resorption while showing synergism with IL-1β and TNFα [122, 124]. Osteoblasts stimulated by IL-1β, TNFα, and IL-6 become a source thereof and may also produce MMPs by adversely affecting the cartilage located near it [124]. In addition to the demonstrated role of other cytokines in inducing the production of IL-6, its secretion by osteoblasts, chondrocytes is also affected by prostaglandin E2 [125, 126]. In turn, while analyzing tests on animal models, it can be observed that IL-6 may have a different effect in some situations. An experiment performed on mice lacking the gene for IL-6 showed that they exhibited a tendency to develop much more advanced degenerative changes than healthy mice [127]. During another experiment, it was also demonstrated in mice lacking the gene for IL-6 that intra-articular injection of IL-6 reduces the loss of proteoglycans in the acute phase of chronic joint inflammation and induces the formation of osteophytes [128].
2.4. Interleukin-15 (IL-15)
Interleukin-15 (IL-15) is a glycoprotein that takes the form of four interconnected α-helices with a mass of 14-15 kDa [129, 130]. Its action is mainly based on the stimulation of differentiation and proliferation of T cells and NK cells [131]. It is one of the better documented cytokines involved in the pathogenesis of rheumatoid arthritis (RA) [132, 133]. While analyzing the involvement of IL-15 in the pathogenesis of OA, its increased concentration was found in the synovial fluid in the early stages of the disease [134]. It has also been shown that the increased IL-15 level in the serum correlates with both the sensation of pain and the severity of lesions in the X-ray image [135]. It has also been noted that its presence can stimulate the secretion of certain types of metalloproteinases from the MMPs group [134].
2.5. Interleukin-17 (IL-17)
The family of interleukins-17 (IL-17) is a group of cytokines with inflammatory effect, which draws more and more attention of researchers for its participation in the pathogenesis of OA. It consists of six members (IL-17A-F) that can interact through five types of receptors (IL-17RA-E) [136, 137]. The source of IL-17 is mainly stimulated CD4+ T cells and mast cells that infiltrate the synovial membrane and the entire joint through blood vessels [138–141]. The main cells in the joint that are affected by IL-17 are chondrocytes and FLS exhibiting the expression of IL-17R on their surface [142]. In addition, studies have demonstrated direct cellular immune response of those T cells against membrane antigens of chondrocytes and fibroblasts in the course of OA [143]. The level of IL-17 measured in the serum and the synovial fluid of patients is elevated and shows a positive correlation with the radiographic image of lesions in OA [144]. IL-17 has been shown to inhibit the synthesis of proteoglycans by chondrocytes and promotes the production of enzymes of the MMPs group [145–147]. Furthermore, IL-17 influences the secretion of other cytokines and compounds negatively affecting the cartilage, such as IL-1β, TNFα, IL-6, NO, and PGE2 [148–150]. The effect of IL-17 on the secretion of VEGF by both chondrocytes and FLS is also characteristic; it favours the excessive development of blood vessel network within the synovial membrane, leading to its hypertrophy [94, 142]. It was also confirmed recently that the polymorphism the gene IL-17A G-197A in certain populations may correlate with the susceptibility to the development of OA [151].
2.6. Interleukin-18 (IL-18)
Interleukin-18 (IL-18) is another representative of the IL-1F family [9]. It is generated as a precursor form of the pro-IL-18, consisting of 192 amino acid residues, which is transformed into a biologically active form after the activation of Caspase 1 or proteinase 3, comprising 157 amino acid residues [152, 153]. It has been noted that the level of Caspase 1 is elevated both in the articular cartilage and synovium in OA patients which greatly promotes the formation of IL-1β and IL-18 [154]. The production of IL-18 in the joint is determined by chondrocytes, osteoblasts, FLS, and macrophages [155–157]. Its increased concentration is evident in the synovial fluid, synovium, cartilage, and blood serum and shows a positive correlation with the degree of severity of the disease seen in radiographic images [158–160]. The effect of IL-18 is mediated by the IL-18Rα receptor, which bears structural and functional similarity to IL-1R. As in the case of IL-1β, efficient signal transmission by IL-18 requires not only the presence of IL-18Rα but also the additional recruitment of the IL-18Rβ (IL-18RAcP) chain [161, 162]. Intraventricular signal transmission through a heterodimeric receptor complex IL-18 does not show significant differences from the mechanism of signal transduction by the IL-1β receptor complex [163]. Studies have shown that polymorphism of genes encoding both IL-18R and the IL-18 interleukin in some variants may predetermine both the development of OA and lumbar disc degeneration (LDD) [164, 165]. IL-18 affects chondrocytes by inducing the upregulation of IL-18Rα on their surface and stimulation excess synthesis of metalloproteinases MMP-1, MMP-3, and MMP-13 [166]. In addition to increasing the concentration of cartilage degrading enzymes, there is an inhibition of production of proteoglycans, aggrecan, and type II collagen; moreover, chondrocytes exhibit morphological changes typical of cells entering apoptosis [167–169]. IL-18 affects chondrocytes and synovial cells, increasing the production of a range of compounds and enzymes, such as the IL-18 in an autocrine manner, IL-6, iNOS, PGE2, COX-2, and VEGF [155, 170–173].
3. Anti-Inflammatory Cytokines
The main representatives of the group of anti-inflammatory cytokines involved in the pathogenesis of OA are IL-4, IL-10, and IL-13. In this chapter, the authors focus on presenting the effect of only those of the above cytokines, whose biological activity is the most important in the light of the relevant problems (Figure 4). However, it should be noted that IL-4, IL-10, and IL-13 belong to a broader group of anti-inflammatory cytokines and are not the only representatives.
Figure 4.
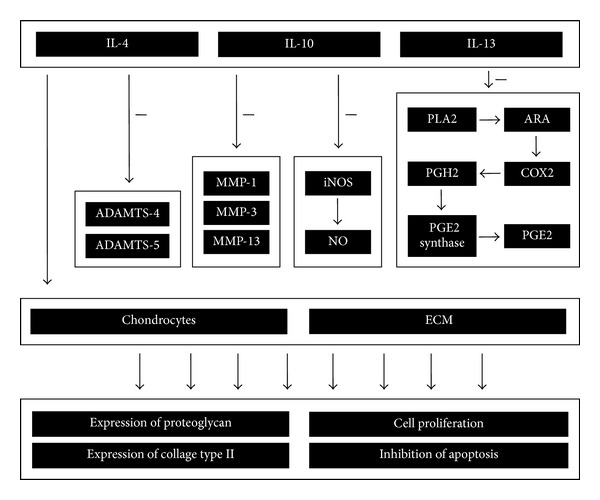
Schematic of the anti-inflammatory and chondroprotective effect of IL-4, IL-10, and IL-13 on articular cartilage during the course of OA.
3.1. Interleukin-4 (IL-4)
Interleukin-4 (IL-4) is a protein composed of 129 amino acids, which takes the form of four interconnected α-helices additionally stabilized by three disulfide bonds [174–176]. IL-4 is a ligand whose biological activity is mediated through a receptor system dedicated to both IL-4 and IL-13 [177]. There are two types of receptors forming heterodimers which are structurally a combination of two or three receptor chains, such as IL-4Rα, IL-13Rα1, and IL-2Rγc [178]. The dimerization of IL-4Rα and IL-2Rγc (type 1 complex) enables the attachment of IL-4, while the interaction between IL-4Rα and IL-13Rα1 (type 2 complex) enables efficient attachment of both IL-4 and IL-13 [177–179].
After the formation of the receptor type 1 complex, an intracellular signal transduction takes place by gradual phosphorylation of the IL-4Rα/JAK1/STAT3/STAT6 cascade, leading to the expression of several proinflammatory genes [180, 181]. The observed polymorphism of the genes encoding both IL-4 and IL-4Rα may predetermine the development of OA in the joints of the hand, knee and hip joint gives reason for claiming that the interaction between IL-4 and its dedicated receptor complex is one of the elements of the pathogenesis of the disease [182–184]. The production of IL-4 is primarily determined by T cells (Th2) infiltrating the synovium of the joint by blood vessels [185, 186]. It was further found that the level of soluble IL-4Rα (sIL-4R) is elevated in the serum of patients suffering from OA as compared to the healthy control group [187]. The increased concentration of IL-4 is also observed in the synovial fluid and synovial cells [185, 188, 189]. IL-4 is associated with a strong chondoprotective effect. In a number of studies it was found that IL-4 has an inhibiting effect on the degradation of proteoglycans in the articular cartilage, by inhibiting the secretion of MMPs metalloproteinases, as well as reducing the variation in the production of proteoglycans that are visible in the course of OA [190–193]. Interestingly, in the course of OA, chondrocytes showed decreased susceptibility to the effects of IL-4 which may be responsible for the rapid degeneration of the articular cartilage [194–196]. In addition, IL-4 alone or in combination with IL-10 exhibits properties inhibiting the apoptosis of both the chondrocytes and FLS [191, 197, 198]. Considering the effect of IL-4 on cell cultures of chondrocytes and FLS treated with it, there is a decrease of synthesis of inflammatory cytokines such as IL-1β, TNFα, and IL-6 [191, 197, 199]. Simultaneously, IL-4 can induce upregulation of the expression of TNFα receptors, such as TNF-R1, and TNF-R2 [200]. In addition to a direct decrease in the secretion of inflammatory cytokines, there is also a decrease in the secretion of other inflammatory mediators such as PGE2, COX-2, PLA2, and iNOS [191, 197, 200, 201]. It seems reasonable to cite a study, which analyzed the effect of rmIL-4 (recombinant mouse IL-4) in combination with rmGM-CSF for the dendritic cells CD11b+F4/80+iDC in a mouse model, where inflammatory joint disease was induced artificially [202]. The observed results seem to confirm the positive effect of IL-4 and GM-CSF on restoring the balance between the secretion of proinflammatory and anti-inflammatory factors in the immune cells in mice with CIA (collagen-induced arthritis). CD11b+F4/80+iDC cells exposure to said effect showed an increased ability to synthesize the anti-inflammatory IL-10 and TGF-β as well as an inhibitory or excitatory effect relative to other cells of the immune system. It has been noted that proliferation of Th17 lymphocytes has been reduced and that they showed the ability to synthesize and secrete proinflammatory IL-17. Macrophages showed a reduced ability to produce and secrete IL-1 and TNFα. In turn, the regulatory T cells were stimulated to increase the production of IL-10 and TGF-β. The test mice showed reduced morphological markers of inflammation of the synovial membrane and articular cartilage and decreased immunological activity of the thymus, relative to the control group (unexposed to rmIL-4 rmGM-CSF). Although the mouse model with CIA corresponds mainly to RA in the world literature, the dependencies between the effect of IL-4 and GM-CSF observed for cells of the immune system may be a reference to some of the processes occurring in OA. In the context of other studies, this gives us a rough idea about the anti-inflammatory effect of IL-4 in the course of OA, although it certainly requires further study.
