

Abstract
The timing of therapeutic intervention in traumatic brain injury (TBI) is critical. Although immediate cell death cascades have become established in adult TBI, the pathophysiology underlying neonatal TBI is poorly understood. The objective of the present study was to determine the role of cytokine regulation following TBI in neonatal rats. Seven-day-old Sprague-Dawley rats were subjected to TBI using the controlled cortical impact (CCI) injury model. Age-matched littermates that did not receive TBI served as the controls. Immediately following TBI, rats were euthanized, and the brains were divided into the ipsilateral and contralateral hemispheres then flash frozen. A BioRad 23-Plex panel was used to measure cytokine levels. Surprisingly, the data revealed that 18 of the 23 cytokines analyzed were significantly downregulated in the hemisphere contralateral to the TBI impacted hemisphere. IL-5, IL-6 and MIP-3a were significantly suppressed in both ipsilateral and contralateral hemispheres of neonatal TBI rats compared to the control rats. A parallel study processing the plasma of the same cohort of neonatal rats revealed no difference in the same cytokines analyzed in the brain tissue, suggesting highly localized cytokine suppression in the brain during early injury. In stark contrast to the reported early pro-inflammatory response exhibited in adult TBI, the present neonatal TBI study demonstrated a reversed cytokine profile of downregulation. These results suggest a robust, immediate anti-inflammatory response mounted by the contralateral hemisphere of the young brain.
Keywords: Traumatic Brain Injury, Neonatal Traumatic Brain Injury, Cytokine Expression, Neuroinflammation
1. Introduction
A traumatic brain injury (TBI) can be defined as a force to the head or to the brain which disrupts proper neurological functions of the brain. Rapid brain acceleration-deceleration, as seen in shaken baby syndrome, can cause diffuse damage or focal damage at the point of impact (coup) and at the opposite pole (countercoup). In addition, traumatic axonal injury (TAI) is present in which axons and blood vessels are sheared, causing contusions, subdural or epidural hematomas, and intracerebral or subarachnoid hemorrhages (Altimier, 2008; Case, 2007).
TBI contributes to a substantial number of deaths and cases of permanent disability annually. To date, numerous studies have detailed both the effects and potential treatments of adult TBI. Many pharmacological and rehabilitative therapies have been tested in adult models (Chung et al., 2013; Wheaton et al., 2011). However, research focusing on neonatal TBI is sparse despite the large young patient population prone to TBI. Indeed, very young children (age 0–4 years) and the elderly (over 65 years) are more likely to sustain a TBI (Andelic et al., 2008; Faul et al., 2010; Levchakov et al., 2006). Falls and drops are the leading cause of TBI-linked death in young children (Karasu et al., 2009). They account for 64% of TBI-related emergency department visits and 42% of hospitalizations. Motor vehicle accidents account for 40% of all TBI-related deaths in young children (Faul et al., 2010), and shaken baby syndrome accounts for a significant portion of TBI in young children as well, especially those under 6 months (Barr et al., 2006; Paiva et al., 2011). Moreover, very few studies detail evidence-based therapy for children who suffered TBI in infancy (Ashton, 2010). Traditional views maintain that the plasticity from young brains enable younger TBI victims to experience a greater recovery. Contrary to these beliefs, more recent research suggests that very early TBI can have a significant negative impact on brain development (Anderson et al., 2005, 2009; Crowe, 2012; Maxwell, 2012); however, studies are not conclusive as to exact cell death mechanisms closely associated with neurodevelopmental impairments (Anderson et al., 2005, 2009).
In experimental adult rodent TBI, the brain attempts to repair itself via endogenous repair mechanisms including cell proliferation shortly after TBI (Kaneko et al., 2013; Sun et al., 2009). However, these mechanisms cannot sufficiently remedy secondary cell death and severe inflammatory response (Kaneko et al., 2013; Woodcock and Morganti-Kossmann, 2013). In chronic studies, research indicates neuroinflammation, which can be observed up to 17 years post TBI in humans (Giunta et al., 2012), as a major secondary cell death pathway culminating in neuron loss, impeded cell proliferation and an upregulation of activated microglia cells. All these neuroinflammatory responses can interfere with endogenous repair mechanisms (Acosta et al., 2013; Woodcock and Morganti-Kossmann, 2013). In fact, waves of cytokines and chemokines mediate this inflammatory response throughout the brain (Das et al., 2012; Giunta et al., 2012; Hernandez-Ontiveros et al., 2013; Waters et al., 2013) and facilitate the activation and recruitment of immune cells to the injury (Das et al., 2012; Woodcock and Morganti-Kossmann, 2013). Whether such neuroinflammation accompanies neonatal TBI and the cell death cascades associated with this inflammation remains a needed field of research examination. Investigation of these neuroinflammatory mechanisms will lead to both a greater understanding of the pathology of TBI and insights in potential therapy to arrest the disease evolution (Rovegno et al., 2012).
