

Abstract
Cell adhesion and migration play important roles in physiological and pathological states, including embryonic development and cancer invasion and metastasis. The type I transmembrane protein with epidermal growth factor and two follistatin motifs 2 (TMEFF2) is expressed mainly in brain and prostate and its expression is deregulated in prostate cancer. We have previously shown that TMEFF2 can function as a tumor suppressor by inhibiting cell migration and invasion of prostate cells. However, the molecular mechanisms involved in this inhibition are not clear. In this study we demonstrate that TMEFF2 affects cell adhesion and migration of prostate cancer cells and that this effect correlates with changes in integrin expression and RhoA activation. Deletion of a 13 basic-rich amino acid region in the cytoplasmic domain of TMEFF2 prevented these effects. Overexpression of TMEFF2 reduced cell attachment and migration on vitronectin and caused a concomitant decrease in RhoA activation, stress fiber formation and expression of αv, β1 and β3 integrin subunits. Conversely, TMEFF2 interference in 22Rv1 prostate cancer cells resulted in increased integrin expression. Results obtained with a double TRAMP/TMEFF2 transgenic mouse also indicated that TMEFF2 expression reduced integrin expression in the mouse prostate. In summary, the data presented here indicate an important role of TMEFF2 in regulating cell adhesion and migration that involves integrin signaling and is mediated by its cytoplasmic domain.
Keywords: TMEFF2, integrin, cell migration, cell attachment, prostate cancer
1. INTRODUCTION
Prostate cancer is the most commonly diagnosed cancer and the second leading cause of cancer deaths in American men [1]. While organ-confined prostate cancer is successfully treated by surgical methods, no curative treatment is available for the metastatic form of the disease, which is responsible for the mortality associated with this disease. Prostate cancer cells are known to metastasize to numerous organs, with the bone, liver, and lymph nodes being the most common [2]; however, the molecular mechanisms that drive the metastatic cascade in prostate cancer are poorly understood. Understanding these mechanisms and the molecules involved in the metastatic cascade is critical to developing strategies for maximizing the efficacy of prostate cancer treatment.
Integrins are members of a family of transmembrane glycoprotein receptors that mediate cell-cell and the interactions with the extracellular matrix (ECM) [3]. By interacting with cytoskeletal-associated proteins, integrins provide a link between the extracellular environment and the cytoskeleton inside the cells. Integrins are heterodimers composed of non-covalently associated α and β subunitsthat can recognize and bind multiple ECM ligands, triggering a variety of signal transduction events that modulate diverse cellular processes including proliferation, survival, gene expression, adhesion and migration [3, 4]. Evidence of altered integrin signaling has been demonstrated in several types of cancer, including prostate cancer. These changes correlate with tumor growth, invasion, and metastatic potential [5, 6].
Several integrins, α2β1, α3β1, α5β1, α6β1, αvβ1, αIIbβ3, and αvβ3 are expressed in prostate cancer cells [6, 7]. Of those, αvβ3 and β1 seem to play important roles in bone metastases, the main site of metastatic prostate cancer [7–9]. While not typically expressed in epithelial cells [8], integrin αvβ3 is expressed in prostate cancer. Its expression correlates with disease progression, metastatic potential [5, 6–8], and with prostate cancer cell adhesion to vitronectin, a major extracellular component of mature bone [10]. Integrin β1 -is upregulated in specimens from prostate cancer patients, and antibodies against β1 integrins inhibit binding of PC3 prostate cancer cells to human bone marrow endothelial cells [7, 11], suggesting that β1 integrin mediates bone metastasis.
Integrins signal bidirectionally [12–15]. Intracellular stimuli (inside-out signaling) can promote a conformational change in the integrin that lead to higher affinity for its ligand [12–14]. Ligand binding (outside-in signaling) ultimately leads to integrin activation and clustering into large mature focal adhesions (FA) which generate downstream signaling events in a temporal fashion [12, 15]. One of the changes that take place early following integrin activation are cytoskeletal rearrangements that regulate stress fiber formation and promote cell spreading and initiation of migration [16]. These events involve integrin-mediated modulation of specific cellular kinases and of RhoA activity [14, 17]. RhoA is essential to remodeling actin fibers, regulation of actomyosin contractility, and rear cell detachment during motility [18]. RhoA is also activated via G-protein coupled receptors (GPCRs) that couple to the heterotrimeric G12/13 proteins, highlighting a cooperative relationship between integrin and GPCR signaling. Cross talk has also been described between integrins and growth factor receptor signaling to affect cell spreading, migration, growth and survival [19, 20].
TMEFF2 is an evolutionarily conserved type I transmembrane protein expressed in the embryo, and selectively in the adult brain and prostate [21–23]. A role for TMEFF2 in prostate cancer was suggested by studies indicating that TMEFF2 expression is altered in a significant fraction of primary and metastatic prostate tumors [22, 23]. We have described that TMEFF2 functions as a tumor suppressor, and that this role correlates, at least in part, with its ability to interact with SARDH to modulate cellular levels of sarcosine [24]. TMEFF2 overexpression blocked basal and sarcosine-induced cellular invasion of prostate epithelial RWPE cells, while TMEFF2 knockdown in 22Rv1 prostate cancer cells promoted increased cellular migration/invasion [25]. While these results highlight a role for TMEFF2 in invasion of prostate cells, the molecular mechanism(s) involved in this process are not known. Here we report that TMEFF2 expression inhibits spreading and migration of RWPE2 prostate cancer cells on vitronectin. This inhibition correlates with a defect in FA and stress fibers formation and in RhoA activation and requires the presence of the cytoplasmic tail of TMEFF2. Importantly, TMEFF2 downregulates the expression of several integrins in RWPE2 cells indicating that the motility effects observed are integrin-mediated. The results presented point to an important role of TMEFF2 in modulating integrin signaling and prostate cell motility.