3.2. Interleukin-10 (IL-10)
Interleukin-10 (IL-10) is a cytokine structurally related to interferons, which is in the form of a homodimer wherein every monomer is a polypeptide chain consisting of 160 amino acid residues, taking the structure of 6 interconnected α-helices [203–205]. Initiation of IL-10 is made possible by binding it to the receptor IL-10R, which is a heterodimer composed of subunits IL-10R1 and IL-10R2 [206, 207]. The first step of activation of the receptor involves binding IL-10 to the IL-10R1 subunit, which then changes its conformation in such a way as to allow binding the IL-10 and IL-10R1 complex to the IL-10R2 subunit [206, 207]. At this point a cascade of intracellular signal transmission is started. This leads to the activation of the tyrosine kinase JAK1 (associated with the IL-10R1 subunit) and TYK2 (associated with IL-10R2) [208]. This in turn implies sending the cellular signal further by the transcriptional activators STAT3, which upon binding to SBE (STAT-binding elements) of the cell nucleus stimulate the synthesis of the products of genes dependent on IL-10 [209]. IL-10 is another cytokine that shows chondoprotective effect in the course of OA. Chondrocytes express both the cytokine IL-10 and the receptor IL-10R [210]. It has been proven that IL-10 is involved in stimulating the synthesis of type II collagen and aggrecan. Following the administration of IL-10 in vitro conditions, both healthy articular cartilage and one in the course of OA demonstrated an increase in proteoglycan synthesis and its percentage share in the extracellular matrix. Furthermore, the synovium of patients with osteoarthritis in the context of hemophilia, which showed a tendency to excessive production of TNFα and IL-1β following the in vitro administration of IL-10 significantly reduced the average secretion of the mentioned proinflammatory cytokines by 60% and 83%, respectively [211]. It has been shown that IL-10 is responsible for inhibiting the production of MMPs family of metalloproteinases [212]. It was confirmed that IL-10 (similar to IL-4) inhibits the apoptosis of chondrocytes [213]. These properties of IL-10 are likely the result of stimulation of the synthesis of IL-1β antagonist, which is IL-1Ra and the tissue inhibitor of metalloproteinases-1 (TIMP-1) as well as growth factors [214, 215]. The study of the effect of IL-10 on the stimulation of chondrocyte proliferation in a mouse model shows further signaling pathways of IL-10 [216]. It has been proved that IL-10 activates the kinase pathway SMAD1/SMAD5/SMAD8 and ERK1/2 MAP and induces the expression of bone morphogenetic proteins 2 and 6 (BMP-2, BMP-6). BMP proteins belong to the TGF-β family and play a crucial role in chondrogenesis [217]. They are part of the signaling pathway that affects many genes and proteins responsible for the regulation of the mesenchymal cell transformation in chondrocytes, such as NKX-3.2/SOX9, SOX5, and SOX6 [218, 219]. In addition to determining the transformation of multipotent cells towards chondrocytes, BMP proteins stimulate their proliferation both directly and through the IHH/PTHrP-dependent pathway [220, 221]. The involvement of IL-10 in the induction of differentiation and proliferation of chondrocytes through the BMP signaling pathway was thus confirmed. The study authors deduce that the observed mechanisms may be partly responsible for the chondoprotective effects of IL-10 in OA. In another study, the cells of the haematopoietic system were observed after stimulation by LPS. The aim was to figure out how LPS stimulation affects the synthesis and level increase of IL-10 in blood samples from patients with OA and healthy subjects. Based on the results of IL-10 levels and information on the health status of the subjects, a specific prediction model of OA was developed. Persons whose blood samples showed no significant increase in the level of IL-10 relative to the starting point (the lowest quartile of results) were running even a 3-fold risk of developing OA compared to those whose levels of IL-10 after stimulation were in the highest quartile [222]. IL-10 reduces the effect of TNFα on synovial fibroblasts sampled from patients with OA [200]. This is manifest in a significant decrease in their secretion of PGE2, COX-2, and PLA2. Interestingly, IL-10 does not have those properties with respect to the fibroblasts not treated with TNFα. It was confirmed that IL-10 reduces the expression of TNFα receptors and thus their ability to bind to the surface of fibroblasts. The anti-inflammatory and chondroprotective nature of IL-10 is composed of results of interesting studies of the influence of exercise on the secretion of cytokines in joint fluid and periarticular tissues in patients with OA of the knee joints [223, 224]. In these studies, patients were divided into a study group, which has exercised knee joints affected by OA for 3 h and a control group, which had to refrain from exercise during the same time. It is worth noting that in the control group there were patients with the same degree of severity of OA of the knee joints. Knee joints affected by OA were connected to catheters in both the study and control groups. Catheters, placed into the joints and periarticularly, were used to examine the levels of IL-10 and other cytokines before exercise, during exercise, and after exercise relative to the control group. A significant increase in the level of IL-10 in the synovial fluid and periarticular tissues was observed in patients in the study group as compared to the control group, in which the levels of IL-10 observed before and after exercise showed no significant difference. On the basis of these observations, it seems reasonable to advocate the medicinal effect of physical exercise on joints affected by OA. In truth, it is not clear what mechanism leads to an increased secretion of IL-10. Some authors argue that this is due to the inductive effect of increased intra-articular pressure on cellular secretion [225, 226].
3.3. Interleukin-13 (IL-13)
Interleukin-13 (IL-13) is a compound that takes the structure of four interconnected α-helices, which is very similar in its effect to IL-4 [227, 228]. Similar to IL-4, the action of IL-13 as a ligand is mediated through a receptor system that combines both cytokines [177]. In this case it requires the recruitment of a ligand, namely, IL-13, and in addition the chains IL-4Rα and IL-13Rα1 to create a type 2 complex [177, 178]. With this combination, IL-13 has the capacity to transfer the intracellular signal both by the cascade IL-4Rα/JAK2/STAT3 and IL-13Rα1/TYK2/STAT1/STAT6 [180]. The anti-inflammatory and chondoprotective effects of IL-13 on the cells of the immune response, articular cartilage, and synovium in OA have been fairly well documented [229, 230]. The anti-inflammatory effect of IL-13 in the context of OA seems most important with respect to fibroblasts included in the synovium. Note, however, that the anti-inflammatory properties based on the inhibition of the secretion of inflammatory cytokines relate to a whole range of cells such as macrophages, monocytes, B cells, NK cells, and endothelial cells [231]. In one of the most important studies into the effects of IL-13 on OA, synovium samples were taken from 16 patients with OA during the knee endoprosthesis implantation procedure (TKR) [232]. Patients were diagnosed based on the criteria of the ACR Diagnostic Subcommittee for OA, revealing stage III or IV of the disease according to radiological imaging [233]. The synovial samples taken were administered proinflammatory LPS (control samples), followed by IL-13 (test samples). After 72 h incubation, laboratory tests were performed including binding tests, northern blotting, and ELISA. It has been shown that, compared to the control samples, IL-13 showed inhibitory effects on the synthesis of proinflammatory IL-1β, TNFα, and MMP-3 with a simultaneous increase in the level of IL-1Ra. In the studies synovium cells, the amount of mRNA for IL-1β was reduced, while the mRNA level for IL-1Ra was increased relative to control samples. Moreover, synovial fibroblasts showed a reduction in binding between IL-1β and its receptor, which was caused by the increased production and action of IL-1Ra. It has been demonstrated that IL-13 has the ability to inhibit the proinflammatory effect of TNFα relative to fibroblasts from patients with OA [190]. A significant decrease in PGE2 synthesis by blocking the synthesis of COX-2 was observed. Interestingly, compared to IL-4 and IL-10, IL-13 does not affect the production level of PLA2. This may indicate the selective effect of IL-13 that only involves COX-2 and, more specifically, the expression of its gene. The study proved that IL-13 has the ability to decrease the nuclear concentration of transcription factors C/EBP, which may affect the inhibition of the synthesis of COX-2. In this context, fibroblasts may exhibit properties similar to osteoblasts [234]. The analysis of these results indicates the potential utility of IL-13 in the treatment of OA, as a compound that inhibits the inflammatory processes, protects chondrocytes, reduces the secretion of inflammatory cytokines and metalloproteinases, while stimulating the synthesis of IL-1Ra.
4. Summary
OA is a set of complex and difficult-to-identify processes leading to progressive degeneration of joints. But, contrary to what it may seem to be, it is not only a set of catabolic effects. Apart from them, anabolic anti-inflammatory processes also occur continually. Evident imbalance in metabolism observed in OA is the final manifestation of the dysregulation within the so-called cytokine network between anti-inflammatory and inflammatory cytokines. In this paper, current knowledge on the impact of the most important cytokines in the pathogenesis of OA has been presented, based on world literature survey. Cytokines were categorized with respect to their biological effect as inflammatory and anti-inflammatory. However, this criterion is not so obvious in particular conditions. Respective cytokines should be analysed together with the nature and structure of specific receptors and the intracellular signalling pathways triggered by their activation in the specific tissue. Thus, particular attention was paid to the potential impact of cytokines on articular cartilage, synovium and the cells of the immune system. In the context of developing optimal treatment tactics for OA, the authors of this paper have tried to summarize and comment on the most important information resulting from the collected above (published) data.