2. Results
Twenty-three different cytokines were analyzed in this study: IL-1a, IL-b, IL-2, IL-4, IL-6, IL-10, GM-CSF, INF-g, TNF-a, EPO, G-CSF, IL-5, GRO/KC, IL-7, IL-12p70, IL-13, IL-17A, IL-18, M-CSF, MIP-1a, MIP-3A, RANTES and VEGF. Analysis of cytokine levels in the harvested brains exposed sizable downregulation of 18 out of the 23 cytokines in the contralateral hemisphere of TBI inflicted rats relative to the controls. These cytokines included IL-5, IL-6 and MIP-3a which were downregulated in both hemispheres (Figure 1).
Figure 1. Global decrease in IL-6, IL-5 and MIP-3a is observed following TBI.
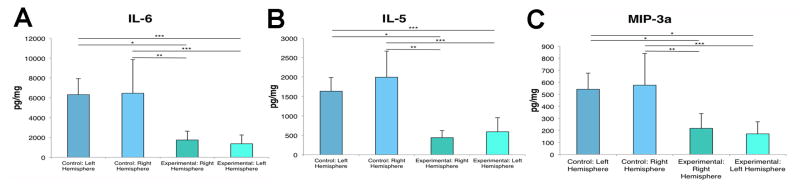
ANOVA revealed that IL-6 (A), IL-5 (B) and MIP-3a (C) were significantly decreased in both the contralateral and ipsilateral hemispheres of the brain following TBI compared to the control brains. Bars represent mean ± SD; (*) p<0.05, (**) p<0.005, (***) p<0.001.
IL-10, IL-18 and IL-17A were significantly downregulated in both the contralateral and ipsilateral hemispheres of the TBI inflicted brains compared to the right hemisphere of the control brains and significantly downregluated in the left hemisphere compared to the left hemisphere of the controls (Figure 2). IL-1a, IL-4, IL-B, EPO and IL-12p70 levels decreased in the ipsilateral hemisphere of the brain following TBI compared to right hemisphere of the control brains (Figure 3). IL-2 was significantly downregulated in the ipsilateral hemisphere of the brain with TBI compared to the control brains (Figure 4). GM-CSF and G-CSF were both significantly decreased throughout the TBI inflicted brains compared to the right hemisphere of the control brains (Figure 5).
Figure 2. Global decrease in IL-10, IL-18 and IL-17A is observed following TBI.
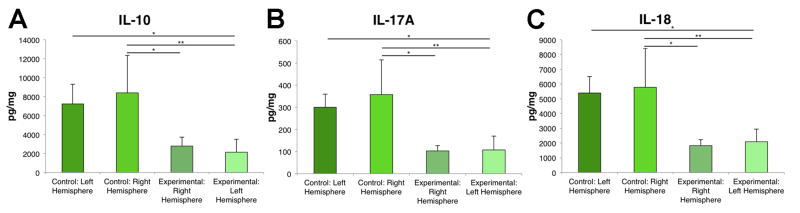
ANOVA revealed that IL-10 (A), IL-17A (B) and IL-18 (C) were significantly decreased in both the contralateral and ipsilateral hemispheres of the brain following TBI compared to the control brains. IL-10, IL-18 and IL-17A were significantly decreased in the left (TBI) hemisphere compared to the left hemisphere of the control animals. Bars represent mean ± SD; (*) p<0.05, (**) p<0.005.
Figure 3. IL-1a, IL-4, IL-B, EPO and IL-12p70 are decreased in the TBI-impacted hemisphere.