2. MATERIALS AND METHODS
2.1. Cell culture and plasmids
The 22Rv1, RWPE1 and RWPE2 cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). The human prostate epithelial cell line RWPE1 and its Ki-ras transformed tumorigenic derivative, RWPE2, were cultured in KSF medium (Invitrogen, Carlsbad, CA). The human prostate carcinoma cell line 22Rv1 was maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA). TMEFF2 full length and ΔGA expression constructs were previously described [24, 26]. Development of a system for inducible expression of FL_TMEFF2 and TMEFF2_ΔGA in RWPE2 cells was achieved using the Clontech’s Tet-On Advanced system (Clontech, Mountain View, CA) essentially as described before for RWPE1 cells [24]. To inducibly express TMEFF2, cultures were grown in the presence of doxycycline (250 ng/ml; Sigma, St. Louis, MO). 22Rv1 cells transduced with pLKO.1 vectors containing shRNA to TMEFF2 or scramble control were described before [25].
2.2. Reagents
Antibodies recognizing TMEFF2 from cell lysates were purchased from Abcam (Cambridge, MA) and Sigma (St. Louis, MO). For TMEFF2 detection from mouse tissue lysates a SDIX, custom antibody was utilized (SDIX, Newark, DE). Other antibodies utilized in this study are: SV40 T-antigen (Abcam; Cambridge, MA), ITGAV, ITGB1 and ITGB3 (BD Biosciences, Franklin Lakes, NJ), ITGB3 (for mouse tissue lysates, Sigma, St. Louis, MO), ITGA5 (Millipore, Billerica, MA), ribosomal protein S6, β-actin, phospho-FAK, and FAK (Cell Signaling, Danvers, MA). Antibodies used for the immunofluorescence studies are listed in section 2.5. The U0126 MAPK inhibitor or the inactive analog U0124 were purchased from Sigma (St. Louis, MO). Bovine serum albumin (BSA) was from Sigma (St. Louis, MO).
2.3. Mice
Animals were maintained in accordance with the Institutional Animal Care and Use Committee of East Carolina University. Transgenic TMEFF2 mice (129/Sv background, Lineberger Cancer Center Transgenic mouse facility) and maintained in a C57BL/6 background (backcrossed for over seven generations to C57BL/6). TRAMP mice (FVB background) were purchased from the Jackson laboratory (Bar Harbor, ME; stock number 008215) and crossed to C57Bl/6J (stock number 000664). The F1 generation derived from this cross, was then crossed to the transgenic TMEFF2 mouse and the TRAMP/TMEFF2 and TRAMP progeny were selected after genotyping by PCR using tail genomic DNA. The TRAMP mice were genotyped as specified by the Jackson Laboratory (Bar Harbor, ME). The TMEFF2 mice were genotyped using primers 5′-GGAATTGCTCTGGTTATGATG-3′ and 5′-CAAATGTGGTATGGCTGATTATG-3′.
2.4. Cell migration assays
Cell migration was measured using either a wound healing assay or Boyden chambers. Both assays were performed in the presence of 1 μg/ml aphidicolin (Sigma, St. Louis, MO) to prevent proliferation. For wound healing assays, 70 μl of cells (from a 3–7×105 cells/ml suspension) were loaded into each well of a culture plate insert (ibidi, Verona, WI). After 24 hours incubation, the insert was removed to allow cell migration into the wound. Wound healing process was monitored by taking pictures at 0, 10, 24, or 48 hours after removal of the insert, using an EVOS FL cell imaging system (Life Technologies, Carlsbad CA). When specified, 2 μg/ml of CT04 (Cytoskeleton, Denver, CO) was added to the fresh medium after insert removal.
Cell migration was also assayed using Boyden chambers with non-coated 8 μM pore size membranes (BD Biosciences, Franklin Lakes, NJ). Cells (5 × 104) were suspended in 200 μl of serum-free medium and loaded into the upper chamber. The lower chamber was filled with 500 μl of medium supplemented with 20% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) that was used as a chemoattractant. After 20 hours of incubation, the cells that had migrated to the lower surface of the membrane were fixed with 70% ethanol for 10 minutes, followed by staining with 0.1% crystal violet (Sigma, St. Louis, MO) and photographed. To assay cell migration towards vitronectin or fibronectin the following modifications were implemented: i) the lower surface of the membrane in the Boyden chambers was treated with 10 μg/ml of the indicated ECM proteins or BSA as control (prepared in PBS) for 16 hours at 4 °C; ii) Cells (0.5–1 × 104) were suspended in 200 μl of KSF base medium and loaded into the upper chamber; iii)The lower chamber was also filled with KSF medium; and iv) The culture was maintained for 1–2 days.
2.5. Cell spreading assay and immunofluorescence
Round cover glass (Fisher Scientific, Hampton, NH) were coated with collagen (40 μg/ml; BD Biosciences, Franklin Lakes, NJ), laminin (10 μg/ml; Sigma, St. Louis, MO), fibronectin (40 μg/ml; Millipore, Billerica, MA), or vitronectin (2 μg/ml; Promega, Fitchburg, WI) by incubation at 4 °C overnight and rinsed twice with PBS. Once coated, the cover glass were placed inside the wells of 12-well culture plates and 40,000 cells were loaded onto each well. Following 3 hours incubation, pictures were taken for 10 random fields for each cover glass using an EVOS FL cell imaging system (Life Technologies, Carlsbad, CA) and spread/round cells were counted. For immunofluorescence analysis, cells were fixed with 4% paraformaldehyde in PBS (Affymetrix USB, Cleveland, OH) for 10 min, permeabilized with 0.5% Triton X-100 (Bio-Rad, Hercules, CA) in PBS (PBS-T) for 5 min, and blocked with 5% normal goat serum in PBS-T for 30 min. Samples were then incubated with anti-vinculin antibody (Sigma, St. Louis, MO) at 4 °C overnight followed by goat anti-mouse Alexa Fluor 488 (Invitrogen, Carlsbad, CA) and rhodamine phalloidin (Cytoskeleton, Denver, CO) for 1 h. Nuclei were counterstained with DAPI (Santa Cruz Biotechnology, Santa Cruz, CA). For immunofluorescence analysis with rhodamine phalloidin only staining, cells were seeded in 8 well glass chamber slides (Lab-Tek, Scotts Valley, CA) and the primary antibody and the corresponding secondary antibody were omitted. Immunofluorescent images were taken on an Optiphot-2 fluorescent microscope (Nikon) with an AxioCam MRm digital camera (Zeiss).