Due to their effects within the joint, inflammatory cytokines have a primarily destructive impact on articular cartilage. It is a multilevel impact that involves not only the induction of aging and apoptosis of chondrocytes, but also a decrease in the synthesis of the key components of ECM, such as proteoglycans, aggrecan, and type II collagen. In addition, inflammatory cytokines contribute to the increased synthesis and release of many proteolytic enzymes that decompose articular cartilage, which include the metalloproteinases family MMPs and ADAMTS. Apart from their effect on chondrocytes, synovial cells and other articular, and periarticular tissues, inflammatory cytokines affect the cells of the immune system migrating to the site of inflammation. Due to the action of inflammatory cytokines, all of these cells tend to produce excessive amounts of inflammatory PGE2, COX-2, phospholipase A2, NO, and free radicals. It is worth noting that inflammatory cytokines stimulate cells to synthesize other inflammatory cytokines, namely, IL-1β, TNFα, IL-6, IL-8, and chemokines CCL5 (RANTES). They can thereby also promote their own production, thus demonstrating an autocrine, paracrine, and self-propelling effect on the inflammation process.
The action of anti-inflammatory cytokines mainly involves inhibiting the synthesis of inflammatory cytokines, particularly IL-1β and TNFα. Observed effects of anti-inflammatory cytokines include increased proteoglycan synthesis, inhibited apoptosis of chondrocytes, decreased synthesis and secretion of metalloproteinases, and decreased level of PGE2. However, these properties should be treated as an inhibitory effect on inflammatory cytokines rather than direct chondoprotective capabilities. It should be added that it has been proven that the action of anti-inflammatory cytokines is mainly performed relating to cells stimulated by inflammatory cytokines, whereas no significant differences are noted in the metabolism of cells not subject to such stimulation. Such a relationship has been observed, for example, regarding synovial fibroblasts obtained from patients with OA [200]. However, certain properties of anti-inflammatory cytokines go beyond the simplest idea of antagonism with respect to inflammatory cytokines. For example, IL-10 stimulates the synthesis of growth factors of the BMP family (BMP-2, BMP-6) and affects the regulatory proteins such as NKX-3.2/SOX9, SOX5, and SOX6 which induces and regulates the process of chondrogenesis. In turn, rmIL-4 in a mouse model stimulates regulatory T cells to synthesize IL-10 and the growth factor TGF-β, which at least corresponds to the properties of inflammatory cytokines in the context of stimulating their own synthesis [202]. The dominant observation concerning the effects of inflammatory and anti-inflammatory cytokines in the course of OA is that the inflammatory response within the affected joints is never controlled completely. Many authors have confirmed that the effect of anti-inflammatory cytokines and the treatment based on them cannot positively stop the progression of the disease by blocking the promotion of catabolic pathways [200]. So far, there have been no comprehensive answers to this issue. According to the standpoint of the authors of this study, the insufficient effect of anti-inflammatory cytokines in the joint may be determined by the characteristics of their action. Relative to healthy joint tissue, anti-inflammatory cytokines show no significant anabolic effect likely to create a sufficient biochemical advantage against the effects of inflammatory cytokines. It is only in a joint affected by the disease in the presence of inflammatory mediators that the role of anti-inflammatory cytokines becomes noticeable. The effect of anti-inflammatory cytokines therefore shows no primary prophylactic capability; however, it has the properties of a secondary response to combat the symptoms of the disease. Such blocking effect of the activity of inflammatory cytokines on the “action-reaction” principle, already in the early stages of the disease places the anticatabolic processes as secondary in relation to the catabolic effect and is therefore late or delayed.
In search of a better understanding of the pathomechanism of OA, one should pay attention to the quantitative changes that occur in the so-called cytokine network. The abundance or deficiency of any element can cause disturbances of the whole structure and ultimately its collapse. In OA, there is a lot of missing “building blocks” that contribute to preservation of the metabolic balance in the joint. The main figure in this group is primarily TGF-β, a cytokine and growth factor responsible for stimulating the production of proteoglycan, type II collagen, and chondrogenesis [235, 236]. Its presence in the joints is observed in healthy subjects, as opposed to patients with OA, where the amounts of TGF-β are low or even undetectable [237]. Some authors believe that reducing the level of this cytokine is responsible for sensitizing the cartilage to the effect of inflammation mediators and for the progress of the disease [235]. The explanation for the reduced amount of TGF-β in the course of OA and thus also its biological activity may involve intercepting the cell signalling pathways by inflammatory cytokines, although this certainly requires further investigation [238, 239]. Another factor, which may contribute to the unrestrained progression of OA, is the cell type responsible for the production of specific cytokines. Namely, in the OA model, the production of anti-inflammatory cytokines is determined primarily by cells of the immune system, which are largely infiltrating cells. In turn, inflammatory cytokines such as IL-1β or TNFα are synthesized and secreted by the cells of the immune system, as well as locally by articular cartilage cells [240]. This dual model of release seems to create favourable conditions for the development of OA. Add a slightly visible influence of anti-inflammatory cytokines on stimulating their own secretion and thus the lack of a “self-supplying” mechanism (as opposed to inflammatory cytokines), and we will see the image of the disease, in which it is not possible to bring about the advantages of inflammatory processes. Such a scheme of the progress disease quite accurately corresponds to the image of OA progression. Currently we are trying to seek appropriate treatment likely to stop the progress of OA or at least slow it down to a satisfactory degree. The tactics used by researchers are to influence cell transduction pathways that are crucial to the induction of inflammatory process. Therefore, research is underway, aimed at obtaining more accurate knowledge of the effect of inflammatory and anti-inflammatory cytokines. The emerging picture shows the multiplicity, complexity, and the multilevel nature of processes taking place with the participation of cytokines in the course of OA. Their discovery and understanding is a big challenge for today's medicine; however, it seems absolutely crucial to finally develop effective methods of treatment.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Bijlsma JWJ, Berenbaum F, Lafeber FPJG. Osteoarthritis: an update with relevance for clinical practice. The Lancet. 2011;377(9783):2115–2126. doi: 10.1016/S0140-6736(11)60243-2. [DOI] [PubMed] [Google Scholar]
- 2.Madry H, Cucchiarini M. Advances and challenges in gene-based approaches for osteoarthritis. The Journal of Gene Medicine. 2013;15(10):343–355. doi: 10.1002/jgm.2741. [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB, Otero M. Inflammation in osteoarthritis. Current Opinion in Rheumatology. 2011;23(5):471–478. doi: 10.1097/BOR.0b013e328349c2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lange-Brokaar BJ, Ioan-Facsinay A, van Osch GJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis Cartilage. 2012;20(12):1484–1499. doi: 10.1016/j.joca.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 5.Vangsness CT, Jr., Burke WS, Narvy SJ, MacPhee RD, Fedenko AN. Human knee synovial fluid cytokines correlated with grade of knee osteoarthritis—a pilot study. Bulletin of the NYU Hospital for Joint Diseases. 2011;69(2):122–127. [PubMed] [Google Scholar]
- 6.Mueller MB, Tuan RS. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: identifying molecular targets. PM&R. 2011;3(6, supplement 1):S3–S11. doi: 10.1016/j.pmrj.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clinical Orthopaedics and Related Research. 2004;(427, supplement):S27–S36. doi: 10.1097/01.blo.0000144854.66565.8f. [DOI] [PubMed] [Google Scholar]
- 8.Mobasheri A. The future of osteoarthritis therapeutics: emerging biological therapy. Current Opinion in Rheumatology. 2013;15(12):p. 385. doi: 10.1007/s11926-013-0385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol. 2013;25(6):389–393. doi: 10.1016/j.smim.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Auron PE, Webb AC, Rosenwasser LJ, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:7907–7911. doi: 10.1073/pnas.81.24.7907. The Journal of Immunology, vol. 178, no. 9, pp. 5413–5417, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piccioli P, Rubartelli A. The secretion of IL-1β and options for release. Seminars in Immunology. 2013;25(6):425–429. doi: 10.1016/j.smim.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Melchiorri C, Meliconi R, Frizziero L, et al. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis & Rheumatology. 1998;41(12):2165–2174. doi: 10.1002/1529-0131(199812)41:12<2165::AID-ART11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 13.Massicotte F, Lajeunesse D, Benderdour M, et al. Can altered production of interleukin-1β, interleukin-6, transforming growth factor-β and prostaglandin E2 by isolated human subchondral osteoblasts identity two subgroups of osteoarthritic patients. Osteoarthritis and Cartilage. 2002;10(6):491–500. doi: 10.1053/joca.2002.0528. [DOI] [PubMed] [Google Scholar]