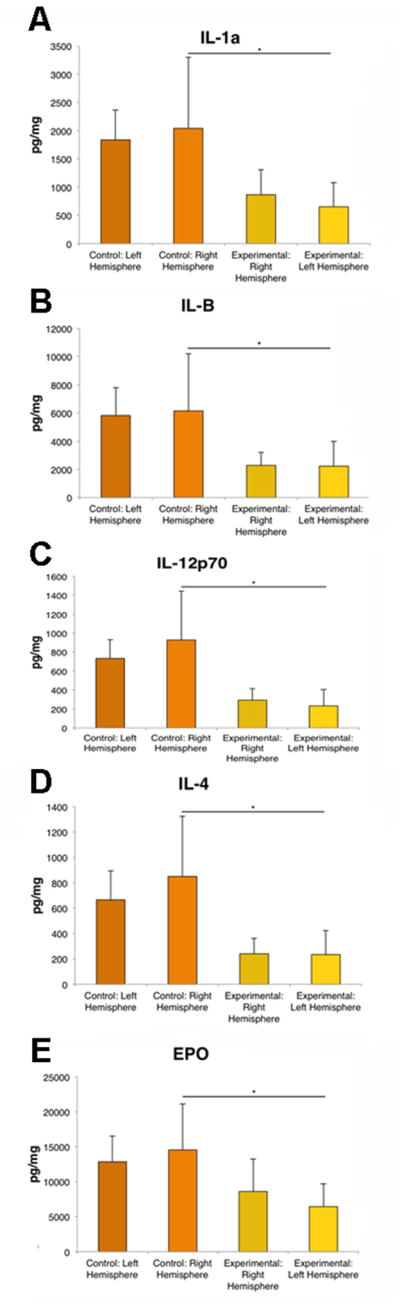
ANOVA revealed that IL-1a (A), IL-B (B), IL-12p70 (C), IL-4 (D), and EPO (E) were significantly decreased in ipsilateral hemisphere of the brain following TBI compared to the control brains. Bars represent mean ± SD; (*) p<0.05.
Figure 4. IL-2 is downregulated in the ipsilateral hemisphere following TBI.
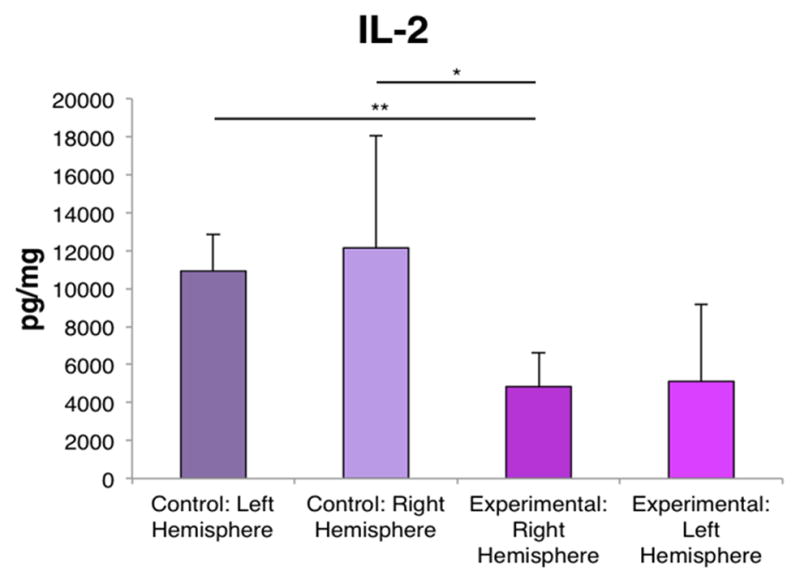
ANOVA revealed that IL-2 was significantly decreased in the ipsilateral hemisphere of the brain following TBI compared to the control brains. Bars represent mean ± SD; (*) p<0.05, (**) p<0.005.
Figure 5. GM-CSF and G-CSF are decreased following TBI.