2.6. G-LISA RhoA activation assay
Cells were incubated in serum free EpiLife CF/PRF medium (Invitrogen, Carlsbad, CA) for 3 d and then stimulated with 10% FBS for 2 min. RhoA activity was determined using a colorimetric G-LISA RhoA activation assay biochem kit (Cytoskeleton, Denver, CO) according to the manufacturer’s instructions.
2.7. Western blotting
Western blot analysis was conducted as described before [24] using cell lysates or lysates from individual mouse prostate lobes. Briefly, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail (Sigma, St. Louis, MO), sodium orthovanadate and beta-glycerophosphate. Lobes were lysed in the same buffer using a glass homogenizer. Equal amounts of proteins were resolved by SDS-PAGE and transferred to Immobilon transfer membrane (Millipore, Billerica, MA). Blots were blocked in 5% non-fat milk or bovine serum albumin (BSA; Sigma, St. Louis, MO) and probed with the appropriate antibodies. Immunoreactive bands were visualized using SuperSignal West Pico or Femto chemiluminescent substrate (Thermo Scientific, Waltham, MA).
2.8. RT2 Profiler PCR Array
The Human Focal Adhesion RT2 Profiler PCR Array (SABiosciences, Valencia, CA) was used to determine cellular adhesion-related gene expression affected by TMEFF2 according to the manufacturer’s instructions. Briefly, total RNA was extracted from cells expressing TMEFF2 or the vector, as a control, with RNeasy mini kit and cDNA was synthesized with RT2 First Strand Kit (Qiagen, Valencia, CA). The cDNA was combined with RT2 SYBR Green Mastermix (Qiagen, Valencia, CA) and dispensed into the RT2 Profiler PCR Array. Real-time PCR was performed on an iQ5 instrument (Bio-Rad, Hercules, CA).
2.8. Statistical analysis
Data are presented as mean ± SD. Student’s t test (paired, two-tailed) was used to compare two groups of independent samples. P values under 0.05 or 0.01 were considered significant.
3. RESULTS
3.1. The cytoplasmic domain of TMEFF2 is necessary for its tumor suppression role
The transmembrane and cytoplasmic domains of TMEFF1 and TMEFF2, the two members of the TMEFF family of proteins, are very well conserved at the amino acid level, and contain potential GPCR-signaling motifs in the membrane and cytoplasmic domains [27]. For TMEFF1, the relevance of these domains is underscored by data pointing to different functions of the protein depending on their presence [28].
TMEFF2 overexpression in prostate epithelial RWPE1 cells inhibits the intrinsic and sarcoine-induced migration potential of these cells [24]. To examine the role of the TMEFF2 cytoplasmic domain, we used a deletion mutant lacking 13 consecutive basic-rich amino acids in the C-terminus of the protein (TMEFF2_ΔGA). Full length TMEFF2 FL_TMEFF2) or TMEFF2_ΔGA were expressed in RWPE1 and RWPE2 cells and their effect on cell migration was analyzed. Expression of the different forms of TMEFF2 was achieved using cell lines that express FL_TMEFF2 or TMEFF2_ΔGA under the control of a doxycycline-inducible promoter (Figure 1A and B). Control cells were transduced with the transactivator construct only (vector).
Figure 1.

TMEFF2 inhibits migration in RWPE1 and RWPE2 cells. A, B) Cells were grown in the presence of 250 ng/ml doxycycline to induce the expression of FL_TMEFF2 or TMEFF2_ΔGA. Overexpression of these proteins in RWPE1 (A) and RWPE2 (B) cells after doxycycline treatment was assessed by western blot. The slow mobility band corresponds to a previously described highly glycosylated form of the protein [22]. The reason why this from is over-represented when the TMEFF2_ΔGA mutant is expressed is not clear. C, D) Cell migration of RWPE1 (C) or RWPE2 (D) cells expressing the different forms of TMEFF2, was determined using a wound healing assay, 48 (RWPE1) or 10 hours (RWPE2) after the wound was made. Quantification of relative migration (fold over the vector) is presented as mean ± SD of three independent experiments.
Results obtained using a wound-healing assay indicated that, as opposed to cells expressing the full length TMEFF2 protein, RWPE1 or RWPE2 cells expressing the TMEFF2_ΔGA protein did not significantly show inhibition of migration when compared with cells transfected with the vector control only (Figure 1C, 1D and Supplementary Fig. S1A). Similar results were obtained using Boyden chambers (Supplementary Fig. S1B). These results suggest that the cytoplasmic tail of TMEFF2 is necessary for its ability to inhibit migration of prostate epithelial and cancer cells. Interestingly, a tendency towards slower migration was observed in cells expressing TMEFF2_ΔGA when compared with cells expressing the vector control. This may reflect more than one mechanism to inhibit migration. All together these results suggest that the cytoplasmic tail of TMEFF2 is important for its tumor suppressor function.
It is worth noting that while in RWPE2 cells the expression of the TMEFF2_ΔGA mutant seems lower than expression from the FL_TMEFF2, variable levels of expression have been frequently observed from experiment to experiment and this does not seem to be the reason why TMEFF2_ΔGA does not have an effect on migration. In fact, as shown in Figure 1A, the expression level of the FL_TMEFF2 and TMEFF2_ΔGA was similar in RWPE1 cells but the TMEFF2_ΔGA did not affect migration in this cell line.