- 14.Ribel-Madsen S, Bartels EM, Stockmarr A, et al. A synoviocyte model for osteoarthritis and rheumatoid arthritis: response to Ibuprofen, betamethasone, and ginger extract-a cross-sectional in vitro study. Arthritis. 2012;2012:9 pages. doi: 10.1155/2012/505842.505842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Annals of the Rheumatic Diseases. 1993;52(12):870–875. doi: 10.1136/ard.52.12.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szekanecz Z, Koch AE. Cell-cell interactions in synovitis. Endothelial cells and immune cell migration. Arthritis Research. 2000;2(5):368–373. doi: 10.1186/ar114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohn DH, Sokolove J, Sharpe O, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Research and Therapy. 2012;14(1, article R7) doi: 10.1186/ar3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Symons JA, Young PR, Duff GW. Soluble type II interleukin 1 (IL-1) receptor binds and blocks processing of IL-1β precursor and loses affinity for IL-1 receptor antagonist. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(5):1714–1718. doi: 10.1073/pnas.92.5.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boraschi D, Tagliabue A. The interleukin-1 receptor family. Seminars in Immunology. 2013;25(6):394–407. doi: 10.1016/j.smim.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 20.Palmer G, Guerne P-A, Mezin F, et al. Production of interleukin-1 receptor antagonist by human articular chondrocytes. Arthritis Research. 2002;4(3):226–231. doi: 10.1186/ar411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caron JP, Fernandes JC, Martel-Pelletier J, et al. Chondroprotective effect of intraarticular injections of interleukin-1 receptor antagonist in experimental osteoarthritis: suppression of collagenase-1 expression. Arthritis and Rheumatism. 1996;39(9):1535–1544. doi: 10.1002/art.1780390914. [DOI] [PubMed] [Google Scholar]
- 22.Martel-Pelletier J, McCollum R, DiBattista J, et al. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes: identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis and Rheumatism. 1992;35(5):530–540. doi: 10.1002/art.1780350507. [DOI] [PubMed] [Google Scholar]
- 23.Sadouk MB, Pelletier J-P, Tardif G, Kiansa K, Cloutier J-M, Martel- Pelletier J. Human synovial fibroblasts coexpress IL-1 receptor type I and type II mRNA: the increased level of the IL-1 receptor in osteoarthritic cells is related to an increased level of the type I receptor. Laboratory Investigation. 1995;73(3):347–355. [PubMed] [Google Scholar]
- 24.Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochimica et Biophysica Acta—Molecular Cell Research. 2002;1592(3):265–280. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, Akira S. TLR signaling. Seminars in Immunology. 2007;19(1):24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Roman-Blas JA, Jimenez SA. NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis and Cartilage. 2006;14(9):839–848. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-κB signaling: multiple angles to target OA. Current Drug Targets. 2010;11(5):599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shakibaei M, Schulze-Tanzil G, John T, Mobasheri A. Curcumin protects human chondrocytes from IL-1β-induced inhibition of collagen type II and β1-integrin expression and activition of caspase-3: an immunomorphological study. Annals of Anatomy. 2005;187(5-6):487–497. doi: 10.1016/j.aanat.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Stöve J, Huch K, Günther K-P, Scharf H-P. Interleukin-1β induces different gene expression of stromelysin, aggrecan and tumor-necrosis-factor-stimulated gene 6 in human osteoarthritic chondrocytes in vitro . Pathobiology. 2000;68(3):144–149. doi: 10.1159/000055915. [DOI] [PubMed] [Google Scholar]
- 30.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis & Rheumatology. 2000;43(4):801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Research. 2002;4(3):157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meszaros E, Malemud CJ. Prospects for treating osteoarthritis: enzyme-protein interactions regulating matrix metalloproteinase activity. Therapeutic Advances in Chronic Disease. 2012;3(5):219–229. doi: 10.1177/2040622312454157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. Journal of Cellular Biochemistry. 2011;112(12):3507–3514. doi: 10.1002/jcb.23298. [DOI] [PubMed] [Google Scholar]
- 34.Koshy PJT, Lundy CJ, Rowan AD, et al. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis and Rheumatism. 2002;46(4):961–967. doi: 10.1002/art.10212. [DOI] [PubMed] [Google Scholar]
- 35.Ye Z, Chen Y, Zhang R, et al. c-Jun N-terminal kinase—c-Jun pathway transactivates Bim to promote osteoarthritis. Canadian Journal of Physiology and Pharmacology. 2014;92(2):132–139. doi: 10.1139/cjpp-2013-0228. [DOI] [PubMed] [Google Scholar]
- 36.López-Armada MJ, Caramés B, Lires-Deán M, et al. Cytokines, tumor necrosis factor-α and interleukin-1β, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthritis and Cartilage. 2006;14(7):660–669. doi: 10.1016/j.joca.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Héraud F, Héraud A, Harmand MF. Apoptosis in normal and osteoarthritic human articular cartilage. Annals of the Rheumatic Diseases. 2000;59(12):959–965. doi: 10.1136/ard.59.12.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aigner T, McKenna L, Zien A, Fan Z, Gebhard PM, Zimmer R. Gene expression profiling of serum- and interleukin-1β-stimulated primary human adult articular chondrocytes - A molecular analysis based on chondrocytes isolated from one donor. Cytokine. 2005;31(3):227–240. doi: 10.1016/j.cyto.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Guerne P-A, Carson DA, Lotz M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro . Journal of Immunology. 1990;144(2):499–505. [PubMed] [Google Scholar]
- 40.Lotz M, Terkeltaub R, Villiger PM. Cartilage and joint inflammation: regulation of IL-8 expression by human articular chondrocytes. Journal of Immunology. 1992;148(2):466–473. [PubMed] [Google Scholar]
- 41.Alaaeddine N, Olee T, Hashimoto S, Creighton-Achermann L, Lotz M. Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis & Rheumatology. 2001;44(7):1633–1643. doi: 10.1002/1529-0131(200107)44:7<1633::AID-ART286>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Pulsatelli L, Dolzani P, Piacentini A, et al. Chemokine production by human chondrocytes. Journal of Rheumatology. 1999;26(9):1992–2001. [PubMed] [Google Scholar]
- 43.Baugé C, Attia J, Leclercq S, Pujol JP, Galéra P, Boumédiene K. Interleukin-1beta up-regulation of Smad7 via NF-kappaB activation in human chondrocytes. Arthritis & Rheumatology. 2008;58(1):221–226. doi: 10.1002/art.23154. [DOI] [PubMed] [Google Scholar]
- 44.El Mansouri FE, Chabane N, Zayed N, et al. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis and Rheumatism. 2011;63(1):168–179. doi: 10.1002/art.27762. [DOI] [PubMed] [Google Scholar]
- 45.Gilman SC, Chang J, Zeigler PR, Uhl J, Mochan E. Interleukin-1 activates phospholipase A2 in human synovial cells. Arthritis and Rheumatism. 1988;31(1):126–130. doi: 10.1002/art.1780310118. [DOI] [PubMed] [Google Scholar]
- 46.Hardy MM, Seibert K, Manning PT, et al. Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis and Rheumatism. 2002;46(7):1789–1803. doi: 10.1002/art.10356. [DOI] [PubMed] [Google Scholar]
- 47.Afonso V, Champy R, Mitrovic D, Collin P, Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine. 2007;74(4):324–329. doi: 10.1016/j.jbspin.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Bodmer J-L, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends in Biochemical Sciences. 2002;27(1):19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- 49.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53(1):45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 50.Tang P, Hung M-C, Klostergaard J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry. 1996;35(25):8216–8225. doi: 10.1021/bi952182t. [DOI] [PubMed] [Google Scholar]
- 51.Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s) Microscopy Research and Technique. 2000;50(3):184–195. doi: 10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 52.MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cellular Signalling. 2002;14(6):477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 53.Grell M, Douni E, Wajant H, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83(5):793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 54.Westacott CI, Barakat AF, Wood L, et al. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthritis and Cartilage. 2000;8(3):213–221. doi: 10.1053/joca.1999.0292. [DOI] [PubMed] [Google Scholar]
- 55.Webb GR, Westacott CI, Elson CJ. Osteoarthritic synovial fluid and synovium supernatants up-regulate tumor necrosis factor receptors on human articular chondrocytes. Osteoarthritis and Cartilage. 1998;6(3):167–176. doi: 10.1053/joca.1998.0109. [DOI] [PubMed] [Google Scholar]
- 56.Steenvoorden MMC, Bank RA, Ronday HK, Toes REM, Huizinga TWJ, DeGroot J. Fibroblast-like synoviocyte-chondrocyte interaction in cartilage degradation. Clinical and Experimental Rheumatology. 2007;25(2):239–245. [PubMed] [Google Scholar]
- 57.Alaaeddine N, DiBattista JA, Pelletier J-P, et al. Osteoarthritic synovial fibroblasts possess an increased level of tumor necrosis factor-receptor 55 (TNF-R55) that mediates biological activation by TNF-α . Journal of Rheumatology. 1997;24(10):1985–1994. [PubMed] [Google Scholar]
- 58.Pennica D, Lam VT, Weber RF, et al. Biochemical characterization of the extracellular domain of the 75- kilodalton tumor necrosis factor receptor. Biochemistry. 1993;32(12):3131–3138. doi: 10.1021/bi00063a027. [DOI] [PubMed] [Google Scholar]
- 59.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 60.Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and Is required for TNF-mediated gene induction. Molecular Cell. 2009;36(5):831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 61.Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nature Immunology. 2011;12(12):1143–1149. doi: 10.1038/ni.2159. [DOI] [PubMed] [Google Scholar]