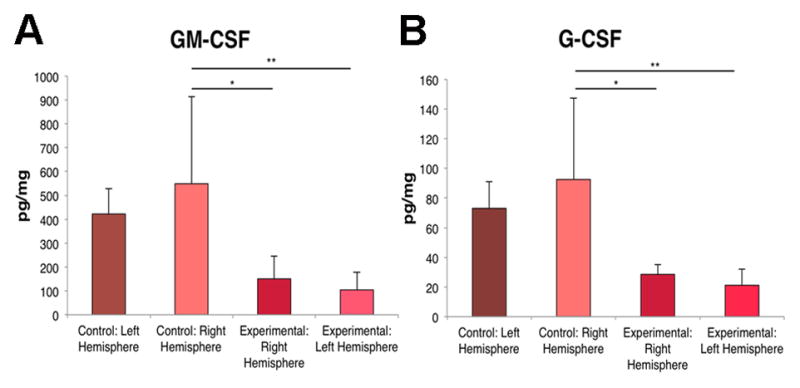
ANOVA revealed that GM-CSF (A) and G-CSF (B) were significantly decreased in both ipsilateral and contralateral hemispheres of the TBI brains compared to the control brains. Bars represent mean ± SD; (*) p<0.05, (**).
The five remaining cytokines did not signficantly differ between the TBI and control rats. These results indicate an overwhelming downregulation of the entire range of cytokines analyzed. Of note, plasma of the same cohort of neonatal rats revealed no significant change in the cytokine levels analyzed in the brain tissue.
3. Discussion
A significant downregulation of 18 out of the 23 cytokines analyzed was observed in the contralateral hemisphere of the brain following TBI in neonatal rats (n=10 per treatment group). The other five cytokines did not significantly differ between the experimental and control groups; therefore, we observed a clear trend of cytokine downregulation in the TBI-afflicted neonatal brain. In addition, analysis of the blood plasma showed no significant changes in the cytokine levels observed in the brain tissue, indicating localized downregulation of inflammatory cytokines in the TBI brain.
Although neonatal TBI remains a relatively unexplored frontier, in adults the cytokine response to regulate inflammation post TBI is well characterized (Das et al., 2012; Giunta et al., 2012; Hernandez-Ontiveros et al., 2013; Utagawa et al., 2008; Waters et al., 2013; Woodcock and Morganti-Kossmann, 2013). We found, however, that the cytokine response in neonatal studies is distinctly different; a profile of downregulation was observed instead of the upregulation found in adults (Lloyd et al., 2008; Shojo et al., 2010; Utagawa et al., 2008). Few studies have examined cytokine levels in neonates. A study with mice reported lower baseline cytokine levels before injury in younger—although not neonatal—organisms as opposed to older ones (Timaru-Kast et al., 2012). These reduced levels suggest an anti-inflammatory disposition in younger organisms relative to their older counterparts, supporting our findings. In neonatal hypoxia/ischemia, a shift from pro-inflammatory cytokines to anti-inflammatory cytokines was observed as the injury progressed (Shrivastava et al., 2013), backing the anti-inflammatory response we observed in the neonatal brain following TBI. However, this response following TBI is much more immediate and robust. Further research into the exact molecular mechanisms of the response remains an open area of investigation.
The present study showed that exemplary pro-inflammatory cytokines IL-1 and IL-6 (Woodcock and Morganti-Kossmann, 2013) were significantly suppressed following TBI in neonates. In contrast to the early upregulation of inflammatory response markers in adult TBI (Kaneko et al., 2013; Woodcock and Morganti-Kossmann, 2013), cytokine levels were downregulated in the neonatal brain, suggesting the contalateral hemisphere mounts a robust endogenous anti-inflammatory response. This response is localized to the brain, evidenced by the absence of altered plasma cytokine levels. In adults, chronic inflammation causes neuron loss, impedes endogenous repair mechanisms and exacerbates the immune response (Acosta et al., 2013; Woodcock and Morganti-Kossmann, 2013). These data suggest the downregulation of cytokines found in neonates allows endogenous repair mechanisms to work unhindered by checking the inflammatory response of the immune system. As a result, there should be a more successful increase in cell proliferation and differentiation in neonates to mitigate the detrimental effects of TBI. As expected, a more complete recovery from TBI is seen in younger humans (Anderson et al., 2005; Niedzwecki et al., 2008); however the exact response and effects across young age groups from neonates to older children warrants further study. While neonatal TBI is largely unexamined, the anti-inflammatory response stands in direct contrast to the well-characterized inflammatory response found in adult TBI and opens up an exciting opportunity for research.