3.2. TMEFF2 promotes cell rounding and reduces cell spreading
In order for cells to migrate they must attach and spread. In these studies we observed that TMEFF2 promotes cell rounding of RWPE2 and several other cell lines (Figure 2A and not shown), suggesting defective attachment and spreading of TMEFF2 expressing cells, perhaps accounting for the inhibitory effect of TMEFF2 on migration. To test this hypothesis, attachment/spreading to tissue culture plates of RWPE2 cells expressing the inducible FL_TMEFF2, TMEFF2_ΔGA proteins or the empty vector, as a control, was examined. As shown in Figure 2B, when grown in the presence of doxycycline to induce FL_TMEFF2 expression, a two-fold increase in the number of round cells was observed when compared to cells expressing the empty vector or the TMEFF2_ΔGA. These rounded cells had few lamellipodia protrusions (Figure 2C), indicating that the initial stages of spreading were affected. Cell attachment/spreading to plates coated with the ECM proteins vitronectin, fibronectin, laminin and collagen type I was also analyzed. RWPE2 cells expressing the empty vector control or the TMEFF2_ΔGA behaved similarly, with over 90% of cells attaching/spreading to fibronectin and collagen type I and 80–85% to vitronectin and laminin (Figure 3A). However, when compared to cells expressing the empty vector or the TMEFF2_ΔGA, expression of FL_TMEFF2 significantly reduced the ability of cells to attach/spread on the vitronectin coated surface (only 58% of the cells; Figure 3A). This suggests that TMEFF2 blocks spreading on vitronectin coated surfaces, and this could ultimately affect migration. Further, results obtained using a transwell assay, indicated that migration of RWPE2 cells expressing FL_TMEFF2 was significantly reduced in vitronectin but not in fibronectin, when compared to cells expressing TMEFF2_ΔGA or the empty vector control (Figure 3B). Migration towards BSA coated transwell membranes was negligible independent of the cells examined (not shown). All together, these results suggest that TMEFF2 inhibits attachment/spreading on vitronectin and that this inhibitory effect requires the presence of the cytoplasmic tail.
Figure 2.

TMEFF2 inhibits RWPE2 cell spreading on tissue culture plates. A) Representative images of cell morphology of RWPE2 cells expressing different forms of the TMEFF2 protein, or the empty vector, on tissue culture surface. Arrows point to rounded cells. Scale bars, 100 μm. B) Quantification of round cells at 3 h after seeding 40,000 cells/well in a 12-well plate. Data are presented as mean ± SD of three independent experiments. C) Representative images of cell morphology of RWPE2 cells expressing the inducible FL_TMEFF2 before and after the addition of doxycycline. Note that FL_TMEFF2 expression promotes cell rounding and those cells lack lamellipodia.
Figure 3.

Effect of TMEFF2 in different ECM substrates. A) TMEFF2 significantly inhibits cell spreading on vitronectin-coated surfaces. RWPE2 cells expressing FL_TMEFF2, TMEFF2_ΔGA, or the vector were plated onto cover glass coated with the different ECM substrates. Percentages of spread cells were analyzed 3 h after seeding. Data are presented as mean ± SD of five independent experiments. B) TMEFF2 inhibits RWPE2 cell migration towards vitronectin but not fibronectin as measured in a Boyden chamber assay. Shown are representative images of RWPE2 cells expressing the different TMEFF2 constructs after 48 h migration (top), and quantification of those images by densitometric analysis using ImageJ (bottom). Data are presented as mean ± SD of three independent experiments.
3.3. TMEFF2 reduces stress fiber and focal adhesion formation and activation
Since the presence of focal adhesions (FA) correlates with cell attachment and spreading [29] the ability of FL_TMEFF2 to inhibit formation of these macromolecular structures was examined. RWPE2 cells expressing FL_TMEFF2, TMEFF2_ΔGA or the empty vector control were grown on cover glass coated with fibronectin or vitronectin and stained with rhodamine-phalloidin to detect polymerized actin and vinculin antibodies to detect FA. As described above, expression of FL_TMEFF2, but not the mutant lacking the cytoplasmic tail, resulted in an increase in rounded cells when cells were grown on vitronectin. These rounded cells revealed a cortical actin cytoskeleton, without stress fibers, and a lack of FAs as demonstrated by diffuse vinculin staining (Figure 4A; middle row). Conversely, the normal attachment/spreading of FL_TMEFF2 expressing RWPE2 cells to fibronectin correlated with stress fiber formation and the presence of FA at the ends of the fibers (Figure 4A, Supplementary Fig. S2). Expression of the TMEFF2_ΔGA mutant, did not affect stress fiber or FA formation when grown on either vitronectin or fibronectin (Figure 4A bottom row) when compared to cells expressing the vector control (Figure 4A, upper row). Finally, phosphorylation of focal adhesion kinase (FAK) at tyrosine 397 (Y397) was analyzed by western blot. FAK phosphorylation is required for FA formation and cell migration. The results, shown in figure 4B, indicated that FL_TMEFF2 but not TMEFF2_ΔGA inhibited FAK-Y397 phosphorylation. All together these results indicate that TMEFF2 interferes with FA formation/activation and, as a consequence, it inhibits cell spreading and migration. The cytoplasmic tail of TMEFF2 is required for these effects.
Figure 4.
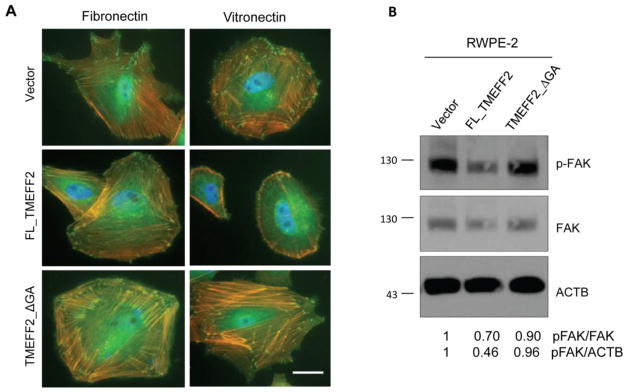
TMEFF2 reduces stress fiber and focal adhesion formation and activation. A) RWPE2 cells expressing FL_TMEFF2, TMEFF2_ΔGA, or the vector were cultured on cover glass coated with fibronectin or vitronectin for 3 h and then stained with anti-vinculin (green), rhodamine phalloidin (orange), and DAPI (blue). Scale bar, 20 μm. B) Immunoblotting of phosphorylated FAK (Y397), FAK and β-actin (ACTB) in RWPE2 cells. Numbers under the western blots represent the densitometry quantifications using ImageJ.
3.4. TMEFF2 inhibits RhoA activation
RhoA activation promotes stress fiber formation and maturation of focal adhesions. Since the results obtained indicated that expression of TMEFF2 affects these processes, the ability of TMEFF2 to affect RhoA activation was examined. Rho is active in the GTP-bound state, which is catalyzed by the Dbl family of guanine nucleotide exchange factors (GEFs) [30]. The amount of activated RhoA was determined using the G-LISA kit in RWPE2 cells expressing the different forms of TMEFF2 and compared to the activity of cells expressing the vector as control. Cells were grown in serum-free media and then stimulated with fetal bovine serum (FBS). The results indicated that TMEFF2 expression significantly reduced the level of active RhoA to 78% of that in the control cells (Figure 5). RhoA activity in cells expressing TMEFF2_ΔGA did not differ significantly from either the FL_TMEFF2 expressing or empty vector control expressing cells. Changes in RhoA activation correlated with the formation of stress fibers, since expression of FL_TMEFF2, but not TMEFF2_ΔGA, also inhibited stress fiber formation (Figure 5). These results indicate that TMEFF2 decreases RhoA activation in FBS-stimulated conditions and this may in part be mediated by the cytoplasmic tail of TMEFF2. Since RhoA plays a role in migration, it is possible that the observed TMEFF2-mediated inhibition of migration is, at least partly, due to its effect on RhoA activation. In support of this, treatment of RWPE2 cells with CT04, a Rho inhibitor, dramatically reduced stress fiber formation and inhibited cellular migration by 50% (Supplementary Fig S3A).
Figure 5.

TMEFF2 inhibits Rho activation. RWPE2 cells expressing FL_TMEFF2, TMEFF2_ΔGA, or the vector were incubated in serum-free EpiLife CF/PRF medium for 3 days and then stimulated with 10% FBS for 2 min. Levels of GTP-bound RhoA were determined by G-LISA RhoA activation assays. Data are presented as mean ± SD of five independent experiments (top). Rhodamine phalloidin staining of the cells stimulated with 10% FBS for 10 min (bottom) demonstrating lack of stress fibers in the cells expressing FL_TMEFF2. Scale bar, 25 μm.
3.5. TMEFF2 modulates expression of integrins in part through the MAPK pathway
Integrins are the main ECM receptors. They are involved in motility and migration, and integrin signaling can modulate RhoA activation [17]. To determine whether the effect of TMEFF2 on migration and RhoA activation is mediated by an effect on integrin levels, changes in the expression of a subset of integrins in response to TMEFF2 were analyzed. The human extracellular matrix and adhesion molecules RT2 profiler PCR array was used to investigate differences in expression of 21 different integrin subunits (15 α and 6 β) – other genes important for cell-cell and cell-matrix interactions are also included in the array— in RWPE2 cells. Expression of FL_TMEFF2 but not TMEFF2_ΔGA in RWPE2 cells reduced the levels of some integrin’s mRNA, including αv, β1 and β3 (Supplementary Fig. S4). For those integrins, the observed decrease in mRNA levels corresponded with a decrease in total protein levels as measured by western blot (Figure 6A). Expression of these integrins was also analyzed in 22Rv1/sh_TMEFF2 cells [25], in which expression of endogenous TMEFF2 is reduced by sh_RNA, and in 22Rv1/sh_scramble control cells. As shown in figure 6A, decreasing the level of TMEFF2 by sh_RNA promoted an increase in integrin αv protein levels. Expression of the β1 and β3 integrins was not detected in these cells (Figure 6A and data not shown). These results demonstrate that FL_TMEFF2 inhibits expression of the αv, β1 and β3 integrins, and that the cytoplasmic tail is required for this inhibition. Attempts to rescue the effect on migration mediated by TMEFF2 by transiently overexpressing αv and β3 integrins failed. However, these experiments are complicated by the large number of integrins that appear to be affected by TMEFF2 (see Supplementary Fig. S4) and the rather promiscuous function of the integrins [31].
Figure 6.

TMEFF2 modulates the abundance of αv, β1 and β3 integrins in cell lines and in mouse prostates. A) Total lysates of the indicated cells lines were subjected to immunoblotting with antibodies against the integrins as shown. Note that TMEFF2 did not promote changes in α5 integrin levels and that 22Rv1 cells do not express β3 integrin. B) Total lysates of RWPE2 cells treated with 10μM MAPK inhibitor U0126 or inactive analog U0124 for 24 h were subjected to immunoblotting with integrin β3 antibody. The effectiveness of MAPK inhibition was demonstrated using an antibody against phosphorylated ERK. C) Tissue lysates from the anterior (A), ventral (V) or dorsolateral (DL) lobes of the mouse prostate were subjected to immunoblotting with antibodies against the specified integrins. Arrows indicate the sizes of the individual integrin subunits observed in cell lines. Additional bands may represent precursor proteins, which are also decreased by TMEFF2. β-tubulin (TUBB) or ribosomal protein S6 (RPS6) were used as a loading controls. Numbers under the western blots are the densitometry quantifications of the arrow-pointed bands normalized to the loading control using ImageJ. Note that the complexity of the banding pattern for the β3 integrin in mouse prostate lobes did not allow quantification however, the decrease in the abundance as a result of TMEFF2 expression is clear.
Since we have previously demonstrated that TMEFF2 modulates MAPK signaling in response to growth factors [26], the possibility that TMEFF2 affects integrin expression through this pathway was examined. RWPE2 cells expressing FL_TMEFF2 or the empty vector as a control, were treated with the U0126 MAPK inhibitor or with U0124, a chemical analog that does not have MAPK inhibitory activity, and the effect on integrin expression was determined. As shown in figure 6B, in the presence of the inactive analog U0124, expression of TMEFF2 resulted in a decrease in integrin β3 level that was less pronounced in the presence of U0126 – four fold difference. No effect was observed in the level of integrin αv (data not shown). Western blot analysis with antibodies against phosphorylated ERK indicated that U0126 inhibited the MAPK pathway. There was not a significant inhibition observed with U0124. While the effect of TMEFF2 on integrin expression was not fully abolished by U0126, these results indicate that MAPK signaling plays a role in modulation of integrin expression by TMEFF2.
3.6. TMEFF2 expression inhibits integrin expression in vivo
In humans TMEFF2 is expressed mainly in brain and prostate, however, TMEFF2 is not expressed in the adult mouse prostate [23]. We have generated a transgenic TMEFF2 mouse model that expresses TMEFF2 driven by the probasin promoter with transgene expression restricted to the prostate (Supplementary Fig. S5). As described for the probasin promoter expression pattern [32], TMEFF2 is expressed more in the ventral (VP) and dorsolateral (DLP) prostate with almost no expression in the anterior lobe (AP) of the transgenic mouse (Supplementary Fig. S5; Overcash, RF and Ruiz-Echevarria, MJ, unpublished observations). The TMEFF2 transgenic animals were crossed to a mouse model of prostate cancer designated TRAMP (transgenic adenocarcinoma of mouse prostate) and 15 weeks TMEFF2/TRAMP and TRAMP siblings were selected for analysis of integrin expression. After euthanasia, the prostates were dissected and protein lysates from the anterior, ventral and dorsolateral lobes were prepared and analyzed for the expression of αv, β3 and β1 integrin subunits by western blot analysis. β1 integrin has been previously shown to be upregulated in the TRAMP mouse [33]. As shown in figure 6C, as compared with the TRAMP model, expression of TMEFF2 in the TRAMP mouse led to reduced expression of the αv, β1 and β3, integrins indicating that expression of TMEFF2 reduces integrin expression in vivo. Changes in the level of the α5 integrin subunit were not observed, as it was the case in the prostate cell lines tested.
4. DISCUSSION
Cell migration is an essential step in embryogenesis as well as in the development of metastatic lesions during cancer progression. While the metastatic process is not fully understood, enhanced migration on extracellular matrix (ECM) substrates correlates with increased metastasis. Here we present data to show that TMEFF2, a transmembrane protein with limited expression to embryo and adult brain and prostate, plays a role in cellular adhesion and migration by modulating activation of the small GTPase RhoA, and/or integrin expression.
In this study, we demonstrated that expression of TMEFF2 in prostate cancer cells, or in prostate lobes of a TRAMP/TMEFF2 transgenic mouse, significantly reduced the expression of at least three integrin subunits, αv, β3 and β1. In agreement with this, we observe that TMEFF2 inhibits prostate cancer cell migration in vitronectin; αvβ3 is the major receptor for vitronectin. Importantly, in vitro and in vivo experiments have demonstrated that expression of αvβ3 plays an essential role in the metastasis of prostate cancer to bone, accounting for more than 80% of prostate cancer metastases [2]. The αvβ3 integrin plays numerous roles in prostate cancer metastasis. By modulating engraftment and survival after bone colonization tumor cell expression of this integrin is critical to the success of metastatic lesions. Expressed also in osteoclasts, αvβ3 is also critical to bone resorption and the metastatic growth of the tumor in the bone [9]. Similar results have been observed in breast cancer where expression of αvβ3 in a mammary carcinoma line that metastasizes to the lung, but not to bone, was sufficient to promote its spontaneous metastasis to bone [34, 35]. Expression of αvβ3 has also been associated with metastasis to lungs [36]. Interestingly, preliminary data from our laboratory indicates that formation of metastasis to lungs is reduced in the double TRAMP/TMEFF2 transgenic when compared with the TRAMP mouse (not shown), suggesting that TMEFF2 inhibits metastasis by affecting integrin expression. The results presented here also indicated that TMEFF2 affects expression of the β1 integrin. Interestingly, it has been reported that β1 integrin deletion in a TRAMP mouse increases prostate epithelial cell differentiation and results in more aggressive tumors while having no effect on the frequency of metastases, as determined by visual inspection [37]. Conversely, in our TRAMP/TMEFF2 transgenic animal, in which expression of β1 and other integrins is reduced, we do not observe changes in the latency or grade of the tumors but in the occurrence and number of metastases (Overcash RF. and Ruiz-Echevarria MJ., unpublished observations). It is possible that this reflects differences in the balance of integrin heterodimer formation. Interestingly, it has recently been reported that inactivation of integrin β1 promotes expression of β3 in malignant cells, enhancing metastatic progression [38]. Based on these results, the fact that TMEFF2 reduces the levels of integrins β1 and β3 could provide an explanation to the phenotypic differences observed between the TRAMP mouse with a deletion of integrin β1 and the TRAMP/TMEFF2 transgenic animals.
In prostate cancer cells, expression of TMEFF2 affects cellular migration and invasion [24, 25, and this study]. Overexpression of TMEFF2 inhibited migration ofRWPE1 and RWPE2 cells. Conversely, interference of TMEFF2 expression in prostate cancer 22Rv1 cells promoted increased migration/invasion. Interestingly, the invasive ability of 22Rv1 cells in which expression of TMEFF2 was reduced, was highly susceptible to the anti-folate drug methotrexate [25] suggesting that one-carbon availability is central to the migration/invasion phenotype mediated by changes in TMEFF2. Based on these results, it is reasonable to speculate that TMEFF2, by affecting one carbon metabolism, may affect expression of integrin genes epigenetically, via methylation. Although we have not directly tested that hypothesis, several studies have described epigenetic alterations –DNA methylation and histone modifications –that affect integrin expression during tumor progression [39, 40].
The role of TMEFF2 in prostate cancer is complex, and while the full length membrane bound form functions as a tumor suppressor, a soluble shed form of TMEFF2, the ectodomain, promotes growth [24]. This has led to the hypothesis that the predominant form of TMEFF2, and therefore its role, changes as the disease progresses [24, 26, 41]. It is likely that the full length and the TMEFF2 ectodomain differentially affect integrin expression during disease progression. We have previously demonstrated that TMEFF2 affects Akt and/or ERK activation so that the full-length activates ERK but has no effect on Akt phosphorylation while the ectodomain inhibits ERK phosphorylation concomitantly with Akt activation in response to growth factors [26]. The results presented here suggest that TMEFF2 modulates integrin expression, in part via the MAPK pathway. Other mechanisms need to be identified. Since integrins have been shown to induce Akt [42, 43] and ERK phosphorylation [44], it is also possible that TMEFF2 modulates MAPK and PI3K pathways via its effects on integrin expression establishing a negative feedback loop. This would suggest that TMEFF2 may modulate the cross-talk between integrins and growth factor receptors to control cellular responses including survival, growth, differentiation and migration. In this respect, it is known that signaling through receptor tyrosine kinases (RTKs) is not only regulated by growth factors but also by functional collaboration with integrins, and integrins may activate RTKs in the absence of growth factors [19, 45].
In this study we have demonstrated that expression of TMEFF2 resulted in decreased RhoA activation and stress fiber formation and that the latter effect was also depended on the presence of an intact cytoplasmic domain of TMEFF2. Based on a predicted homology of this domain with G-protein couple receptors (GPCR), initially plausible hypothesis is that TMEFF2 may modulate RhoA activation by, for example, restricting the function of GPCRs that are involved in Gα12/13 or Gαq activation which induce Rho [46], or by promoting the activity of the Rho inhibitory Gαz signaling [47]. If this were the case, TMEFF2 would control migration via two independent mechanisms since RhoA inactivation does not seem to affect integrin expression (Supplementary Fig. S3B). While we have not formally ruled out this possibility, the fact that integrins can control the activation of Rho-GTPases, either by engaging Src-tyrosine kinases, or via crosstalk with growth factor receptor signaling, suggests an alternative mechanism by which TMEFF2 may also control RhoA activation by controlling integrin expression and thus, cell spreading and migration. It is known, that by regulating the balance between Rac1-mediated membrane protrusion and RhoA-mediated contractility, integrins control cytoskeletal-dependent processes involved in cell adhesion and spreading and therefore cell movement and migration [17, 48].
In summary, these results demonstrate that TMEFF2 negatively regulates cell adhesion and migration to the ECM by affecting integrin expression and RhoA activation, and suggest a potentially important role for TMEFF2 as a metastasis inhibitor.
Supplementary Material
HIGHLIGHTS.
TMEFF2 affects cell adhesion and migration of prostate cancer cells on vitronectin
TMEFF2 modulates RhoA activation, stress fiber and focal adhesion formation
TMEFF2 modulates integrin expression in prostate cancer cells and in a mouse model
Acknowledgments
This work was supported in part by a grant from the National Cancer Institute (1R15CA155873). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study.
The authors are thankful to Dr. Matusik for providing the probasin promoter vectors that we utilized to make the TMEFF2 transgenic mouse. The authors acknowledge Tom Green technical help with the characterization of the custom TMEFF2 antibody and Calvin Justus for helpful insights with the spreading/attachment experiments.
Footnotes
Conflict of interest: none declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013 Jan 17;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004 Dec 15;64(24):9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 3.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002 Sep 20;110(6):673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 4.Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2010 Jan;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel HL, Li J, Kogan S, Languino LR. Integrins in prostate cancer progression. Endocr Relat Cancer. 2008 Sep;15(3):657–64. doi: 10.1677/ERC-08-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fornaro M, Manes T, Languino LR. Integrins and prostate cancer metastases. Cancer Metastasis Rev. 2001;20(3–4):321–31. doi: 10.1023/a:1015547830323. [DOI] [PubMed] [Google Scholar]
- 7.Lee YC, Jin JK, Cheng CJ, Huang CF, Song JH, Huang M, et al. Targeting constitutively activated beta1 integrins inhibits prostate cancer metastasis. Mol Cancer Res. 2013 Apr;11(4):405–17. doi: 10.1158/1541-7786.MCR-12-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 2002 May-Jun;4(3):191–4. doi: 10.1038/sj.neo.7900224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene. 2007 Sep 13;26(42):6238–43. doi: 10.1038/sj.onc.1210429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seiffert D. Detection of vitronectin in mineralized bone matrix. J Histochem Cytochem. 1996 Mar;44(3):275–80. doi: 10.1177/44.3.8648088. [DOI] [PubMed] [Google Scholar]
- 11.Scott LJ, Clarke NW, George NJ, Shanks JH, Testa NG, Lang SH. Interactions of human prostatic epithelial cells with bone marrow endothelium: Binding and invasion. Br J Cancer. 2001 May 18;84(10):1417–23. doi: 10.1054/bjoc.2001.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009 Jan 15;122:159–63. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–45. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 14.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012 Oct;24(5):600–6. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004 Oct;5(10):816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 16.DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003 Oct;15(5):572–82. doi: 10.1016/s0955-0674(03)00109-1. [DOI] [PubMed] [Google Scholar]
- 17.Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009 Apr 15;122:1059–69. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 18.Sah VP, Seasholtz TM, Sagi SA, Brown JH. The role of Rho in G protein-coupled receptor signal transduction. Annu Rev Pharmacol Toxicol. 2000;40:459–89. doi: 10.1146/annurev.pharmtox.40.1.459. [DOI] [PubMed] [Google Scholar]
- 19.Eliceiri BP. Integrin and growth factor receptor crosstalk. Circ Res. 2001 Dec 7;89(12):1104–10. doi: 10.1161/hh2401.101084. [DOI] [PubMed] [Google Scholar]
- 20.Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol. 2011;27:291–320. doi: 10.1146/annurev-cellbio-092910-154017. [DOI] [PubMed] [Google Scholar]
- 21.Gery S, Sawyers CL, Agus DB, Said JW, Koeffler HP. TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene. 2002 Jul 18;21(31):4739–46. doi: 10.1038/sj.onc.1205142. [DOI] [PubMed] [Google Scholar]
- 22.Glynne-Jones E, Harper ME, Seery LT, James R, Anglin I, Morgan HE, et al. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int J Cancer. 2001 Oct 15;94(2):178–84. doi: 10.1002/ijc.1450. [DOI] [PubMed] [Google Scholar]
- 23.Afar DE, Bhaskar V, Ibsen E, Breinberg D, Henshall SM, Kench JG, et al. Preclinical validation of anti-TMEFF2-auristatin E-conjugated antibodies in the treatment of prostate cancer. Mol Cancer Ther. 2004 Aug;3(8):921–32. [PubMed] [Google Scholar]
- 24.Chen X, Overcash R, Green T, Hoffman D, Asch AS, Ruiz-Echevarria MJ. The tumor suppressor activity of the transmembrane protein with epidermal growth factor and two follistatin motifs 2 (TMEFF2) correlates with its ability to modulate sarcosine levels. J Biol Chem. 2011 May 6;286(18):16091–100. doi: 10.1074/jbc.M110.193805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green T, Chen X, Ryan S, Asch AS, Ruiz-Echevarria MJ. TMEFF2 and SARDH cooperate to modulate one-carbon metabolism and invasion of prostate cancer cells. Prostate. 2013 Oct;73(14):1561–75. doi: 10.1002/pros.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Ruiz-Echevarria MJ. TMEFF2 modulates the AKT and ERK signaling pathways. Int J Biochem Mol Biol. 2013 Jul 29;4(2):83–94. [PMC free article] [PubMed] [Google Scholar]
- 27.Horie M, Mitsumoto Y, Kyushiki H, Kanemoto N, Watanabe A, Taniguchi Y, et al. Identification and characterization of TMEFF2, a novel survival factor for hippocampal and mesencephalic neurons. Genomics. 2000 Jul 15;67(2):146–52. doi: 10.1006/geno.2000.6228. [DOI] [PubMed] [Google Scholar]
- 28.Chang C, Eggen BJ, Weinstein DC, Brivanlou AH. Regulation of nodal and BMP signaling by tomoregulin-1 (X7365) through novel mechanisms. Dev Biol. 2003 Mar 1;255(1):1–11. doi: 10.1016/s0012-1606(02)00075-1. [DOI] [PubMed] [Google Scholar]
- 29.Dubash AD, Menold MM, Samson T, Boulter E, García-Mata R, Doughman R, et al. Chapter 1. Focal adhesions: new angles on an old structure. Int Rev Cell Mol Biol. 2009;277:1–65. doi: 10.1016/S1937-6448(09)77001-7. [DOI] [PubMed] [Google Scholar]
- 30.Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001 Dec;26(12):724–32. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 31.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006 Oct 1;119:3901–3. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg NM, DeMayo FJ, Sheppard PC, Barrios R, Lebovitz R, Finegold M, et al. The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice. Mol Endocrinol. 1994 Feb;8(2):230–9. doi: 10.1210/mend.8.2.8170479. [DOI] [PubMed] [Google Scholar]
- 33.Goel HL, Breen M, Zhang J, Das I, Aznavoorian-Cheshire S, Greenberg NM, et al. beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005 Aug 1;65(15):6692–700. doi: 10.1158/0008-5472.CAN-04-4315. [DOI] [PubMed] [Google Scholar]
- 34.Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, et al. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006;8(2):R20. doi: 10.1186/bcr1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider JG, Amend SR, Weilbaecher KN. Integrins and bone metastasis: Integrating tumor cell and stromal cell interactions. Bone. 2011 Jan;48(1):54–65. doi: 10.1016/j.bone.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan X, Jia SF, Zhou Z, Langley RR, Bolontrade MF, Kleinerman ES. Association of alphavbeta3 integrin expression with the metastatic potential and migratory and chemotactic ability of human osteosarcoma cells. Clin Exp Metastasis. 2004;21(8):747–53. doi: 10.1007/s10585-005-0599-6. [DOI] [PubMed] [Google Scholar]
- 37.Moran-Jones K, Ledger A, Naylor MJ. β1 integrin deletion enhances progression of prostate cancer in the TRAMP mouse model. Sci Rep. 2012 Jul 24;2:526. doi: 10.1038/srep00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Mol Biol Cell. 2013 Nov;24(21):3449–59. doi: 10.1091/mbc.E12-10-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park J, Song SH, Kim TY, Choi MC, Jong HS, Kim TY, et al. Aberrant methylation of integrin alpha4 gene in human gastric cancer cells. Oncogene. 2004 Apr 22;23(19):3474–80. doi: 10.1038/sj.onc.1207470. [DOI] [PubMed] [Google Scholar]
- 40.Yang X, Pursell B, Lu S, Chang TK, Mercurio AM. Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J Cell Sci. 2009 Jul 15;122:2473–80. doi: 10.1242/jcs.049148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali N, Knaüper V. Phorbol ester-induced shedding of the prostate cancer marker transmembrane protein with epidermal growth factor and two follistatin motifs 2 is mediated by the disintegrin and metalloproteinase-17. J Biol Chem. 2007 Dec 28;282(52):37378–88. doi: 10.1074/jbc.M702170200. [DOI] [PubMed] [Google Scholar]
- 42.Velling T, Stefansson A, Johansson S. EGFR and beta1 integrins utilize different signaling pathways to activate akt. Exp Cell Res. 2008 Jan 15;314(2):309–16. doi: 10.1016/j.yexcr.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 43.Zeller KS, Idevall-Hagren O, Stefansson A, Velling T, Jackson SP, Downward J, et al. PI3-kinase p110alpha mediates beta1 integrin-induced akt activation and membrane protrusion during cell attachment and initial spreading. Cell Signal. 2010 Dec;22(12):1838–48. doi: 10.1016/j.cellsig.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 44.Stupack DG, Cheresh DA. Get a ligand, get a life: Integrins, signaling and cell survival. J Cell Sci. 2002 Oct 1;115:3729–38. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 45.Ross RS. Molecular and mechanical synergy: Cross-talk between integrins and growth factor receptors. Cardiovasc Res. 2004 Aug 15;63(3):381–90. doi: 10.1016/j.cardiores.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 46.Dutt P, Kjoller L, Giel M, Hall A, Toksoz D. Activated galphaq family members induce rho GTPase activation and rho-dependent actin filament assembly. FEBS Lett. 2002 Nov 20;531(3):565–9. doi: 10.1016/s0014-5793(02)03625-6. [DOI] [PubMed] [Google Scholar]
- 47.Dutt P, Jaffe AB, Merdek KD, Hall A, Toksoz D. Galphaz inhibits serum response factor-dependent transcription by inhibiting rho signaling. Mol Pharmacol. 2004 Dec;66(6):1508–16. doi: 10.1124/mol.104.002949. [DOI] [PubMed] [Google Scholar]
- 48.Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration--the actin connection. J Cell Sci. 2009 Jan 15;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.