- 62.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81(4):495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 63.Hsu H, Huang J, Shu H-B, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 64.Varfolomeev E, Goncharov T, Fedorova AV, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. Journal of Biological Chemistry. 2008;283(36):24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 Regulates an NF-κB-Independent Cell-Death Switch in TNF Signaling. Current Biology. 2007;17(5):418–424. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Molecular Cell. 2006;22(2):245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Z, Connell MC, MacEwan DJ. TNFR1-induced NF-κB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cellular Signalling. 2007;19(6):1238–1248. doi: 10.1016/j.cellsig.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 68.Campbell J, Ciesielski CJ, Hunt AE, et al. A novel mechanism for TNF-α regulation by p38 MAPK: involvement of NF-κB with implications for therapy in rheumatoid arthritis. Journal of Immunology. 2004;173(11):6928–6937. doi: 10.4049/jimmunol.173.11.6928. [DOI] [PubMed] [Google Scholar]
- 69.Schütze S, Machleidt T, Adam D, et al. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. Journal of Biological Chemistry. 1999;274(15):10203–10212. doi: 10.1074/jbc.274.15.10203. [DOI] [PubMed] [Google Scholar]
- 70.Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83(7):1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 71.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78(4):681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 72.Matsuzawa A, Tseng P-H, Vallabhapurapu S, et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321(5889):663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodríguez M, Cabal-Hierro L, Carcedo MT, et al. NF-κB signal triggering and termination by tumor necrosis factor receptor 2. Journal of Biological Chemistry. 2011;286(26):22814–22824. doi: 10.1074/jbc.M111.225631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oregón-Romero E, Vázquez-Del Mercado M, Navarro-Hernández RE, et al. Tumor necrosis factor receptor 2 M196R polymorphism in rheumatoid arthritis and osteoarthritis: relationship with sTNFR2 levels and clinical features. Rheumatology International. 2006;27(1):53–59. doi: 10.1007/s00296-006-0159-7. [DOI] [PubMed] [Google Scholar]
- 75.Ji B, Shi J, Cheng X, et al. Association analysis of two candidate polymorphisms in the tumour necrosis factor-α gene with osteoarthritis in a Chinese population. International Orthopaedics. 2013;37(10):2061–2063. doi: 10.1007/s00264-013-1931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han L, Song JH, Yoon JH, et al. TNF-a and TNF-β polymorphisms are associated with susceptibility to osteoarthritis in a Korean population. Korean Journal of Pathology. 2012;46(1):30–37. doi: 10.4132/KoreanJPathol.2012.46.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang W, Lu Y, Tian Q-Y, et al. The growth factor progranulin binds to tnf receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332(6028):478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jian J, Zhao S, Tian Q, et al. Progranulin directly binds to the CRD2 and CRD3 of TNFR extracellular domains. FEBS Letters. 2013;587(21):3428–3436. doi: 10.1016/j.febslet.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu C-J. Progranulin: a promising therapeutic target for rheumatoid arthritis. FEBS Letters. 2011;585(23):3675–3680. doi: 10.1016/j.febslet.2011.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu CJ, Bosch X. Progranulin: a growth factor, a novel TNFR ligand and a drug target. Pharmacology & Therapeutics. 2012;133(1):124–132. doi: 10.1016/j.pharmthera.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jian J, Konopka J, Liu C. Insights into the role of progranulin in immunity, infection, and inflammation. Journal of Leukocyte Biology. 2013;93(2):199–208. doi: 10.1189/jlb.0812429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo F, Lai Y, Tian Q, Lin EA, Kong L, Liu C. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis and Rheumatism. 2010;62(7):2023–2036. doi: 10.1002/art.27491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Konopka J, Richbourgh B, Liu C. The role of PGRN in musculoskeletal development and disease. Frontiers in Bioscience. 2014;19:662–671. doi: 10.2741/4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thurner L, Zaks M, Preuss KD, et al. Progranulin antibodies entertain a proinflammatory environment in a subgroup of patients with psoriatic arthritis. Arthritis Research & Therapy. 2013;15(6, article R211) doi: 10.1186/ar4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thurner L, Preuss KD, Fadle N, et al. Progranulin antibodies in autoimmune diseases. Journal of Autoimmunity. 2013;42:29–38. doi: 10.1016/j.jaut.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 86.Thurner L, Stöger E, Fadle N, et al. Proinflammatory progranulin antibodies in inflammatory bowel diseases. doi: 10.1007/s10620-014-3089-3. Digestive Diseases and Sciences. In press. [DOI] [PubMed] [Google Scholar]
- 87.Henderson B, Pettipher ER. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor α in the rabbit: evidence for synergistic interactions between cytokines in vivo . Clinical and Experimental Immunology. 1989;75(2):306–310. [PMC free article] [PubMed] [Google Scholar]
- 88.Saklatvala J. Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 1986;322(6079):547–549. doi: 10.1038/322547a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Séguin CA, Bernier SM. TNFalpha suppresses link protein and type II collagen expression in chondrocytes: role of MEK1/2 and NF-kappaB signaling pathways. Journal of Cellular Physiology. 2003;197(3):356–369. doi: 10.1002/jcp.10371. [DOI] [PubMed] [Google Scholar]
- 90.Lefebvre V, Peeters-Joris C, Vaes G. Modulation by interleukin 1 and tumor necrosis factor α of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochimica et Biophysica Acta—Molecular Cell Research. 1990;1052(3):366–378. doi: 10.1016/0167-4889(90)90145-4. [DOI] [PubMed] [Google Scholar]
- 91.Xue J, Wang J, Liu Q, Luo A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Molecular Medicine Reports. 2013;8(6):1755–1760. doi: 10.3892/mmr.2013.1729. [DOI] [PubMed] [Google Scholar]
- 92.Joos H, Wildner A, Hogrefe C, Reichel H, Brenner RE. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Research & Therapy. 2013;15(5, article R119) doi: 10.1186/ar4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.López-Armada MJ, Caramés B, Martín MA, et al. Mitochondrial activity is modulated by TNFα and IL-1β in normal human chondrocyte cells. Osteoarthritis and Cartilage. 2006;14(10):1011–1022. doi: 10.1016/j.joca.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 94.Honorati MC, Cattini L, Facchini A. VEGF production by osteoarthritic chondrocytes cultured in micromass and stimulated by IL-17 and TNF-α . Connective Tissue Research. 2007;48(5):239–245. doi: 10.1080/03008200701541767. [DOI] [PubMed] [Google Scholar]
- 95.Martel-Pelletier J, Pelletier J-P, Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Seminars in Arthritis and Rheumatism. 2003;33(3):155–167. doi: 10.1016/s0049-0172(03)00134-3. [DOI] [PubMed] [Google Scholar]
- 96.Jones SW, Brockbank SMV, Clements KM, et al. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthritis and Cartilage. 2009;17(1):124–131. doi: 10.1016/j.joca.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 97.Hammacher A, Ward LD, Weinstock J, Treutlein H, Yasukawa K, Simpson RJ. Structure-function analysis of human IL-6: identification of two distinct regions that are important for receptor binding. Protein Science. 1994;3(12):2280–2293. doi: 10.1002/pro.5560031213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bender S, Haubeck H-D, Van de Leur E, et al. Interleukin-1β induces synthesis and secretion of interleukin-6 in human chondrocytes. FEBS Letters. 1990;263(2):321–324. doi: 10.1016/0014-5793(90)81404-c. [DOI] [PubMed] [Google Scholar]
- 99.Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Research and Therapy. 2006;8, article R187 doi: 10.1186/ar2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ishimi Y, Miyaura C, Jin CH, et al. IL-6 is produced by osteoblasts and induces bone resorption. Journal of Immunology. 1990;145(10):3297–3303. [PubMed] [Google Scholar]
- 101.Ushiyama T, Chano T, Inoue K, Matsusue Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Annals of the Rheumatic Diseases. 2003;62(2):108–112. doi: 10.1136/ard.62.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Distel E, Cadoudal T, Durant S, Poignard A, Chevalier X, Benelli C. The infrapatellar fat pad in knee osteoarthritis: an important source of interleukin-6 and its soluble receptor. Arthritis and Rheumatism. 2009;60(11):3374–3377. doi: 10.1002/art.24881. [DOI] [PubMed] [Google Scholar]
- 103.Kaneko S, Satoh T, Chiba J, Ju C, Inoue K, Kagawa J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines, Cellular and Molecular Therapy. 2000;6(2):71–79. doi: 10.1080/13684730050515796. [DOI] [PubMed] [Google Scholar]
- 104.Doß F, Menard J, Hauschild M, et al. Elevated IL-6 levels in the synovial fluid of osteoarthritis patients stem from plasma cells. Scandinavian Journal of Rheumatology. 2007;36(2):136–139. doi: 10.1080/03009740701250785. [DOI] [PubMed] [Google Scholar]
- 105.Pearle AD, Scanzello CR, George S, et al. Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthritis and Cartilage. 2007;15(5):516–523. doi: 10.1016/j.joca.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 106.Stannus O, Jones G, Cicuttini F, et al. Circulating levels of IL-6 and TNF-α are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthritis and Cartilage. 2010;18(11):1441–1447. doi: 10.1016/j.joca.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 107.Livshits G, Zhai G, Hart DJ, et al. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford Study. Arthritis and Rheumatism. 2009;60(7):2037–2045. doi: 10.1002/art.24598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rose-John S, Neurath MF. IL-6 trans-signaling: the heat is on. Immunity. 2004;20(1):2–4. doi: 10.1016/s1074-7613(04)00003-2. [DOI] [PubMed] [Google Scholar]
- 109.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63(6):1149–1157. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 110.Saito M, Yoshida K, Hibi M, Taga T, Kishimoto T. Molecular cloning of a murine IL-6 receptor-associated signal transducer, gp130, and its regulated expression in vivo . Journal of Immunology. 1992;148(12):4066–4071. [PubMed] [Google Scholar]
- 111.Narazaki M, Yasukawa K, Saito T, et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood. 1993;82(4):1120–1126. [PubMed] [Google Scholar]