That the anti-inflammatory response in neonatal brain may be a therapeutic target for TBI is an appealing venue for developing neuroprotective strategies. A caveat though is that the error bars in the right hemisphere (with TBI) group appear larger than the other treatment groups. Such variability following injury suggests varying levels of neuroplasticity in individual animals, in this case newborn pups, which are closely detected within the injured brain region, i.e., right hemisphere with TBI. Although our observations indicate a general robust neuroplasticity in neonates, monitoring the individual subject’s response to TBI will facilitate optimal treatment regimens. Another caution we raised here is that two of the cytokines, namely IL-4 and IL-10, analyzed here are anti-inflammatory, which at first glance appeared to be down-regulated as well. However, an additional assay for these specific cytokines compared with the adult TBI brain using the same timing post-TBI (i.e., 10 minutes after TBI insult and decapitation) revealed that the TBI neonate IL4 and IL10 still appeared upregulated compared to non-detectable levels in adult TBI brain. Altogether, these results indicate that specific cytokines with anti-inflammatory properties remained elevated in TBI neonates supporting our claim of robust neuroplasticity in the young animals compared to adults.
Future therapies designed to assist this endogenous repair mechanism may prove to be a beneficial avenue of exploration for neonatal TBI treatment. Neuroinflammation-mediated secondary cell death in chronic TBI suggests that a treatment boosting the anti-inflammatory response mounted by the contralateral hemisphere may prove effective in combating neonatal TBI by ameliorating secondary injury (Gatson et al., 2013). However, a greater understanding of the cytokine inflammatory regulation after neonatal TBI can lead to more effective therapies for adult TBI as well. Evidence suggests that if the anti-inflammatory response we found in neonates can be trigged or artificially reproduced in adults, it will ameliorate many of the detrimental effects of TBI resulting from neuroinflammation. Reduced inflammation also induces greater recovery as a result of uninhibited endogenous repair mechanisms. Stem cell therapy has proved effective in ameliorating the damage from ischemic stroke, possibly by boosting these endogenous repair mechanisms (Tajiri et al., 2012), and translational research on stem cell therapy for neonatal ischemic stroke is already available (Pabon and Borlongan, 2013). Therefore, we propose that a stem cell-based approach could potentially boost the endogenous repair mechanisms and the anti-inflammatory response following neonatal TBI. The present study and future studies detailing the neuroinflammatory response accompanying neonatal TBI may lead to potential therapies for TBI-afflicted individuals of all ages. The rapid reduction in the cytokine levels in TBI neonates supports the neonatal brain’s high capacity to mount neuroplasticity but also suggests the immature brain’s ability to exert an anti-inflammatory response, which has been implicated as a major early pro-survival mechanism associated with TBI and other neurological disorders. Our results suggest that the neonatal brain appears to trigger such anti-inflammatory pathway much more rapidly than the adult brain. We nonetheless acknowledged that the specific cellular and molecular pathways associated with this robust anti-inflammatory response by the neonatal brain warrant additional studies.
In the present study, we primarily examined the immediate brain cytokine response in neonatal TBI. Data are only indicative of the direct response of the brain. Indeed, the plasma from the same neonatal TBI animals revealed no significant alterations in any of the cytokine analyzed here. These results suggest the presence of highly localized cytokine supression in the brain during the early injury phase which was not reflected in peripheral circulation. Furthermore, the present neonatal TBI study reveals a reversed cytokine trend of downregulation, opposite to the pro-inflammatory response present in adult TBI. Nonetheless, our results do not reveal if the anti-inflammatory response is sufficient and stable over long-term to abrogate the detrimental effects of TBI, or if supplementary anti-inflammatory treatment in neonates can enhance recovery. Future directions to consider include chronic studies to examine the long-term anti-inflammatory response in neonates as well as in vivo experiments in a neonatal TBI model. Our study suggests that neonates will likely exhibit improved recovery and less chronic inflammation relative to adults after TBI, but these speculations warrant further studies in order to determine the robustness of the neonatal endogenous repair mechanism and whether adjunctive anti-inflammatory regimen is required for abrogating neonatal TBI in the clinic.
4. Experimental Procedure
Seven-day old male Sprague-Dawley rats (n = 10 per group) were subjected to TBI using the controlled cortical impact (CCI) injury model (Pittsburgh Precision Instruments, Inc, USA). Age-matched littermates that did not receive TBI served as the controls. Animals were anesthetized using 1–2% isoflurane, and once deep anesthesia was achieved, individual animals were fixed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). Anesthesia was maintained via gas mask. After exposing the skull, a 4.0 mm craniectomy was performed over the right frontoparietal cortex (−2.0 mm anteroposterior and +2.0 mm mediolateral to bregma). The pneumatically operated TBI device, outfitted with a convex tip diameter of 3.0 mm for rats, impacted the brain at a velocity of 6.0 m/s, reached a depth of 1.0 mm to achieve a moderate TBI model below the dura mater layer (Glover et al., 2012), and then remained in the brain for 150 ms. The impactor rod was angled 15° to the vertical so that it remained perpendicular to the tangential plane of brain curvature at the impact surface. A linear variable displacement transducer (Macrosensors, Pennsauken, NJ) connected to the impactor measured velocity and duration to verify consistency. This CCI approach produced skull fracture overlying the frontal cortex (Glover et al., 2012). Immediately following TBI (i.e., 10 minutes from TBI insult and the time the animal was decapitated), rats were euthanized and the brains were divided into the ipsilateral and contralateral hemispheres then flash frozen. A BioRad 23-Plex protein assay panel (Bio-Rad, Hercules, CA) was used to measure cytokine levels. Through the use of the 23-Plex multiplex bead-based suspension array system, coupled reactions of the specific beads isolated the desired biomarkers. The entire sample was then thoroughly washed to eliminate the unbounded, unwanted proteins. A promptly added biotinylated detection antibody formed the final detection complex. Analysis was performed according to system and panel instructions. Measurement of cytokines was conducted by comparing expression levels in TBI neonatal brains relative to control non-injured, age-matched control brains.
Highlights.
We examined neonatal TBI in seven-day-old Sprague-Dawley rats
We analyzed cytokine levels in the brains directly following TBI
A trend of downregulation is evident in cytokine levels after neonatal TBI
Neonatal TBI is accompanied by a localized anti-inflammatory response as opposed to the neuroinflammation seen in adult TBI
Therapies designed to augment the anti-inflammatory response may prove effective in abrogating TBI in neonates
Acknowledgments
This research was supported by the Department of Neurosurgery and Brain Repair funds.
Footnotes
Conflict of Interests: CVB is supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke 1R01NS071956-01, Department of Defense W81XWH-11-1-0634, James and Esther King Foundation for Biomedical Research Program, SanBio Inc., KMPHC and NeuralStem Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Acosta SA, et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One. 2013;8:53376. doi: 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altimier L. Shaken Baby Syndrome. J Perinat Neonatal Nurs. 2008;22:77–8. doi: 10.1097/01.JPN.0000311877.32614.69. [DOI] [PubMed] [Google Scholar]
- 3.Andelic N, et al. Incidence of hospital-treated traumatic brain injury in the Oslo population. Neuroepidemiology. 2008;30:120–128. doi: 10.1159/000120025. [DOI] [PubMed] [Google Scholar]
- 4.Anderson V, et al. Functional plasticity or vulnerability after early brain injury? Pediatrics. 2005;116:1374–1382. doi: 10.1542/peds.2004-1728. [DOI] [PubMed] [Google Scholar]
- 5.Anderson V, et al. Childhood brain insult: can age at insult help us predict outcome? Brain. 2009;132:456. doi: 10.1093/brain/awn293. [DOI] [PubMed] [Google Scholar]
- 6.Ashton R. Practitioner review: beyond shaken baby syndrome: what influences the outcomes for infants following traumatic brain injury? J Child Psychol Psychiatry. 2010;51:967–980. doi: 10.1111/j.1469-7610.2010.02272.x. [DOI] [PubMed] [Google Scholar]
- 7.Barr RG, Trent RB, Cross J. Age-related incidence curve of hospitalized Shaken Baby Syndrome cases: convergent evidence for crying as a trigger to shaking. Child Abuse Negl. 2006;30:7–16. doi: 10.1016/j.chiabu.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Case ME. Abusive head injuries in infants and young children. Leg Med. 2007;9:83–87. doi: 10.1016/j.legalmed.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 9.Chung CS, et al. Cognitive rehabilitation for executive dysfunction in adults with stroke or other adult non-progressive acquired brain damage. Cochrane Database Syst Rev. 2013;30:CD008391. doi: 10.1002/14651858.CD008391.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowe LM, et al. Timing of traumatic brain injury in childhood and intellectual outcome. J Pediatr Psychol. 2012;37:745–754. doi: 10.1093/jpepsy/jss070. [DOI] [PubMed] [Google Scholar]