- 112.Jostock T, Müllberg J, Özbek S, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. European Journal of Biochemistry. 2001;268(1):160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 113.Boulanger MJ, Chow D-C, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interieukin-6/IL-6 α-receptor/gp130 complex. Science. 2003;300(5628):2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 114.Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine and Growth Factor Reviews. 1997;8(4):241–252. doi: 10.1016/s1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 115.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Reviews of Physiology, Biochemistry and Pharmacology. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 116.Honsawek S, Deepaisarnsakul B, Tanavalee A, et al. Association of the IL-6 -174G/C gene polymorphism with knee osteoarthritis in a Thai population. Genetics and Molecular Research. 2011;10(3):1674–1680. doi: 10.4238/vol10-3gmr1161. [DOI] [PubMed] [Google Scholar]
- 117.Pola E, Papaleo P, Pola R, et al. Interleukin-6 gene polymorphism and risk of osteoarthritis of the hip: a case-control study. Osteoarthritis and Cartilage. 2005;13(11):1025–1028. doi: 10.1016/j.joca.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 118.Porée B, Kypriotou M, Chadjichristos C, et al. Interleukin-6 (IL-6) and/or soluble IL-6 receptor down-regulation of human type II collagen gene expression in articular chondrocytes requires a decrease of Sp1·Sp3 ratio and of the binding activity of both factors to the COL2A1 promoter. Journal of Biological Chemistry. 2008;283(8):4850–4865. doi: 10.1074/jbc.M706387200. [DOI] [PubMed] [Google Scholar]
- 119.Rowan AD, Koshy PJ, Shingleton WD, et al. Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis & Rheumatology. 2001;44(7):1620–1632. doi: 10.1002/1529-0131(200107)44:7<1620::AID-ART285>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 120.Cawston TE, Curry VA, Summers CA, et al. The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis & Rheumatology. 1998;41(10):1760–1771. doi: 10.1002/1529-0131(199810)41:10<1760::AID-ART8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 121.Sui Y, Lee JH, DiMicco MA, et al. Mechanical injury potentiates proteoglycan catabolism induced by interleukin-6 with soluble interleukin-6 receptor and tumor necrosis factor α in immature bovine and adult human articular cartilage. Arthritis and Rheumatism. 2009;60(10):2985–2996. doi: 10.1002/art.24857. [DOI] [PubMed] [Google Scholar]
- 122.Steeve KT, Marc P, Sandrine T, Dominique H, Yannick F. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine and Growth Factor Reviews. 2004;15(1):49–60. doi: 10.1016/j.cytogfr.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 123.Chenoufi H-L, Diamant M, Rieneck K, Lund B, Stein GS, Lian JB. Increased mRNA expression and protein secretion of interleukin-6 in primary human osteoblasts differentiated in vitro from rheumatoid and osteoarthritic bone. Journal of Cellular Biochemistry. 2001;81(4):666–678. doi: 10.1002/jcb.1104. [DOI] [PubMed] [Google Scholar]
- 124.Sakao K, Takahashi KA, Arai Y, et al. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. Journal of Bone and Mineral Metabolism. 2009;27(4):412–423. doi: 10.1007/s00774-009-0058-6. [DOI] [PubMed] [Google Scholar]
- 125.Wang P, Zhu F, Konstantopoulos K. Prostaglandin E2 induces interleukin-6 expression in human chondrocytes via cAMP/protein kinase A- and phosphatidylinositol 3-kinase-dependent NF-κB activation. American Journal of Physiology—Cell Physiology. 2010;298(6):C1445–C1456. doi: 10.1152/ajpcell.00508.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu X-H, Kirschenbaum A, Yao S, Levine AC. The role of the interleukin-6/gp130 signaling pathway in bone metabolism. Vitamins and Hormones. 2006;74:341–355. doi: 10.1016/S0083-6729(06)74014-6. [DOI] [PubMed] [Google Scholar]
- 127.de Hooge ASK, van de Loo FAJ, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis and Cartilage. 2005;13(1):66–73. doi: 10.1016/j.joca.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 128.van de Loo FAJ, Kuiper S, van Enckevort FHJ, Arntz OJ, van den Berg WB. Interleukin-6 reduces cartilage destruction during experimental arthritis: a study in interleukin-6-deficient mice. American Journal of Pathology. 1997;151(1):177–191. [PMC free article] [PubMed] [Google Scholar]
- 129.Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends in Pharmacological Sciences. 2012;33(1):35–41. doi: 10.1016/j.tips.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Perera LP. Interleukin 15: its role in inflammation and immunity. Archivum Immunologiae et Therapia Experimentalis. 2000;48(6):457–464. [PubMed] [Google Scholar]
- 131.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annual Review of Immunology. 1999;17:19–49. doi: 10.1146/annurev.immunol.17.1.19. [DOI] [PubMed] [Google Scholar]
- 132.Baslund B, Tvede N, Danneskiold-Samsoe B, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis and Rheumatism. 2005;52(9):2686–2692. doi: 10.1002/art.21249. [DOI] [PubMed] [Google Scholar]
- 133.Mcinnes IB, Al-Mughales J, Field M, et al. The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nature Medicine. 1996;2(2):175–182. doi: 10.1038/nm0296-175. [DOI] [PubMed] [Google Scholar]
- 134.Scanzello CR, Umoh E, Pessler F, et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis and Cartilage. 2009;17(8):1040–1048. doi: 10.1016/j.joca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 135.Sun JM, Sun LZ, Liu J, Su BH, Shi L. Serum interleukin-15 levels are associated with severity of pain in patients with knee osteoarthritis. Disease Markers. 2013;35(3):203–206. doi: 10.1155/2013/176278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell Signaling. 2011;23(7):1069–1075. doi: 10.1016/j.cellsig.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang X, Angkasekwinai P, Dong C, Tang H. Structure and function of interleukin-17 family cytokines. Protein and Cell. 2011;2(1):26–40. doi: 10.1007/s13238-011-1006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annual Review of Immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 139.Pawłowska J, Mikosik A, Soroczynska-Cybula M, et al. Different distribution of CD4 and CD8 t cells in synovial membrane and peripheral blood of rheumatoid arthritis and osteoarthritis patients. Folia Histochemica et Cytobiologica. 2009;47(4):627–632. doi: 10.2478/v10042-009-0117-9. [DOI] [PubMed] [Google Scholar]
- 140.Ishii H, Tanaka H, Katoh K, Nakamura H, Nagashima M, Yoshino S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthritis and Cartilage. 2002;10(4):277–281. doi: 10.1053/joca.2001.0509. [DOI] [PubMed] [Google Scholar]
- 141.Suurmond J, Dorjee AL, Boon MR, et al. Mast cells are the main interleukin-17-positive cells in anti-citrullinated protein antibody-positive and -negative rheumatoid arthritis and osteoarthritis synovium. Arthritis Research & Therapy. 2011;13(5, article R150) doi: 10.1186/ar3466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 142.Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis and Cartilage. 2006;14(4):345–352. doi: 10.1016/j.joca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 143.Alsalameh S, Mollenhauer J, Hain N, Stock K-P, Kalden JR, Burmester GR. Cellular immune response toward human articular chondrocytes: T cell reactivities against chondrocyte and fibroblast membranes in destructive joint diseases. Arthritis and Rheumatism. 1990;33(10):1477–1486. doi: 10.1002/art.1780331004. [DOI] [PubMed] [Google Scholar]
- 144.Chen B, Deng Y, Tan Y, Qin J, Chen LB. Association between severity of knee osteoarthritis and serum and synovial fluid interleukin 17 concentrations. Journal of International Medical Research. 2014;42(1):138–144. doi: 10.1177/0300060513501751. [DOI] [PubMed] [Google Scholar]
- 145.Lubberts E, Joosten LA, van de Loo FA, van den Gersselaar LA, van den Berg WB. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis & Rheumatology. 2000;43(6):1300–1306. doi: 10.1002/1529-0131(200006)43:6<1300::AID-ANR12>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 146.Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP. Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK) Arthritis & Rheumatology. 1999;42(11):2399–2409. doi: 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 147.Benderdour M, Tardif G, Pelletier J-P, et al. Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1β . Journal of Rheumatology. 2002;29(6):1262–1272. [PubMed] [Google Scholar]
- 148.Attur MG, Patel RN, Abramson SB, Amin AR. Interleukin-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis and Rheumatism. 1997;40(6):1050–1053. doi: 10.1002/art.1780400609. [DOI] [PubMed] [Google Scholar]
- 149.LeGrand A, Fermor B, Fink C, et al. Interleukin-1, tumor necrosis factor alpha, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin E2 production in explants of human osteoarthritic knee menisci. Arthritis and Rheumatism. 2001;44(9):2078–2083. doi: 10.1002/1529-0131(200109)44:9<2078::AID-ART358>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 150.Honorati MC, Bovara M, Cattini L, Piacentini A, Facchini A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthritis and Cartilage. 2002;10(10):799–807. doi: 10.1053/joca.2002.0829. [DOI] [PubMed] [Google Scholar]
- 151.Han L, Lee HS, Yoon JH, et al. Association of IL-17A and IL-17F single nucleotide polymorphisms with susceptibility to osteoarthritis in a Korean population. Gene. 2014;533(1):119–122. doi: 10.1016/j.gene.2013.09.113. [DOI] [PubMed] [Google Scholar]
- 152.Okamura H, Tsutsui H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378(6552):88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 153.Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;386(6625):619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 154.Saha N, Moldovan F, Tardif G, Pelletier JP, Cloutier JM, Martel-Pelletier J. Interleukin-1beta-converting enzyme/caspase-1 in human osteoarthritic tissues: localization and role in the maturation of interleukin-1beta and interleukin-18. Arthritis & Rheumatology. 1999;42(8):1577–1587. doi: 10.1002/1529-0131(199908)42:8<1577::AID-ANR3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 155.Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. Journal of Immunology. 1999;162(2):1096–1100. [PubMed] [Google Scholar]
- 156.Udagawa N, Horwood NJ, Elliott J, et al. Interleukin-18 (interferon-γ-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-γ to inhibit osteoclast formation. Journal of Experimental Medicine. 1997;185(6):1005–1012. doi: 10.1084/jem.185.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Möller B, Kukoc-Zivojnov N, Kessler U, et al. Interferon-gamma induces expression of interleukin-18 binding protein in fibroblast-like synoviocytes. Rheumatology. 2003;42(3):442–445. doi: 10.1093/rheumatology/keg146. [DOI] [PubMed] [Google Scholar]
- 158.Peng C-Z, Cao J-M, Xiao T, et al. Concentration of IL-18 and PGE2 in synovial fluid in patients with osteoarthritis and its significance. Journal of Central South University. 2006;31(6):862–865. [PubMed] [Google Scholar]
- 159.Wang Y, Xu D, Long L, Deng X, Tao R, Huang G. Correlation between plasma, synovial fluid and articular cartilage Interleukin-18 with radiographic severity in 33 patients with osteoarthritis of the knee. doi: 10.1007/s10238-013-0251-8. Clinical and Experimental Medicine. In press. [DOI] [PubMed] [Google Scholar]
- 160.Denoble AE, Huffman KM, Stabler TV, et al. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(5):2088–2093. doi: 10.1073/pnas.1012743108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Torigoe K, Ushio S, Okura T, et al. Purification and characterization of the human interleukin-18 receptor. Journal of Biological Chemistry. 1997;272(41):25737–25742. doi: 10.1074/jbc.272.41.25737. [DOI] [PubMed] [Google Scholar]
- 162.Born TL, Thomassen E, Bird TA, Sims JE. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. Journal of Biological Chemistry. 1998;273(45):29445–29450. doi: 10.1074/jbc.273.45.29445. [DOI] [PubMed] [Google Scholar]
- 163.Thomassen E, Bird TA, Renshaw BR, Kennedy MK, Sims JE. Binding of interleukin-18 to the interleukin-1 receptor homologous receptor IL-1Rrp1 leads to activation of signaling pathways similar to those used by interleukin-1. Journal of Interferon and Cytokine Research. 1998;18(12):1077–1088. doi: 10.1089/jir.1998.18.1077. [DOI] [PubMed] [Google Scholar]
- 164.Hulin-Curtis SL, Bidwell JL, Perry MJ. Evaluation of IL18 and IL18R1 polymorphisms: genetic susceptibility to knee osteoarthritis. International Journal of Immunogenetics. 2012;39(2):106–109. doi: 10.1111/j.1744-313X.2011.01060.x. [DOI] [PubMed] [Google Scholar]
- 165.Omair A, Lie BA, Reikeras O, Brox JI. An association study of interleukin 18 receptor genes (IL18R1 and IL18RAP) in lumbar disc degeneration. The Open Orthopaedics Journal. 2012;6:164–171. doi: 10.2174/1874325001206010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dai S-M, Shan Z-Z, Nishioka K, Yudoh K. Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: direct effect on chondrocytes may not be pivotal. Annals of the Rheumatic Diseases. 2005;64(5):735–742. doi: 10.1136/ard.2004.026088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Joosten LAB, Smeets RL, Koenders MI, et al. Interleukin-18 promotes joint inflammation and induces interleukin-1-driven cartilage destruction. American Journal of Pathology. 2004;165(3):959–967. doi: 10.1016/S0002-9440(10)63357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Inoue H, Hiraoka K, Hoshino T, et al. High levels of serum IL-18 promote cartilage loss through suppression of aggrecan synthesis. Bone. 2008;42(6):1102–1110. doi: 10.1016/j.bone.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 169.John T, Kohl B, Mobasheri A, Ertel W, Shakibaei M. Interleukin-18 induces apoptosis in human articular chondrocytes. Histology and Histopathology. 2007;22(5):469–482. doi: 10.14670/HH-22.469. [DOI] [PubMed] [Google Scholar]
- 170.Futani H, Okayama A, Matsui K, et al. Relation between interleukin-18 and PGE2 in synovial fluid of osteoarthritis: a potential therapeutic target of cartilage degradation. Journal of Immunotherapy. 2002;25(supplement 1):S61–S64. doi: 10.1097/00002371-200203001-00009. [DOI] [PubMed] [Google Scholar]
- 171.Li Y, Jiang J-M, Yang D-H, Wang F-L, Mao Z-X. Determination of the concentrations of interleukin-18 and other cytokines in the synovial fluid in patients with osteoarthritis. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(4):729–731. [PubMed] [Google Scholar]
- 172.Wang F-L, Jiang J-M, Wang F, Fu Z-Z, Zhang Z-F. Expressions of interleukin 18 and prostaglandin E2 and their correlation in the synoviocytes of patients with osteoarthritis. Nan Fang Yi Ke Da Xue Xue Bao. 2010;30(4):731–733. [PubMed] [Google Scholar]
- 173.Cho M-L, Jung YO, Moon Y-M, et al. Interleukin-18 induces the production of vascular endothelial growth factor (VEGF) in rheumatoid arthritis synovial fibroblasts via AP-1-dependent pathways. Immunology Letters. 2006;103(2):159–166. doi: 10.1016/j.imlet.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 174.Powers R, Garrett DS, March CJ, Frieden EA, Gronenborn AM, Clore GM. The high-resolution, three-dimensional solution structure of human interleukin-4 determined by multidimensional heteronuclear magnetic resonance spectroscopy. Biochemistry. 1993;32(26):6744–6762. doi: 10.1021/bi00077a030. [DOI] [PubMed] [Google Scholar]
- 175.Wlodawer A, Pavlovsky A, Gustchina A. Crystal structure of human recombinant interleukin-4 at 2.25 A resolution. FEBS Letters. 1992;309(1):59–64. doi: 10.1016/0014-5793(92)80739-4. [DOI] [PubMed] [Google Scholar]
- 176.Carr C, Aykent S, Kimack NM, Levine AD. Disulfide assignments in recombinant mouse and human interleukin 4. Biochemistry. 1991;30(6):1515–1523. doi: 10.1021/bi00220a011. [DOI] [PubMed] [Google Scholar]
- 177.Mueller TD, Zhang J-L, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochimica et Biophysica Acta—Molecular Cell Research. 2002;1592(3):237–250. doi: 10.1016/s0167-4889(02)00318-x. [DOI] [PubMed] [Google Scholar]
- 178.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annual Review of Immunology. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 179.LaPorte SL, Juo ZS, Vaclavikova J, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132(2):259–272. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radical Biology & Medicine. 2013;54:1–16. doi: 10.1016/j.freeradbiomed.2012.10.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. Journal of Allergy and Clinical Immunology. 2000;105(6, part 1):1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 182.Forster T, Chapman K, Loughlin J. Common variants within the interleukin 4 receptor α gene (IL4R) are associated with susceptibility to osteoarthritis. Human Genetics. 2004;114(4):391–395. doi: 10.1007/s00439-004-1083-0. [DOI] [PubMed] [Google Scholar]
- 183.Vargiolu M, Silvestri T, Bonora E, et al. Interleukin-4/interleukin-4 receptor gene polymorphisms in hand osteoarthritis. Osteoarthritis and Cartilage. 2010;18(6):810–816. doi: 10.1016/j.joca.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 184.Yigit S, Inanir A, Tekcan A, et al. Significant association of interleukin-4 gene intron 3 VNTR polymorphism with susceptibility to knee osteoarthritis. Gene. 2014;537(1):6–9. doi: 10.1016/j.gene.2013.12.060. [DOI] [PubMed] [Google Scholar]
- 185.Schlaak JF, Pfers I, Meyer Zum Büschenfelde K-H, Märker-Hermann E. Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clinical and Experimental Rheumatology. 1996;14(2):155–162. [PubMed] [Google Scholar]
- 186.Ishii H, Tanaka H, Katoh K, Nakamura H, Nagashima M, Yoshino S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthritis and Cartilage. 2002;10(4):277–281. doi: 10.1053/joca.2001.0509. [DOI] [PubMed] [Google Scholar]
- 187.Silvestri T, Pulsatelli L, Dolzani P, Facchini A, Meliconi R. Elevated serum levels of soluble interleukin-4 receptor in osteoarthritis. Osteoarthritis and Cartilage. 2006;14(7):717–719. doi: 10.1016/j.joca.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 188.Wagner S, Fritz P, Einsele H, Sell S, Saal JG. Evaluation of synovial cytokine patterns in rheumatoid arthritis and osteoarthritis by quantitative reverse transcription polymerase chain reaction. Rheumatology International. 1997;16(5):191–196. doi: 10.1007/BF01330295. [DOI] [PubMed] [Google Scholar]
- 189.Demaziere A, Leek R, Athanasou NA. Histological distribution of the interleukin-4 receptor (IL4R) within the normal and pathological synovium. Revue du Rhumatisme et des Maladies Osteo-Articulaires. 1992;59(3):219–224. [PubMed] [Google Scholar]
- 190.Yeh L-A, Augustine AJ, Lee P, Riviere LR, Sheldon A. Interleukin-4, an inhibitor of cartilage breakdown in bovine articular cartilage explants. Journal of Rheumatology. 1995;22(9):1740–1746. [PubMed] [Google Scholar]
- 191.van Meegeren ME, Roosendaal G, Jansen NW, et al. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthritis and Cartilage. 2012;20(7):764–772. doi: 10.1016/j.joca.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 192.van Lent PLEM, Holthuysen AEM, Slöetjes A, Lubberts E, van den Berg WB. Local overexpression of adeno-viral IL-4 protects cartilage from metallo proteinase-induced destruction during immune complex-mediated arthritis by preventing activation of pro-MMPs. Osteoarthritis and Cartilage. 2002;10(3):234–243. doi: 10.1053/joca.2001.0501. [DOI] [PubMed] [Google Scholar]
- 193.Doi H, Nishida K, Yorimitsu M, et al. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin b expression by rat normal chondrocytes. Acta Medica Okayama. 2008;62(2):119–126. doi: 10.18926/AMO/30956. [DOI] [PubMed] [Google Scholar]
- 194.Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Differential responses of chondrocytes from normal and osteoarthritic human articular cartilage to mechanical stimulation. Biorheology. 2002;39(1-2):97–108. [PubMed] [Google Scholar]
- 195.Millward-Sadler SJ, Wright MO, Lee H-S, et al. Integrin-regulated secretion of interleukin 4: a novel pathway of mechanotransduction in human articular chondrocytes. Journal of Cell Biology. 1999;145(1):183–189. doi: 10.1083/jcb.145.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Millward-Sadler SJ, Wright MO, Davies LW, Nuki G, Salter DM. Mechanotransduction via integrins and interleukin-4 results in altered aggrecan and matrix metalloproteinase 3 gene expression in normal, but not osteoarthritic, human articular chondrocytes. Arthritis & Rheumatology. 2000;43(9):2091–2099. doi: 10.1002/1529-0131(200009)43:9<2091::AID-ANR21>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 197.Schuerwegh AJ, Dombrecht EJ, Stevens WJ, Van Offel JF, Bridts CH, De Clerck LS. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthritis and Cartilage. 2003;11(9):681–687. doi: 10.1016/s1063-4584(03)00156-0. [DOI] [PubMed] [Google Scholar]
- 198.Relić B, Guicheux J, Mezin F, et al. IL-4 and IL-13, but not IL-10, protect human synoviocytes from apoptosis. Journal of Immunology. 2001;166(4):2775–2782. doi: 10.4049/jimmunol.166.4.2775. [DOI] [PubMed] [Google Scholar]
- 199.Rai MF, Graeve T, Twardziok S, Schmidt MFG. Evidence for regulated interleukin-4 expression in chondrocyte-scaffolds under in vitro inflammatory conditions. PLoS ONE. 2011;6(10) doi: 10.1371/journal.pone.0025749.e25749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Alaaeddine N, Di Battista JA, Pelletier JP, et al. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis & Rheumatology. 1999;42(4):710–718. doi: 10.1002/1529-0131(199904)42:4<710::AID-ANR14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 201.Yorimitsu M, Nishida K, Shimizu A, et al. Intra-articular injection of interleukin-4 decreases nitric oxide production by chondrocytes and ameliorates subsequent destruction of cartilage in instability-induced osteoarthritis in rat knee joints. Osteoarthritis and Cartilage. 2008;16(7):764–771. doi: 10.1016/j.joca.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 202.Fu J, Zhang L, Song S, et al. Effect of bone marrow-derived 10 CD11b+F4/80+immature dendritic cells on the balance between pro-inflammatory and anti-inflammatory cytokines in DBA/1 mice with collagen-induced arthritis. Inflammation Research. 2014;63(5):357–367. doi: 10.1007/s00011-014-0707-7. [DOI] [PubMed] [Google Scholar]
- 203.Vieira P, De Waal-Malefyt R, Dang M-N, et al. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(4):1172–1176. doi: 10.1073/pnas.88.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tan JC, Indelicato SR, Narula SK, Zavodny PJ, Chou C-C. Characterization of interleukin-10 receptors on human and mouse cells. Journal of Biological Chemistry. 1993;268(28):21053–21059. [PubMed] [Google Scholar]
- 205.Zdanov A, Schalk-Hihi C, Gustchina A, Tsang M, Weatherbee J, Wlodawer A. Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon γ . Structure. 1995;3(6):591–601. doi: 10.1016/s0969-2126(01)00193-9. [DOI] [PubMed] [Google Scholar]
- 206.Liu Y, Wei SH-Y, Ho AS-Y, Malefyt RDW, Moore KW. Expression cloning and characterization of a human IL-10 receptor. Journal of Immunology. 1994;152(4):1821–1829. [PubMed] [Google Scholar]
- 207.Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. The EMBO Journal. 1997;16(19):5894–5903. doi: 10.1093/emboj/16.19.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1α and STAT3 complexes in human T cells and monocytes. Journal of Immunology. 1995;155(3):1079–1090. [PubMed] [Google Scholar]
- 209.Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. Journal of Interferon and Cytokine Research. 1999;19(6):563–573. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- 210.Iannone F, De Bari C, Dell’ Accio F, et al. Interleukin-10 and interleukin-10 receptor in human osteoarthritic and healthy chondrocytes. Clinical and Experimental Rheumatology. 2001;19(2):139–145. [PubMed] [Google Scholar]
- 211.Jansen NWD, Roosendaal G, Hooiveld MJJ, et al. Interleukin-10 protects against blood-induced joint damage. British Journal of Haematology. 2008;142(6):953–961. doi: 10.1111/j.1365-2141.2008.07278.x. [DOI] [PubMed] [Google Scholar]
- 212.Wang Y, Lou S. Direct protective effect of interleukin-10 on articular chondrocytes in vitro . Chinese Medical Journal. 2001;114(7):723–725. [PubMed] [Google Scholar]
- 213.John T, Müller RD, Oberholzer A, et al. Interleukin-10 modulates pro-apoptotic effects of TNF-α in human articular chondrocytes in vitro . Cytokine. 2007;40(3):226–234. doi: 10.1016/j.cyto.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 214.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer J-M. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. Journal of Clinical Investigation. 1995;96(5):2304–2310. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chadban SJ, Tesch GH, Foti R, Atkins RC, Nikolic-Paterson DJ. Interleukin-10 is a mesangial cell growth factor in vitro and in vivo . Laboratory Investigation. 1997;76(5):619–627. [PubMed] [Google Scholar]
- 216.Jung YK, Kim GW, Park HR, et al. Role of interleukin-10 in endochondral bone formation in mice: anabolic effect via the bone morphogenetic protein/Smad pathway. Arthritis & Rheumatology. 2013;65(12):3153–3164. doi: 10.1002/art.38181. [DOI] [PubMed] [Google Scholar]
- 217.Umulis D, O’Connor MB, Blair SS. The extracellular regulation of bone morphogenetic protein signaling. Development. 2009;136(22):3715–3728. doi: 10.1242/dev.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zeng L, Kempf H, Murtaugh LC, Sato ME, Lassar AB. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes and Development. 2002;16(15):1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chimal-Monroy J, Rodriguez-Leon J, Montero JA, et al. Analysis of the molecular cascade responsible for mesodermal limb chondrogenesis: sox genes and BMP signaling. Developmental Biology. 2003;257(2):292–301. doi: 10.1016/s0012-1606(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 220.Minina E, Wenzel HM, Kreschel C, et al. BMP and lhh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128(22):4523–4534. doi: 10.1242/dev.128.22.4523. [DOI] [PubMed] [Google Scholar]
- 221.Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Developmental Cell. 2002;3(3):439–449. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- 222.Riyazi N, Slagboom E, De Craen AJM, et al. Association of the risk of osteoarthritis with high innate production of interleukin-1β and low innate production of interleukin-10 ex vivo, upon lipopolysaccharide stimulation. Arthritis and Rheumatism. 2005;52(5):1443–1450. doi: 10.1002/art.21014. [DOI] [PubMed] [Google Scholar]
- 223.Helmark IC, Mikkelsen UR, Børglum J, et al. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: a randomized controlled trial. Arthritis Research and Therapy. 2010;12(4, article R126) doi: 10.1186/ar3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lems WF, den Uyl D. Exercise-induced changes in interleukin-10 in patients with knee osteoarthritis: new perspectives? Arthritis Research and Therapy. 2010;12(4, article 131) doi: 10.1186/ar3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Angele P, Yoo JU, Smith C, et al. Cyclic hydrostatic pressure enhances the chondrogenic phenotype of human mesenchymal progenitor cells differentiated in vitro . Journal of Orthopaedic Research. 2003;21(3):451–457. doi: 10.1016/S0736-0266(02)00230-9. [DOI] [PubMed] [Google Scholar]
- 226.Mizuno S, Tateishi T, Ushida T, Glowacki J. Hydrostatic fluid pressure enhances matrix synthesis and accumulation by bovine chondrocytes in three-dimensional culture. Journal of Cellular Physiology. 2002;193(3):319–327. doi: 10.1002/jcp.10180. [DOI] [PubMed] [Google Scholar]
- 227.Eisenmesser EZ, Horita DA, Byrd RA. Secondary structure and backbone resonance assignments for human interleukin-13. Journal of Biomolecular NMR. 2001;19(1):93–94. doi: 10.1023/a:1008336103603. [DOI] [PubMed] [Google Scholar]
- 228.Eisenmesser EZ, Horita DA, Altieri AS, Byrd RA. Solution structure of interleukin-13 and insights into receptor engagement. Journal of Molecular Biology. 2001;310(1):231–241. doi: 10.1006/jmbi.2001.4765. [DOI] [PubMed] [Google Scholar]
- 229.Hart PH, Ahern MJ, Smith MD, Finlay-Jones JJ. Regulatory effects of IL-13 on synovial fluid macrophages and blood monocytes from patients with inflammatory arthritis. Clinical and Experimental Immunology. 1995;99(3):331–337. doi: 10.1111/j.1365-2249.1995.tb05554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.de Waal Malefyt R, Figdor CG, Huijbens R, et al. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes: comparison with IL-4 and modulation by IFN-γ or IL-10. Journal of Immunology. 1993;151(11):6370–6381. [PubMed] [Google Scholar]
- 231.Fernandes JC, Martel-Pelletier J, Pelletier J-P. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39(1-2):237–246. [PubMed] [Google Scholar]
- 232.Jovanovic D, Pelletier J-P, Alaaeddine N, et al. Effect of IL-13 on cytokines, cytokine receptors and inhibitors on human osteoarthritis synovium and synovial fibroblasts. Osteoarthritis and Cartilage. 1998;6(1):40–49. doi: 10.1053/joca.1997.0091. [DOI] [PubMed] [Google Scholar]
- 233.Altman R, Asch E, Bloch D. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Arthritis and Rheumatism. 1986;29(8):1039–1052. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 234.Onoe Y, Miyaura C, Kaminakayashiki T, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. Journal of Immunology. 1996;156(2):758–764. [PubMed] [Google Scholar]
- 235.Blaney Davidson EN, van der Kraan PM, van den Berg WB. TGF-β and osteoarthritis. Osteoarthritis and Cartilage. 2007;15(6):597–604. doi: 10.1016/j.joca.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 236.de Caestecker M. The transforming growth factor-β superfamily of receptors. Cytokine and Growth Factor Reviews. 2004;15(1):1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 237.Blaney Davidson EN, Vitters EL, Van Der Kraan PM, Van Den Berg WB. Expression of transforming growth factor-β (TGFβ) and the TGFβ signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Annals of the Rheumatic Diseases. 2006;65(11):1414–1421. doi: 10.1136/ard.2005.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Verrecchia F, Mauviel A. Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. Journal of Investigative Dermatology. 2002;118(2):211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 239.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 240.Attur MG, Patel IR, Patel RN, Abramson SB, Amin AR. Autocrine production of IL-1β by human osteoarthritis-affected cartilage and differential regulation of endogenous nitric oxide, IL-6, prostaglandin E2, and IL-8. Proceedings of the Association of American Physicians. 1998;110(1):65–72. [PubMed] [Google Scholar]