- 11.Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acutetraumatic brain injury. J Neuroinflammation. 2012;12:236. doi: 10.1186/1742-2094-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatson JW, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74:470–474. doi: 10.1097/TA.0b013e31827e1f51. [DOI] [PubMed] [Google Scholar]
- 13.Giunta B, et al. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. Neuroinflammation. 2012;1:185. doi: 10.1186/1742-2094-9-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glover LE, et al. Immediate, but Not Delayed, Microsurgical Skull Reconstruction Exacerbates Brain Damage in Experimental Traumatic Brain Injury Model. PLoS ONE. 2012;7:e33646. doi: 10.1371/journal.pone.0033646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez-Ontiveros DG, et al. Microglia activation as a biomarker for traumatic brain injury. Front Neurol. 2013;4:30. doi: 10.3389/fneur.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaneko Y, et al. Nestin overexpression precedes caspase-3 upregulation in rats exposed to controlled cortical impact traumatic brain injury. Cell Med. 2013;4:55–63. doi: 10.3727/215517912X639306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karasu A, et al. Epidemiological study in head injury patients. Ulus Travma Acil Cerrahi Dergisi. 2009;15:159–163. [PubMed] [Google Scholar]
- 18.Levchakov A, et al. Computational studies of strain exposures in neonate and mature rat brains during closed head impact. J Neurotrauma. 2006;23:159–163. doi: 10.1089/neu.2006.23.1570. [DOI] [PubMed] [Google Scholar]
- 19.Lloyd E, et al. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;30:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxwell WL. Traumatic brain injury in the neonate, child and adolescent human: an overview of pathology. International Journal of Developmental Neuroscience. 2012;30:167–183. doi: 10.1016/j.ijdevneu.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Niedzwecki CM, et al. Traumatic brain injury: a comparison of inpatient functional outcomes between children and adults. J Head Trauma Rehabil. 2008;23:209–219. doi: 10.1097/01.HTR.0000327253.61751.29. [DOI] [PubMed] [Google Scholar]
- 23.Pabon MM, Borlongan CV. Advances in the Cell-Based Treatment of Neonatal Hypoxic-Ischemic Brain Injury. Future Neurol. 2013;1:193–203. doi: 10.2217/fnl.12.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paiva WS, et al. Traumatic brain injury and shaken baby syndrome. Acta Med Port. 2011;24:805–808. [PubMed] [Google Scholar]
- 25.Rovegno M, et al. Biological mechanisms involved in the spread of traumatic brain damage. Med Intensiva. 2012;36:37–44. doi: 10.1016/j.medin.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Shojo H, et al. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience. 2010;171:1273–1282. doi: 10.1016/j.neuroscience.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Shrivastava K, et al. Temporal Expression of Cytokines and Signal Transducer and Activator of Transcription Factor 3 Activation after Neonatal Hypoxia/Ischemia in Mice. Dev Neurosci. 2013 doi: 10.1159/000348432. In Press. [DOI] [PubMed] [Google Scholar]
- 28.Sun D, et al. Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol. 2009;216:56–65. doi: 10.1016/j.expneurol.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tajiri N, et al. Intravenous Grafts Of Amniotic Fluid-Derived Stem Cells Induce Endogenous Cell Proliferation and Attenuate Behavioral Deficits in Ischemic Stroke Rats. PLoS ONE. 2012;7:e43779. doi: 10.1371/journal.pone.0043779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timaru-Kast R, et al. Influence of Age on Brain Edema Formation, Secondary Brain Damage and Inflammatory Response after Brain Trauma in Mice. PLoS ONE. 2012;7:e43829. doi: 10.1371/journal.pone.0043829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Utagawa A, et al. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol. 2008;211:283–291. doi: 10.1016/j.expneurol.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waters RJ, et al. Cytokine gene polymorphisms and outcome after traumatic brain injury. J Neurotrauma. 2013 doi: 10.1089/neu.2012.2792. [ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol. 2011;31:745–57. doi: 10.1097/JCP.0b013e318235f4ac. [DOI] [PubMed] [Google Scholar]
- 34.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faul M, et al. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. 2010 Retrieved June 27, 2013 from Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf.