

Abstract
Nitric oxide, when combined with superoxide, produces peroxynitrite, which is known to be an important mediator for a number of diseases including various liver diseases. Peroxynitrite can modify tyrosine residue(s) of many proteins resulting in protein nitration, which may alter structure and function of each target protein. Various proteomics and immunological methods including mass spectrometry combined with both high pressure liquid chromatography and 2D PAGE have been employed to identify and characterize nitrated proteins from pathological tissue samples to determine their roles. However, these methods contain a few technical problems such as low efficiencies with the detection of a limited number of nitrated proteins and labor intensiveness. Therefore, a systematic approach to efficiently identify nitrated proteins and characterize their functional roles is likely to shed new insights into understanding of the mechanisms of hepatic disease pathophysiology and subsequent development of new therapeutics. The aims of this review are to briefly describe the mechanisms of hepatic diseases. In addition, we specifically describe a systematic approach to efficiently identify nitrated proteins to study their causal roles or functional consequences in promoting acute and chronic liver diseases including alcoholic and nonalcoholic fatty liver diseases. We finally discuss translational research applications by analyzing nitrated proteins in evaluating the efficacies of potentially beneficial agents to prevent or treat various diseases in the liver and other tissues.
1. Introduction
Nitric oxide (NO) is a common free radical that is synthesized via enzymatic and nonenzymatic reactions in various cells and tissues. NO is also a very important intracellular signaling molecule in all vertebrates and even in plants. NO is enzymatically synthesized through three different isoforms of NOS (nitric oxide synthase), namely, NOS1, NOS2, and NOS3 [1]. Neuronal NOS (nNOS or NOS1) is expressed in the brain in large quantities [2, 3]. NOS2, known as inducible NOS (iNOS), is induced in various tissues in response to proinflammatory cytokines or oxidative stress under pathological conditions or following exposure to toxic agents [4]. NOS3, known as endothelial NO synthase (eNOS), is typically expressed in vascular endothelial cells and associated to plasma membrane [4]. Thus the three NOS isozymes were named either after their constitutive expression in certain tissues (nNOS and eNOS) or after their expression mechanism (iNOS). While both NOS1 and NOS3 can be activated by intercellular calcium and calmodulin, NOS2 can be induced completely with normal levels of calcium [4]. NO is produced by a reaction that is composed of two sequential steps: (1) the hydroxylation of guanidino nitrogen of L-arginine, leading to the generation of the intermediate Nω-hydroxy-L-arginine (NOHA), and (2) NOHA is then oxidized to NO and L-citrulline [5]. NO by itself is not highly reactive since NO is the intermediate molecule between molecular oxygen (O2) and nitrogen (N2) [4]. NO has a very short half-life; however, it can diffuse freely across cell membranes [6]. The classical NO signaling is related to its activation of soluble guanylate cyclase (sGC) and subsequently cyclic guanosine monophosphate- (cGMP-) dependent protein kinases [7]. The less- and nonclassical NO signaling pathways are related to NO binding to cytochrome c oxidase in the mitochondria and the cGMP-independent NO-related posttranslational modifications (PTMs), respectively [7].
There are many forms of NO which are interchangeable and leading to the production of reactive nitrogen species (RNS) such as nitrosonium cation (NO+), nitroxyl radical (NO·), nitroxyl anion (NO−), nitrite (NO2 −), nitrate (NO3 −), and nitrous oxide (N2O). These intermediates provide the NO molecule as a unique signaling molecule that can act within the cell or interact with adjacent cells without the need of a receptor [6]. The generation of secondary oxides of nitrogen such as peroxynitrite (ONOO−) and nitrosothiols (RSNO) can also produce diverse biological effects through interactions with cellular macromolecules (e.g., DNA, lipids, and proteins), as illustrated in Figure 1. Peroxynitrite can stimulate nitration of tyrosine (Tyr) residue(s) and S-nitrosylation of Cys residues, both of which can lead to alterations of protein structure and function [8, and references therein]. Nitrogen dioxide (NO2) can also interact with other oxidants such as superoxide radicals (O2 ·−), H2O2, and transition metal centers in various heme-containing proteins, leading to production of peroxynitrite, which nitrates Tyr residues of various proteins generating 3-nitroTyr (3-NT), which is widely accepted as a foot print of peroxynitrite formation [9–11]. Reactive oxygen species (ROS) including superoxide radicals can be produced from various sources including mitochondrial electron transport chain (ETC) and other cellular enzymes such as NADPH oxidase and myeloperoxidase or eosinophil oxidase in immune cells (i.e., macrophage cells and neutrophils), ethanol-inducible cytochrome P450 2E1 (CYP2E1) and CYP4A isozymes in endoplasmic reticulum (ER), cytosolic xanthine oxidase, and so forth [12–19]. RNS not only interact with Tyr, but also with tryptophan (Trp) [20], lipids [21], and vitamins [22, 23]. Thus, the nitrative modifications of target proteins, DNA, lipids, and vitamins, usually contributing to alterations of their normal functions and the development or progression of tissue injury [4, 24, 25]. Tyr is a common amino acid found in most proteins [26] and is readily an accessible target for protein nitration since it is often exposed on the surface of the protein due to its mild hydrophilic characteristic [27, 28]. Thus, protein nitration data in literature is largely associated with Tyr nitration and functional consequences. The modification of Trp residues in proteins may occur in a more limited number of sites than that of Tyr residues since Trp is less abundant than Tyr in the protein and is usually buried inside with an exception for a few surface exposed Trp residues. Therefore the modification of those Trp residues may alter specific interaction of proteins and/or enzymes with other molecules, which may cause functional dysregulations [20]. However, in this review, we do not discuss much about Trp nitration since its formation, identification, and significance were extensively reviewed elsewhere [20, 29, 30]. It is likely that modification of each amino acid (e.g., nitration of Tyr or Trp or S-nitrosylation of Cys) seems to depend on the local microenvironment such as pH, solvent exposure or accessibility, peptide loop structure, and the presence of other competing amino acids (e.g., Cys near the potentially nitrated Trp or Tyr) [20] or a denitrase present in several tissues [31].
Figure 1.
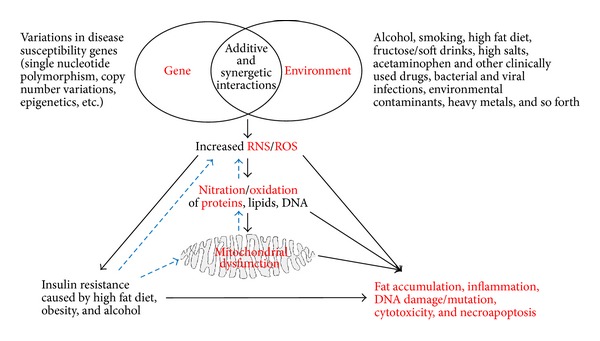
Synergistic interaction between genetic and environmental factors in promoting acute and chronic liver diseases. Additive or synergistic interactions between genetic and environmental factors such as alcohol, smoking, fat diet, and other potentially toxic substances result in increased production of RNS/ROS, which can modify cellular DNA, lipids, and proteins, promoting mitochondria dysfunction and interrupting many important signaling pathways. Continued presence of increased nitroxidative stress (through a vicious cycle shown in blue dotted arrows) contributes to acute and chronic liver diseases including alcoholic fatty liver disease (AFLD) and nonalcoholic fatty liver disease (NAFLD).
Protein Tyr nitration has been reported in correlation with many pathological conditions such as cardiovascular disorder, diabetes, hepatic disease, ischemia-reperfusion (I-R) injury, neurodegenerative diseases, stroke, inflammatory diseases, and cancer, as reported in many human diseases and experimental models including cell culture systems [4, 32]. Despite the numerous reports about the occurrence and consequence of Tyr nitration in various human diseases and in experimental animal models [4, and references therein], there have been a relatively small number of reports that systematically dealt with identification and functional characterization of nitrated proteins in various subcellular organelles, including mitochondria, especially under conditions with increased nitroxidative stress. Fewer reports on protein oxidation, nitrosation, and nitration might be due to the requirement for specific reagents, the lack of suitable methods to systematically identify and purify nitrated proteins, and the relatively late development and advancement of highly sensitive mass spectrometry (MS) instruments. There are many questions that need to be addressed when working with nitrated proteins as follows: (1) what are the sources of ROS/RNS in enhancing protein nitration? (2) which proteins are nitratively modified? (3) are their activities/functions altered following nitrative modifications? (4) what are the overall implications of nitrated proteins in mitochondrial dysfunction or other organelles in various disease states including hepatic liver disease/injury? (5) can the nitrative protein modifications and subsequent dysfunction be prevented with a potential therapeutic agent? and (6) most importantly, do we have a suitable method to efficiently identify the nitrated proteins to study the causal relationship between protein nitration and mitochondrial dysfunction and tissue injury? In this review, we discuss the roles of nitrative stress and protein Tyr nitration in the development and/or progression of various stages of liver disease. We specifically focus on nitration of mitochondrial proteins and their functional implications. We also briefly discuss various methods for identifying nitrated proteins in experimental models with emphasis on the immunoaffinity purification of nitrated proteins followed by their identification by MS analysis. Finally we briefly discuss the utility of studying nitrated proteins in different subcellular fractions in various tissues as well as future translational research opportunities.
2. Functional Consequences of Protein Nitration in Acute and Chronic Liver Diseases
Many pathological conditions usually result from changes in genetic or environmental factors or a synergism between both factors. Increased nitroxidative stress seems to play a critical role in mediating the pathological effects of this synergism or even each factor alone (Figure 1). Under normal physiological conditions, protein nitration and function alterations could be properly managed by the antioxidant host defense system [50, 51] increased nitroxidative stress, four possible outcomes may arise from protein nitration: (A) a decrease or loss, (B) an increase, (C) immunomodulation and immunogenicity, or (D) little or no effect on the biological functions of the modified target proteins. One of the challenges that arise when determining the functional implications of protein nitration is that nitration is not unique to Tyr residues since Trp can be also nitrated. In addition, various amino acids such as Trp, cysteine (Cys), histidine (His), proline (Pro), lysine (Lys), and methionine (Met) can be also oxidized or S-nitrosylated [20, 50]. Another challenge could be to identify and characterize nitrated proteins that are expressed in very low quantities such as various transcription factors and signaling proteins, contributing to tissue injury.
2.1. Inhibition of Nitrated Proteins and Functional Consequences
The decreased function by protein nitration may result from the inhibition or loss of catalytic activity and/or decreased protein levels since protein nitration likely changes the protein secondary structure, hindering the access of the substrate to the activity site [4]. Alternatively, nitrated and/or oxidatively modified proteins that are improperly folded or damaged are known to be removed through ubiquitin-dependent proteolytic degradation mechanism [52, 53] while trypsin, serine proteases, or calpain is not involved in this process [53]. In addition, nitration and/or oxidation may lead to protein aggregation and the consequent loss of its activation and/or functions reported with α-synuclein, a major constituent of the typical protein aggregates observed in several neurodegenerative diseases that are collectively referred to as synucleinopathies [54, 55]. The removal of nitrated proteins can serve as a defense mechanism against nitroxidative stress-related harmful consequences but also plays an essential role in many vital processes including cell division, apoptosis, cell differentiation, DNA repair, membrane transport, oncogenesis, and signal transduction [56, 57]. At the same time, degradation with decreased levels of essential proteins for cell maintenance and survival can also be detrimental to the cells, especially when the degradation rates of proteins essential for energy production, antioxidant or anti-inflammatory defense, urea metabolism, and so forth, exceed those of their regeneration or other compensatory mechanism(s) due to persistence of toxic effects.
By using biotin-N-maleimide (biotin-NM) as a sensitive biotin-switch probe to proficiently identify oxidized proteins, we previously showed that many mitochondrial proteins including the enzymes involved in fat oxidation and energy supply could be oxidatively modified under increased nitroxidative stress and thus inactivated, leading to increased fat accumulation and ATP depletion in the experimental models of alcoholic fatty liver disease (AFLD) and acute I-R liver injury (Figure 1). We expect that protein nitration would also produce similar or more damaging effects in combination with other PTMs. We briefly describe the inhibition of catalytic activities and/or degradations of essential proteins following nitration and their functional implications in three major liver disease/injury models, namely, (1) AFLD, (2) nonalcoholic fatty liver disease (NAFLD), and (3) drug/xenobiotic-induced acute liver injury.
2.1.1. Alcoholic Fatty Liver Disease
The AFLD ranges from simple steatosis with microvesicular fat accumulation to more severe forms including steatohepatitis, fibrosis, cirrhosis, and finally hepatocellular carcinoma following chronic heavy alcohol ingestion [16, 58–62]. Obesity, hyperlipidemia, inflammation, nitroxidative stress, insulin resistance, and so forth are risk factors for AFLD ([63, 64] and references therein). Several mouse models have been used to evaluate the effect of protein nitration on nitroxidative stress-mediated in AFLD. For instance, the role of protein nitration has been studied in mouse strains with ablated genes that are involved in either decreasing or increasing the levels of superoxide and NO. The mouse strains include Cyp2e1(−/−), SOD1(−/−), SOD2(−/−), iNOS(−/−), and TNF-α receptor (TNFR) (−/−), as described below.
Peroxynitrite and protein nitration were suggested to be main causes of acute and chronic alcoholic fatty liver injury models. Expression of iNOS was increased following the exposure to ethanol [15, 16, 37, 65–67]. Administration of the Lieber-DeCarli ethanol liquid diet has significantly increased the levels of steatosis, apoptosis, necrosis, and inflammation in wild-type (WT) mice compared to the corresponding iNOS(−/−) mice. The severity of liver injury was proportional to the levels of hepatic nitration and inhibition of mitochondrial function in alcohol-exposed WT mice, while iNOS(−/−) mice with markedly decreased levels of nitrated proteins were resistant to AFLD [15, 16] and that protein nitration was shown to inhibit complex I (NADH ubiquinone oxidoreductase) and complex V (ATP synthase) activities in acute and chronic alcohol-exposure models [36, 37, 68]. Thus protein nitration seems to play an important role in promoting the ethanol-mediated mitochondrial dysfunction and liver injury since these proteins are essential for normal mitochondrial function while protein nitration might lead to irreversible modification of the respiratory chain proteins, contributing to increasing mitochondrial sensitivity to NO and ultimately AFLD [69]. We and other laboratories clearly showed that nitration of ATP synthase led to significant inhibition of its activity in ethanol fed rats and mice [37, 70]. The suppression of ATP production would certainly increase mitochondrial sensitivity to other oxidative insults and necrotic injury. In an acute binge ethanol model, iNOS expression, serum nitrite/nitrates, increased protein nitration of mitochondrial complex V (ATP synthase), decreased activities of both complex I and V, and mitochondrial DNA depletion were observed in WT mice but not in SOD2(−/−) mice [71]. The authors suggested that these damaging effects were probably due to protein nitration since administration of iNOS inhibitors and peroxynitrite scavengers like uric acid ameliorated the ethanol-induced nitration, inhibition of activity, and mitochondrial depletion of ATP synthase. In addition, mice lacking SOD2, which would scavenge superoxide and thus block the peroxynitrite formation, exhibited prolonged mitochondrial DNA depletion while mice overexpressing SOD2 showed opposite outcomes [36]. Similar to the protective role of mitochondrial SOD2, cytosolic SOD1 also exhibits protective roles against ethanol-mediated hepatic damage. It has also been shown that, in mice deficient in SOD1, the levels of protective hepatic ATP content and SOD2 expression were decreased while oxidative damage and nitro-Tyr formation were elevated in response to ethanol feeding, leading to greater hepatic injury in the SOD1(−/−) mice [61]. This data suggests that mitochondrial dysfunction in the liver following ethanol exposure might originate from the compromise in the cytosolic antioxidant defense mechanism. In alcohol consumption studies, it was suggested that ethanol increased the sensitivity of mouse and rat hepatocytes to the combined effects of ethanol and NO [36, 71–73]. This combined effect of ethanol and NO increases the susceptibility to hypoxia and depression of mitochondria bioenergetics reserve energy state of the liver, leading to energy depletion and ultimately the development of alcohol-mediated hepatic injury [72, 73]. While NO donors inhibited mitochondria respiration and increased mitochondrial dysfunction in ethanol-fed rats more than the controls [73], iNOS(−/−) exhibited less severe liver injury with decreased levels of hypoxia-inducible factor 1-α in the perivenular region of the liver lobule than in the corresponding WT [16, 72]. Taken together, hepatic mitochondria from ethanol-fed mice or rats are more sensitive to NO and RNS while iNOS plays an essential role in determining the response to hypoxic stress in vivo. Since ethanol hepatotoxicity was also significantly prevented through a mechanism that involves a decreased inflammatory response, tumor necrosis factor-α (TNF-α) formation, and fatty liver [15], it was not surprising that TNFR(−/−) mice exhibited significantly less severe ethanol-mediated hepatotoxicity accompanied with markedly lower levels of protein Tyr nitration than those of their WT counterparts.
In addition, the elevated nitroxidative stress and subsequent hepatotoxicity in acute and chronic alcohol exposure models are correlated with the increased hepatic amounts and catalytic activities of CYP2E1, the most relevant cytochrome P450 for the development and progression of AFLD [13, 74–80]. For instance, in the chronic alcohol-fed mouse models, the levels of protein nitration were highest in Cyp2e1 knock-in mice, followed by WT mice and lastly in Cyp2e1(−/−) mice, which exhibited almost basal levels of nitrated proteins [81]. The levels of protein nitration correlated well with the increased levels of hepatic transaminases, steatosis, and necrosis [81]. Since CYP2E1 is highly expressed in the endoplasmic reticulum (ER) and mitochondria [82–85], where iNOS is also present, it is conceivable to predict that protein nitration and oxidation can take place in both organelles and their participation in the development of AFLD either through increased ER stress and/or mitochondrial dysfunction, both of which are known as causative factors of AFLD.
Formation of peroxynitrite and protein nitration might not be the only causes for ethanol-induced hepatic toxicity. For instance, induction of nitrative stress along other forms of oxidation in intestinal epithelium may also serve as a cause for ethanol-induced gut leakiness and endotoxemia, leading to inflammatory liver injury. The intestinal epithelium, under normal conditions, serves as a highly selective barrier so that potentially toxic substances or products (e.g., lipopolysaccharide (LPS)) of the gut bacteria into the circulation [86–88]. Disruption of the intestinal barrier integrity (i.e., leaky gut) may lead to the penetration of luminal bacterial products such as endotoxin into the mucosa, then into the systemic circulation, and initiate local inflammatory processes in the intestine, blood vessels, and even in the liver [88]. Numerous studies in the literature showed that ethanol can compromise the integrity of intestinal epithelium, leading to increased gut leakiness in rodents [89–95] and human alcoholics [90, 96]. Ethanol-induced production of NO via induction of iNOS and subsequent formation of peroxynitrite in intestinal Caco-2 cells lead to barrier dysfunction probably due to oxidation and nitration of cytoskeletal proteins and/or tight junction proteins [88, 90]. This data was also validated in vivo studies in a mouse model fed ethanol for 10 weeks with iNOS inhibitors. Coadministration with iNOS inhibitors attenuated ethanol-mediated NO overproduction, oxidative tissue damage, leaky gut, endotoxemia, and liver injury including steatosis [97].
CYP2E1 protein expression was also increased in Caco-2 intestinal epithelial cells and in both acute and chronic ethanol-exposed mouse models [78, 98]. In addition to increased nitroxidative stress and protein nitration, expression of redox-sensitive circadian clock proteins CLOCK and PER2 was significantly elevated in Caco-2 cells and in mice exposed to ethanol, leading to increasing intestinal hyperpermeability [98]. The usage of a CYP2E1 siRNA in Caco-2 cells or antioxidant N-acetyl cysteine (NAC) in mice exposed to ethanol ameliorated nitroxidative stress including iNOS expression and consequently inhibited the expression of CLOCK and PER2 as well as gut permeability. Further, recent studies from our laboratory showed that iNOS and protein nitration were also increased in intestinal epithelial cells in binge ethanol-exposed WT but not in the corresponding Cyp2e1(−/−) mice, leading to increased endotoxemia and subsequently inflammatory liver damage and apoptosis in ethanol-exposed WT mice [78]. We then showed that inhibition of iNOS induction and protein nitration of intestinal epithelial cells in mice pretreated with a CYP2E1 inhibitor or an antioxidant NAC ameliorated all the damaging effects in intestinal epithelial cells accompanied by decreased serum endotoxin levels and oxidative hepatic injury including steatosis and apoptosis in the ethanol-exposed WT mice. In contrast, these ethanol-mediated events were markedly attenuated in alcohol-exposed Cyp2e1(−/−). These results support at least a partial role of intestinal protein nitration in mediating the alcohol-induced gut leakiness and subsequent hepatic injury. Protein nitration in intestine cells is also likely to affect the cytoskeleton protein architecture [90] and/or intestinal tight gap-junction proteins, leading to alteration of barrier function with increased permeability [78, 97]. Collectively, all these studies suggest that elevated iNOS and CYP2E1 play an important role, at least partially, in producing peroxynitrite and protein nitration in gut leakiness and AFLD, while normal levels of SOD1 and SOD2 seem protective against the development of AFLD since ethanol-mediated hepatic toxicity was worsened upon the inhibition or deletion of these SOD proteins. In addition, the development of peroxynitrite and subsequent protein nitration of essential mitochondrial proteins and other vital proteins in other subcellular organelles (e.g., cytosol) would be expected to play a causal or contributing role in mediating ethanol-mediated hepatotoxicities (Table 1). This data also suggests that in liver exposed to toxic substances/xenobiotics that increase ROS/RNS would sensitize the liver for even stronger and faster development of ethanol-mediated damaging effects, as evidenced with concurrent exposure to LPS, acetaminophen (APAP, a major ingredient of Tylenol), nicotine (a major component of smoking), 3,4-methylendioxymethamphetamine (MDMA) ([38, 99] and references within), high fat diet (HFD), and so forth. However, other forms of PTM including lipid peroxidation, S-nitrosylation including glutathionylation, glycosylation, and glycation including advanced glycation end-products (AGE), might also be involved in AFLD [8, 100] (Figure 1).
Table 1.
List of confirmed nitrated hepatic proteins and functional consequences.
| Nitrated proteins | Activity | Hepatic indication (observed or expected) | References |
|---|---|---|---|
| Carbamoyl phosphate synthase-1 (CPS-1) | Decrease | Hyperammonemia, hepatic encephalopathy | [33] |
| Glutamine synthetase (GS) | Decrease | Hyperammonemia, hepatic encephalopathy related to sepsis | [34] |
| 3-Ketoacyl-CoA thiolase (Thiolase) | Decrease | Decreased β-oxidation of fatty acids with increased hepatic steatosis | [35] |
| Aldehyde dehydrogenase 2 (ALDH2) | Decrease | Accumulation of acetaldehyde and lipid peroxides with increased aldehyde-related liver toxicity | [35] |
| Complex I (NADH ubiquinone oxidoreductase) | Decrease | ROS leakage, contributing to decreased energy production and increased apoptosis or necrosis | [36] |
| Complex V (ATP synthase) | Decrease | Decreased energy production with increased sensitivity toward necrotic liver injury | [11, 35–38] |
| Cytochrome p450 2E1, B6 (CYP2E1, CYP2B6) |
Decrease | Drug metabolism: ROS production and ethanol- and drug-induced liver toxicity | [39] |
| Cytosolic Cu/Zn-SOD (SOD1) | Decrease | Decreased antioxidant defense with increased drug- or toxin-mediated hepatic damage | [35, 40] |
| Mitochondrial Mn-SOD (SOD2) | Decrease | Same as above | [35, 41] |
| Glutathione peroxidase (GPX) | Decrease | Same as above | [35] |
| Glutathione reductase (GR) | Decrease | Increased oxidative stress with elevated levels of oxidized glutathione | [42, 43] |
| AKT, IRβ, IRS-1, IRS-2 | Decrease | Decrease insulin signaling with increased hepatic insulin resistance and fatty liver | [44] |
| CD95 | Decrease | Increased hepatic anti-inflammatory defense | [45] |
| List of nitrated mitochondrial and cytosolic proteins | Not confirmed* | Not confirmed but likely contributing to mitochondrial dysfunction, ER stress, and liver damage | [35] |
| List of nitrated mitochondrial proteins | Not confirmed | Not confirmed but likely contributing to mitochondrial dysfunction and liver damage | [46] |
| List of nitrated proteins in different compartments | Not confirmed | Not confirmed but likely contributing to mitochondrial dysfunction, ER stress, and liver damage | [47] |
| Glutathione-S-transferase (GST) | Increase | Increased hepatic antioxidant defense | [41] |
| Heat shock protein 90 (Hsp90)** | Increase | Conversion to a toxic protein, contributing to increased liver toxicity | [48] |
| Protein phosphatase type 2A (PP2A)** | Increase | Increased microvascular endothelial permeability | [49] |
*With the exception of the five proteins characterized in detail, as in the reference [35].
**Not confirmed in liver, but expected to occur.
2.1.2. Nonalcoholic Fatty Liver Disease
NAFLD, usually caused by chronic ingestion of nonalcoholic substances such as HFD, cholesterol-containing fast food western diet, fructose, and choline-deficient diet, is one of the most common chronic liver diseases in the USA and developed countries [101, 102]. NAFLD often associated with overeating and obesity starts as simple steatosis and can progress to inflammation (nonalcoholic steatohepatitis, NASH), fibrosis, cirrhosis, and even cancer, as similar to the progression of AFLD [13, 103]. Many risk factors are known to promote the progression of NAFLD to NASH, fibrosis, and hepatocarcinoma. The risk factors include increased nitroxidative stress, mitochondria dysfunction, insulin resistance, disturbance of fat homeostasis, gut leakage, cytokine, and immune dysregulation [103, 104]. There are numerous reports in the literature showing that increased levels of iNOS, CYP2E1, and protein nitration play an important role, at least partially, in the development and/or progression of NAFLD, as shown in various experimental models and people with NASH ([74, 105–115], and references therein). In a NASH model fed a HFD for 20 weeks, the overexpression of tissue inhibitor of metalloproteinase (TIMP3) in macrophages ameliorated HFD-induced insulin resistance, adipose inflammation, and NAFLD probably through the inhibition of various parameters of nitroxidative stress including hepatic protein nitration [116]. The role of leptin in mediating the progression of NAFLD to NASH was evaluated in WT and leptin-deficient ob/ob mice fed a HFD for 16 weeks followed by treatment with carbon tetrachloride (CCl4) [117]. HFD-mediated NASH observed in WT was resulted from the induction of iNOS and NADPH oxidase, leading to increased nitroxidative stress, which then activated hepatic Kupffer cells with increased inflammation [117]. In addition, the coadministration of leptin with CCl4 to ob/ob mice significantly elevated nitroxidative stress compared to treatment with CCl4 alone [117]. Finally, the deletion of iNOS or p47 phox subunit of NADPH oxidase ameliorated the peroxynitrite formation and protein nitration [117], suggesting an important role of protein nitration, at least partially, in the leptin-mediated activation of Kupffer cells. Further, metal iron was also found to increase liver injury in diabetic rats, partially, through nitration of glucokinase accompanied with decreased levels of its expression and activity, shedding some new lights on the role of iron in the development of hepatotoxicity in diabetes mellitus [118]. Peroxynitrite and protein nitration were also reported to play a central role in the suppression of the mitochondrial respiratory chain activity, leading to mitochondrial dysfunction in a NASH model using ob/ob mice while these effects were ameliorated by melatonin administration [119]. The source of NO seems to be important in determining the outcome of the increased or decreased NO levels. For instance, in a HFD-mediated NAFLD rat model, simvastatin was found to protect against the development of HFD-induced liver fibrosis via differentially regulating NOS isozymes, where eNOS was found to be elevated while iNOS was decreased [112]. These results suggest that eNOS seems to be protective against NAFLD, while iNOS seems to promote NAFLD [112]. Protein nitration was also reported to play an important role in the development of insulin resistance in NAFLD in WT mice infused for 6 h with a 20% intralipid emulsion [120]. Further, iNOS induction and consequent Tyr-nitration of key insulin signaling proteins such as AKT, insulin receptor-β (IRβ), insulin receptor substrate-1 (IRS-1), and insulin receptor substrate-2 (IRS-2) [120] lead to the interference with Tyr phosphorylation, a signature feature of the insulin signaling pathways. In addition, there was increased serine phosphorylation, which leads to the inhibition of the hepatic insulin signaling pathway, especially when Tyr phosphorylation was inhibited [120]. These events were monitored with minimal changes in the basal levels of the insulin signaling molecules before and after lipid infusion [120]. Thus, a novel mechanism for hepatic insulin resistance caused by circulating lipids from HFD through protein nitration was suggested since these events were significantly ameliorated in iNOS(−/−) mice [120]. Furthermore, nitration of IRS-1 and IRS-2 proteins caused decreased expression of both proteins in obese diabetic rats or when hepa1c1c7 and HepG2 hepatoma cells were exposed to NO donors such as GSNO and SIN-1 in a time- and dose-dependent manner. In contrast, their expressed levels were increased after the hepatoma cells or obese diabetic rats were treated with an iNOS inhibitor, N6-(1-iminoethyl)-L-lysine (L-NIL) compared to their control [44]. The decreased protein levels of IRS-1 and IRS-2 following their nitration in these models of NAFLD might be due to their ubiquitin-dependent degradation, as discussed previously [53, 120]. Taken together, the outcome of these effects would depend not only on the extent of inactivation and/or loss of the nitrated proteins but also on the cellular ability to overcome the loss of these particular nitrated proteins by various cell defense mechanisms. Thus, it is reasonable to conclude from the two aforementioned studies [44, 120] on HFD-mediated hepatic insulin resistance where insulin signaling molecules could be by nitrated without change in their protein levels probably occurring at early stages of the disease process (i.e., following a short-term exposure). Alternatively, insulin resistance could also result from the decreased levels of nitrated insulin signaling molecules via proteolytic degradation, usually observed at later time points following exposure to persistent insults.
Based on the aforementioned reports, future studies on the functional implications of protein nitration should consider the temporal evaluation of protein nitration and the remaining levels of nitrated proteins, since the nitration process is extremely dynamic and that the levels of nitrated proteins could vary dramatically over the course of acute or chronic hepatic insults. This is particularly important since there are many reports suggesting that protein nitration is not chemically stable, as previously believed. By using a reducing agent sodium dithionite as described previously [121], we showed that most of the nitrated cytosolic proteins, determined by immunoblot analysis following 2D gel electrophoresis, completely disappeared in APAP-exposed mouse livers [40]. The disappearance of nitrated protein spots was due to the reduction of nitro-Tyr to amino-Tyr, which was no longer recognized by the antibody to 3-NT. The reduction of nitro-Tyr to amino-Tyr was also reported by a pure chemical reaction from Fe3+-containing heme as in hemoglobin and myoglobin in the presence of a reducing agent [122]. Similar reaction has been also reported under physiological conditions where nitrated deoxynucleo bases were reduced to their amino analogues [123].
One might think that the decreased levels of nitrated proteins over time only result from their proteolytic degradation, as previously discussed; however, this is not necessarily true. For instance, in the presence of protease inhibitors to block proteasomal degradation of nitrated proteins, nitrated BSA disappeared when incubated with the homogenates of spleen or lung tissues, but not with the homogenates of rat liver or kidney, suggesting that different isoforms of denitrase might exist in a tissue-specific manner [124]. Protein with denitrase activity has been detected in a variety of tissues including the liver, heart, lung, brain, spleen, and kidney [31, 125, 126]. Denitrase activity has been suggested to be present in both aqueous and membrane phases in the cells [31]. Indeed, denitration reaction via denitrase was observed with nitrated Histone H1.2 [125], glutamine synthetase [127], calmodulin [128], L-type Ca2+ channel [129], cyclooxygenase [31], and so forth. Although constitutively active denitrase was present in many tissues and partially purified using the nitrated cyclooxygenase as a substrate [31, and references therein], the biochemical and regulatory properties of denitrase enzyme still need additional investigations. Collectively, data from these reports suggested that denitration may serve as an adaptive mechanism by which cells can repair damaged proteins. In addition, the nitration and denitration process can be of particular importance for cell signaling-related events. For a molecule to act as a signal, nitration-denitration has to be a reversible process. Indeed, nitrated cytochrome c was suggested to act as a signal molecule, as shown in cytochrome c overexpressing HeLa cells exposed to peroxynitrite with the spontaneous translocation of nitrated cytochrome c from mitochondria to cytosol and nucleus [130]. It is also very important to temporally study the nitric oxide bioavailability through the course of NAFLD. For example, in a mouse model of NAFLD fed a HFD for 8 and 16 weeks, NO contents were initially increased causing mitochondrial damage with mitochondrial protein alterations but decreased at later stages of the NAFLD [131]. The authors suggested that decreased NO might be involved in the progression of NAFLD to NASH. Again the decreased NO levels at later stages seem to be due to increased levels of arginase-1, depleting the substrate (i.e., L-arginine) of NOS and decreased levels of activated eNOS-PSer1177, which is protective against disease progression [112, 131]. However, in a clinical study using patients with different grades of liver cirrhosis, a positive correlation between the levels of nitrated proteins in plasma, platelets, and liver tissues and the severity of liver cirrhosis was found [132]. Thus, based on the results from previous reports [124, 132] and others, it is still unclear whether protein nitration can directly play a role in the initiation and/or development of NASH progressed from NAFLD. NAFLD is not only induced by pathological conditions such as obesity and diabetes but also by various diets such as HFD, fructose, and (methionine- and) choline-deficient diet [133–136]. Collectively these reports suggest a potential role of nitrated proteins in mediating NAFLD through various mechanisms (Figure 1).
2.1.3. Acute Liver Injury Caused by Drugs and Xenobiotics
There are many enzymes that are expressed in the liver and other extrahepatic tissues while their activities are compromised by Tyr-nitration following exposure to certain drugs (e.g., APAP, zidovudine (AZT), MDMA, tamoxifen, amiodarone, and so forth) or other toxic substances including lipopolysaccharide (LPS). These agents can provoke acute liver injury with or without fat accumulation probably through increased nitroxidative stress-mediated inhibition of the enzymes involved in the fatty acid oxidation pathway (i.e., mitochondrial β-oxidation) [38, 137, 138]. If an enzyme, essential for the energy production or detoxification of certain metabolites, is inhibited under elevated nitroxidative stress, this inhibition is likely to exert deleterious effects on the liver function and/or hepatic disease progression. For instance, mitochondrial carbamoyl phosphate synthetase 1 (CPS1), which is responsible for the detoxification of excess ammonia, was found to be inactivated when it is nitrated at Tyr1450 [33]. This decline in CPS1 activity may result in hyperammonemia and subsequent mitochondrial dysfunction in in vivo situations, contributing to liver damage and possibly injury to the brain. LPS dramatically increased protein nitration along with other parameters of oxidative stress such as lipid peroxidation in the livers of peroxisome proliferator-activated receptor-α (PPARα)(−/−) than in WT, which also exhibited significantly increased nitration compared to its untreated control, leading to elevated hepatic toxicity in PPARα(−/−) than WT [139]. Mitochondrial dysfunction was observed in LPS-exposed PPARα(−/−) mice, as evidenced by the inhibition of complex I and ATP synthase activity, both of which were reported to lose their activities by nitration [36, 139]. Further, LPS exposure decreased the catalytic activity and expressed amount of hepatic mitochondrial glutamine synthetase (GS) in rat liver through nitration at multiple Tyr residues including the active site Tyr160 [34], accompanied by inactivation. Since GS is also responsible for ammonia elimination, a compromise in its activity and function likely contributes to sepsis-induced development of hyperammonemia in patients suffering from hepatic cirrhosis [34]. The disturbance in ammonia metabolism may not only affect the liver but also the brain, leading to a condition known as the hepatic encephalopathy.
Mitochondrial ATP synthase is essential for providing cellular energy (i.e., ATP) for proper maintenance and survival of all living cells. If ATP synthase (mitochondrial complex V) is inhibited, this leads to depletion of an essential energy source, thus contributing to acute necrotic liver injury. We showed that the activity of ATP synthase was significantly inhibited in mice subjected to I-R injury [140], in MDMA-exposed rats [38], in LPS-exposed Ppara(−/−) mice [139], and in mice exposed to a toxic dose of APAP [35]. In these studies, we confirmed its nitration by immunoblot analysis with the anti-3-NT antibody following the immunoprecipitation of ATP synthase. It is noteworthy to mention that, in all of these models, the protein levels of ATP synthase seemed unchanged in response to any of those treatments, suggesting that the inhibition of its activity was mainly due to nitration-mediated inactivation.
Several hepatic enzymes involved in cellular defense were reported to be nitrated and their activities inhibited in livers and other tissues or cultured cells after exposure to toxic insults. APAP can be considered a prime example of the role of protein nitration in drug-induced liver injury (DILI). It has been shown that nitro-Tyr protein adducts formation correlated very well with the areas of hepatic necrosis [141]. The direct evidence of the critical role of peroxynitrite in APAP-mediating hepatic injury has also been provided by the seminal work of Knight et al. [142]. This study showed that the protective effects of GSH, either cotreated with APAP or administered at different intervals up to 2.25 h post-APAP treatment, were resulted from the restoration of cellular GSH levels and the increased efficiency of scavenging peroxynitrite [142]. Similar results were also obtained in glutathione-peroxidase deficient animals [142]. In addition, Cover et al. [143] showed for the first time that APAP can selectively promote mitochondrial protein nitration. This study also provided evidence that peroxynitrite can cause mitochondrial DNA damage directly and/or indirectly via the mitochondrial damage pathway [143]. The events for APAP-induced protein nitration and liver injury seem dependent on the presence of CYP2E1, which is one of the major enzymes involved in the metabolism of APAP [40, and references therein]. Furthermore, in a comprehensive study using a toxic dose of APAP, nitration of cytosolic Cu/Zn-dependent superoxide dismutase (SOD1) eventually led to ubiquitin-mediated degradation and significant inhibition of its activity and protein levels in WT but not in the corresponding Cyp2e1(−/−) mice, as evidenced by immunoprecipitation using the anti-SOD1 antibody followed by immunoblot analysis with either anti-3-NT or antiubiquitin antibody. These results suggest a role of CYP2E1 in protein nitration and subsequent degradation [40]. Further, several reports showed that when isolated cells or proteins were exposed to peroxynitrite, the induction of proteolytic degradation was observed [54, 55, 144, 145]. The proteolytic degradation of bovine SOD1 was also increased by the 20S/26S proteasome when Tyr108 was nitrated [53]. Taken together, protein nitration can contribute to APAP-mediated toxicity via nitration-mediated inactivation and/or proteasomal degradation of essential proteins in antioxidant defense. The inhibition of antioxidant mitochondrial SOD2 also plays a critical role in promoting APAP-induced hepatotoxicity due to its nitration and subsequent inactivation, hampering cell's ability to defend itself against increased nitroxidative stress [146]. In addition, nitration of cytosolic glutathione reductase (GR) at Tyr106 and Tyr114 decreases GR binding to its substrate oxidized glutathione (GSH) and thus markedly decreased its activity with increased amounts of oxidized GSH [42, 43]. Although this reaction was observed in an in vitro system, it is likely that, in case of increased RNS, hepatic GR may become one of the enzymes that can be also inhibited through nitration. Persistently high levels of oxidized GSH may lead to a vicious cycle of oxidative stress development and sensitize the target tissue to an additional insult. All these results suggest that protein nitration also plays an important role, at least partially, in liver injury caused by acute, subchronic, and chronic exposure to a variety of drugs and hepatotoxic substances. It should be noted however that removal of nitrated proteins either via ubiquitin-dependent protein degradation [40, 53] or autophagy-dependent clearance of damaged mitochondria [147] can also be considered a hepatoprotective pathway. Based on these studies, it is likely that the ultimate hepatic effects of a toxic substance could be decided by the delicate balance between the occurrence and removal of nitrated proteins.
2.2. Activation of Nitrated Proteins and Functional Consequences
Another consequence of protein nitration is a gain of normal protein functions that can either be beneficial or harmful. For example, nitration at Tyr33 or Tyr56 of the 90-kDa heat-shock protein (Hsp90) is likely to convert the Hsp90 into a toxic protein through a gain of function. Using an antibody that recognizes the nitrated Hsp90 [48], nitrated Hsp90 immunoreactivity was detected in motor neurons of patients suffering from amyotrophic lateral sclerosis (ALS), in an animal model of ALS, and in an experimental model of spinal cord injury [48]. The authors concluded that nitration of a single important protein Hsp90 can initiate cell death while this protein can also become a potential target for therapeutic intervention [48]. This observation is also of particular interest in understanding the role of CYP2E1 and the mechanism of acute and chronic liver injuries where elevated CYP2E1 is known to play an important role in mediating these injuries. Hsp90 was found to interact with membrane-bound CYP2E1 forming a binary complex, which then transfers the membrane-bound CYP2E1 to the proteasome complex for its degradation [148]. Ethanol was found to compromise this interaction between Hsp90 and CYP2E1, resulting in increased levels of CYP2E1 protein expression, which can damage the liver cells through its nitroxidative stress-mediated events [148]. These results are consistent with the inhibition of Hsp90 by geldanamycin, which increased CYP2E1-mediated toxicity in HepG2 hepatoma cells [149, 150]. Since both acute binge and chronic ethanol administration also promote hepatic protein nitration, it would be interesting to evaluate whether Hsp90 is nitrated in these alcohol-exposure models and whether this nitration leads to hepatic injury directly or indirectly through interference of binding between nitrated Hsp90 and CYP2E1, leading to its elevation.
The antioxidant glutathione S-transferase 1 (GST-1) was reported to be nitrated at Tyr92 and Tyr152. However, nitration at Tyr92 was shown to be critical in functional change as another example of gain in protein function. This conclusion was evidenced by site directed mutagenesis of each Tyr residue of GST-1, overexpression of each GST-1 mutant in LLC-PK1 cells, and then exposure to peroxynitrite followed by monitoring its activity [41]. The activation of GST-1 nitrated at Tyr92 would improve the antioxidant defense. Tyr nitration was also found to be essential for initiating the seeding process followed by protein aggregation in fibrin for blood clot formation [151] and for aggregation of α-synuclein, a component of Lewy bodies in many neurological diseases [54, 55, 152]. In addition, Tyr nitration of the catalytic subunit of protein phosphatase type 2A (PP2A) seems important in reactivating PP2A activity and promoting LPS and interferon-γ induced microvascular endothelial barrier dysfunction with increased albumin permeability. This gain of function by nitrated PP2A was resulted from interference with Tyr phosphorylation of the catalytic subunit, which subsequently suppresses PP2A activity [49]. All these examples, especially the antioxidant GST-1 and PP2A, with increased activities and functions through protein nitration, are of interest and one needs to take extra cautions against premature conclusions that protein nitration is considered only as a pathological risk factor without proper evaluation of the enzyme activity and functional consequence.
2.3. Induction of Immunogenicity by Nitrated Proteins and Potential Implications in Various Liver Diseases
Another consequence of protein nitration is the induction of immunogenicity and immunomodulation. The body system tolerates its endogenous proteins without eliciting an immune response through immunological tolerance. However, once protein secondary structures are modified via various PTMs, the body immune system might not tolerate the modified proteins and thus immune response might ensue. This immune activation may be due to the structural changes following protein nitration or any other PTMs such as oxidation, carbonylation, phosphorylation, and glycation including AGE, acetylation, S-nitrosylation, glutathionylation, and transglutamination. All these PTMs may contribute to the exposure of a new antigenic epitope, leading to stimulation of a cascade of immune response that starts with the activation of B and/or T cells [153]. This immune reaction can be beneficial when under normal conditions where it clears out a harmful complex or can be hazardous if its level becomes abnormal and sustained through a vicious cycle. Thus the aggregated nitrated proteins in various tissues, including the liver, as seen in inflammation, for example, [154], can initiate or exaggerate an inflammatory reaction that might even become chronic, via the stimulation of the immune system against its own proteins, known as an autoimmune response [155]. Indeed, elevated levels of anti-3NT antibodies were reported in the plasma of patients with post-traumatic acute lung injury [156] and in the synovial fluids of patients with rheumatoid arthritis and osteoarthritis as well as in the serum of patients with the autoimmune disease systemic lupus erythematosus [153]. However, Tyr nitration does not always invoke an immune response; instead it might also inhibit the immune response when it seems necessary in some cases. For instance, nitration of T cell receptor—CD8 complex—compromises its ability to bind to the major histocompatibility complex dimers, leading to tolerance of T cells to cancer cells and hence the survival of cancer cells [157, 158]. Thus protein nitration might be involved in immunomodulation, leading to the development of autoimmune diseases and even cancer progression. Since hepatic protein nitration is remarkably elevated in various chronic diseases including inflammatory diseases, aging, and cancers, it would be valuable to carefully evaluate the role of protein nitration in pathophysiology of these diseases. Alternatively, protein nitration can regulate immune function by interfering with Tyr phosphorylation and concurrent activation of various signal transducer and activators of transcription (STAT) proteins, as reported [159, 160].
2.4. Lack of Functional Consequences of Nitrated Proteins
Finally protein nitration might not have any significant effect on protein activity, expression, and consequently function, as reported with Tyr nitration of transferrin and α1-antichymotrypsin in acute respiratory distress syndrome [161]. It is vital to keep in mind, however, that many studies in the literature with regard to the Tyr nitration or denitration of certain proteins and subsequent functional changes were based on in vitro reactions using high concentrations of peroxynitrite or other nitration-inducing substances and in an environment lacking the full host defense system including various antioxidants present in living organisms. Thus, the nitrative effect of any agent on protein function also needs to be validated in in vivo systems. It is still challenging to determine whether catalytic inhibition or degradation of a particular protein is solely resulted from its Tyr nitration as there are many other forms of PTM that can also inhibit its activity.
Table 1 summarizes some nitrated liver proteins with functional consequences with the reported or suggested consequences.
3. Protein Nitration in Mitochondria and Potential Consequences in Various Liver Diseases
Protein Tyr nitration can occur in different cell compartments and play a critical role in hepatic injury. However, based on the aforementioned studies and alterations of key mitochondrial proteins involved in fat and ammonia metabolism, energy supply, antioxidant defense, and so forth (listed in Table 1 and references therein), mitochondrial functional status can become a very important factor in the initiation and/or progression of liver diseases. In addition, NO-dependent inhibition of the mitochondrial respiration was reported in both AFLD and NAFLD [16, 73, 162]. However, most of the reported hepatic studies, with a few exceptions, indicated the pathological role of nitrated proteins in mediating hepatic disease process, relying mainly on the correlation between protein nitration and hepatic injury without in-depth analysis of functional alterations in nitrated mitochondria proteins. Therefore, we briefly describe the formation of protein Tyr nitration and functional consequences that might explain the role of protein nitration in promoting many forms of liver diseases including AFLD and NAFLD.
Under normal physiological conditions, approximately 1%-2% of oxygen leaks out as ROS from the mitochondrial electron transport chain (ETC) [163]. ROS can either combine with protons to produce water [164, 165] or can be handled with mitochondrial antioxidant defense enzymes where these radicals are dismutated by SOD2 and detoxified by glutathione peroxidase (GPX) [165]. Alternatively, these ROS can be involved in various cellular signaling pathways [166]. However, damaged mitochondria in various pathological conditions or exposure to toxic agents such as alcohol, HFD, LPS, abused substances (e.g., MDMA), or other therapeutic drugs (e.g., APAP and AZT) [137, 164, 167, 168], greater amounts of ROS are leaked out from the mitochondrial ETC, possibly at the sites of Complex I and Complex III, as suggested by alcohol-subjected hepatocytes [169]. RNS including NO radicals can also be detected in the mitochondria after NO is produced in cytosol by the NOS isozymes since NO can readily cross the mitochondrial membranes. In addition, mitochondrial RNS can be produced by mitochondria NOS [170]. Thus the excess of ROS and RNS radicals in the mitochondria can lead to the formation of peroxynitrite and hence increased mitochondrial protein Tyr nitration. Mitochondrial protein nitration can also be mediated through the oxidation of Tyr and nitrite via a peroxidase-catalyzed reaction [171, 172]. Mitochondria, which are the major source of cellular ROS, also have low levels of antioxidants such as reduced glutathione (GSH), compared to that in cytosol [173], rendering mitochondria a vulnerable target for nitrative damage. It has also been suggested that rat liver mitochondrial protein turn-over rates were dramatically decreased from days to hours upon exposure to nitrating conditions due to proteolytic degradation [46, 174–176]. Further, mitochondria under nitroxidative stress in animal models or human disease specimens show abnormal and irregular shapes and decreased functions [177, 178]. Nitration of many mitochondrial proteins can further deteriorate mitochondrial functions through inactivation of some critical proteins for cell maintenance and survival. For instance, suppressed activities of antioxidant enzymes such as SOD2, GPX, and GR would lead to decreased antioxidant cell defense, while suppressed ATP synthase may result in decreased ATP production with compromised cellular ability to perform normal functions and increased necrosis. Furthermore, inactivation of thiolase likely leads to the inhibition of the β-oxidation of fatty acids with increased hepatic fat accumulation or inefficient supply of an alternative energy (e.g., ketone bodies produced from fat degradation) when glucose supply is decreased during fasting or not efficiently utilized in many disease states. Suppression of mitochondrial aldehyde dehydrogenase 2 (ALDH2), involved in the metabolism of reactive acetaldehyde and 4-hydroxynonenal, would lead to accumulation of reactive aldehydes including lipid peroxidation products ([179], Table 1 and references therein).
Mitochondrial functions can also be compromised via deletion and/or mutation through oxidative/nitrative modifications of mitochondrial DNA, which is important as they encode 13 polypeptides that are all subunits of the 4 mitochondrial ETC proteins (i.e., Complexes I, III, IV, and V) [168, 180]. Mitochondrial DNA is also extremely sensitive to nitroxidative damage due to its location within the cell close to the inner mitochondrial membrane, and the relatively low levels of protective antioxidant enzymes, histone proteins, or polyamines and DNA repair enzymes in mitochondria compared to other subcellular organelles including nuclei ([138, 181] and references therein). In addition, it has been suggested that the rate of mutation in mitochondrial DNA is 10-fold higher than that in the nuclear DNA [182]. Taken together, oxidative damage and/or deletion of mitochondrial DNA [183, 184] may lead to reduced expression and function of mitochondrial ETC proteins, contributing to greater ROS production, as shown in alcohol-exposed rats [70]. Lipid peroxides can further alter the cell membrane functions and promote fibrosis through activation of stellate cells with elevated production of collagen and proinflammatory cytokines/chemokines that promote recruitment of neutrophils and activation of hepatic macrophage Kupffer cells. All these events contribute to profound deleterious effects on mitochondrial functions with increased steatosis, apoptosis, and necrosis. In addition, activation of autophagy and consequent removal of damaged mitochondria by ethanol [185] or APAP [147] have been suggested to be an adaptive and protective pathway against hepatic damage by both substances. The removal of damaged mitochondria under elevated nitroxidative stress may also be a mechanism by which cells can get rid of the nitrated proteins to protect themselves. Collectively, these studies and mechanisms support the notion that oxidative/nitrative stress produces mitochondrial dysfunction that can play a major role in stimulating the damage in the various hepatic diseases including AFLD and NAFLD and also extrahepatic tissue injury including brain diseases [182, 186–188]. Figure 1 summarizes the different pathways that nitrative stress might cause mitochondrial dysfunction, contributing to tissue injury including AFLD and NAFLD.
4. Detection Methods of Nitrated Proteins and Challenges
Nitrated proteins should be isolated or enriched before they are subjected to gel electrophoresis separation and protein digestion followed by final MS analysis to identify proteins and possibly Tyr-nitrated peptides. Several methods have been reported to detect Tyr nitration including, but not limited to: (1) two-dimensional gel electrophoresis followed by immunoblot analysis with specific anti-3-NT antibody [189], (2) immunoprecipitation with the specific antibody [47, 190] or immunoaffinity chromatography using immobilized specific anti-3-NT antibody on Sepharose followed by capturing nitrated proteins [191], (3) solution isoelectric focusing, and (4) redox proteomic approach via conversion of nitro-Tyr to amino-Tyr followed by its labeling with biotin or dansyl chloride [192] to isolate nitrated proteins [193, 194]. Recent reviews summarized standard methods for the identification and quantification of proteins nitrated at the Tyr residues through purification of nitrated proteins and in-gel or in-solution trypsin digestion followed by gas chromatography (GC)—or liquid chromatography (LC)—tandem MS analyses [51, 195]. The identification of peptides that originate from nitrated proteins can be performed by using matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) MS [51, 195–197], while identification of specific nitrated Tyr residues can be performed by LC-electrospray ionization (ESI)-MS/MS [198]. Despite the fact that all techniques have merits, there are many short outcomes that can be improved especially when it comes to the number of identified proteins and the ability to capture proteins that is not highly expressed or the capturing of nonspecific proteins. Many excellent reviews have been already reported with more details about nitrated protein isolation and characterization protocols and the advantages and disadvantages of each method ([51, 199, 200] and references within).
Our laboratory recently developed a simple approach to efficiently identify and characterize protein nitration and functional consequences. Although the methods of immunoaffinity chromatography were described before [191, 201], the recovery of the nitrated proteins was low. An attempt to improve the method was undertaken by increasing the incubation time of the sample with the immunoaffinity resin to 19 h and by also improving the quality and specificity of captured proteins by using more stringent washing protocol [202]. However, the incubation time in this protocol seems too long and not very practical in purifying sufficient amounts of nitrated proteins for MS analysis especially when nitrated proteins are expressed in very low quantities.
After preparing tissue homogenates in buffer preequilibrated with nitrogen gas for 1 h to remove dissolved oxygen, the levels of Tyr-nitrated proteins in the target sample compared to control should be determined by immunoblot analysis with anti-3-NT antibody. Once different levels of protein Tyr-nitration between the target and control samples are confirmed, whole homogenates should be further fractionated to prepare lysates from specific subcellular organelles (e.g., mitochondria versus cytosol). Nitrated proteins in mitochondrial extracts from treated and control samples, for instance, are then affinity purified by using the commercially available agarose beads coupled with anti-3-NT antibody, as similar to the method [203]. The affinity-purification steps should be repeated several times to collect sufficient amounts of nitrated proteins. The affinity-purified proteins can be concentrated using mini-spin-column concentrators (Millipore). Small aliquots of purified nitrated samples from control and treated or disease group can be separated on 1D gels and stained with silver to confirm the differences before MS analysis for protein identification. Nitration of some selected proteins identified by MS analysis could be individually confirmed by using immunoprecipitation with the specific antibody against each protein followed by immunoblot with anti-3-NT antibody. In addition, the functional consequence of protein nitration should be determined by measuring the catalytic activity of each target protein [35]. It would be ideal to further determine the nitrated Tyr residue(s) and function by site-directed mutagenesis followed by overexpression and activity measurement. Finally, it is always desirable to determine the causal relationship between Tyr-nitration detected earlier and the full-blown pathological states or tissue injury, usually observed at later time points, following exposure to the toxic substances, as described [35, 140]. Using this method of immunoaffinity purification, we were able to identify more than 30 and 70 nitrated cytosolic and mitochondrial proteins, respectively, in APAP-treated mouse liver. Detection and functional consequences of nitration in five selected mitochondrial proteins (Figure 2 and Table 1) were confirmed via immunoprecipitation followed by immunoblot analysis with anti-3-NT antibody and activity measurements in the absence or presence of an antioxidant, which fully prevented APAP-mediated protein nitration and hepatic damage [35]. Tyr-nitrated peptides of many proteins including SOD2 in APAP-exposed mice could not be identified by the MS analysis. However, nitration of four or five Tyr residues including the critical Tyr34 and inactivation was further confirmed by the MS analysis and activity measurement, respectively, after recombinant SOD2 protein was incubated with a nitrating agent tetranitromethane for 20 min [35].
Figure 2.
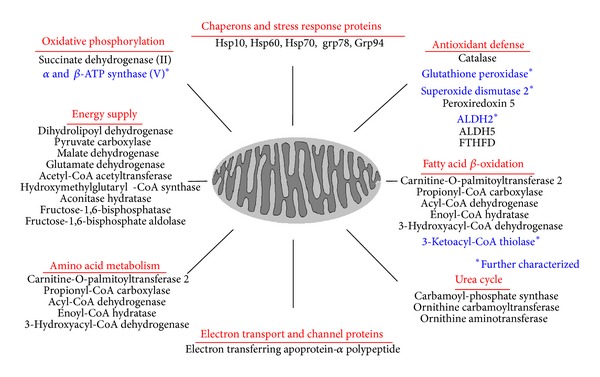
Summary of nitrated mitochondrial proteins in acetaminophen (APAP)-exposed mice. Some recently-identified nitrated mitochondrial proteins in APAP-exposed mouse livers are summarized with respect to the function of each protein identified by mass spectrometry. *Five selected proteins (marked in bold blue) were further characterized for the reversible changes and enzyme activities after APAP exposure in the absence or presence of NAC co-treatment, as described in and adapted from [35].
Based on our recent results, we believe that the immunoaffinity purification followed by MS analysis may represent the best approach among all the aforementioned methods for identifying Tyr nitrated proteins. This approach can be also used to purify sufficient amounts of nitrated proteins for studying the role of protein nitration in various disease models, different organs, and/or different subcellular organelles such as nucleus and endoplasmic reticulum where identification of nitrated proteins can be challenging due to the low yield in isolation of nitrated proteins. However, there are still many challenges with purification and functional characterization of nitrated proteins. Although we and other laboratories reported many nitrated proteins [35, 47, 179, 189–200], and Table 1 it is conceivable that the actual nitrated proteins should be far greater than those we identified in APAP-exposed WT mice. Some nitrated proteins are transiently expressed while the amounts of other nitrated proteins could be decreased possibly through proteolytic degradation upon nitration, as previously mentioned. Therefore, it is difficult to capture and characterize all nitrated proteins analyzed at one time point. By reviewing the literature and based on our own data, we acknowledge that identification of certain nitrated transcription factors, insulin receptors, nuclear receptors, and signaling molecules, for example, could be particularly difficult even after repeated purification steps due to extremely low levels of expression. However, individual characterizations of these factors could be of extreme importance to understand the mechanisms of their well-documented alterations in various disease conditions including AFLD and NAFLD. However, this technical problem could be overcome by immunoprecipitation of the individual protein of interest followed by immunoblot with the specific anti-3-NT antibody and measurement of a functional activity in the absence or presence of a peroxynitrite scavenger. Alternatively, the nitrated proteins of interest could be purified to examine the consequence of their alteration under nitration-inducing conditions. One promising approach could be to employ an in vitro system by using isolated mitochondrial proteins exposed to a nitrating agent or incubated with recombinant NOS. This in vitro method is likely to allow identification of many nitrated mitochondrial proteins. However, the level of peroxynitrite produced by this protocol seems far greater than that generated in in vivo conditions and the in vitro results cannot be used to correlate with samples from animal models or human specimens without extra precaution [46]. Thus there are still many challenges that need to be addressed in future research.
5. Translational Research Using Nitration Proteomics Approaches to Evaluate Beneficial Agents to Prevent or Treat Hepatic and Other Tissue Injuries
Upon understanding the potential mechanism(s) of nitroxidative stress-related liver diseases, the next logical extension would be translational application to identify beneficial agents to either prevent or treat the disease progression. The relative levels of nitrated proteins between controls versus disease samples must be evaluated prior to conducting translational research with some protective agents administered before and after or during disease progression in experimental models. We recently reported this kind of translational application by first characterizing the mechanism of APAP-induced tissue injury followed by efficacy evaluation of an agent against liver damage [35, 40, 78]. Firstly, we demonstrated the importance of CYP2E1 and protein nitration in APAP-mediated hepatotoxicity by comparing the phenotypic changes in WT and CYP2e1(−/−) mice [40]. In this study, we also showed that one of the mechanisms of nitration-mediated tissue injury was suppression (inactivation) of vital proteins such as SOD1 through ubiquitination and subsequent degradation [35, 40, 78]. These results are consistent with the reports by other scientists [204–207]. In continuation, we then immunoaffinity purified and determined identities of many nitrated proteins in both mitochondria and cytosol at earlier time point (e.g., 2 h after APAP treatment). Five critical enzymes SOD2, GPX, ATP synthase, ALDH2, and thiolase were selected for further characterization with respect to their nitration and activity changes at 2 h post-APAP treatment in the absence or presence of NAC. The results revealed that NAC treatment restored APAP-induced changes in transaminase activities, liver histology, and protein nitration to normal levels. NAC also reversed the inactivation of the five enzymes via Tyr nitration, as evidenced by immunoprecipitation with a specific antibody to each protein followed by immunoblot with anti-3-NT antibody. All these results demonstrate the causal roles of protein nitration in promoting full-blown liver damage usually observed at later time points (e.g., 16 or 24 h after APAP treatment) [35]. The combined results of the two studies [35, 40] about the critical role of protein nitration in APAP-induced liver damage and beneficial effect of NAC are consistent with the reports by other laboratories [208, 209]. Thus, our approaches not only allow the characterization of many nitrated mitochondrial proteins but also provide an opportunity for translational research in evaluating the benefits and mechanisms by which NAC protects from APAP-induced liver injury.
This kind of approach can also be applied to future translational studies on various experimental models or human disease specimens where protein nitration seems to play a key role in promoting disease development while the efficacies of beneficial agents can be evaluated, as exemplified in a model of I-R-related acute liver injury. In this study, the benefit of a peroxynitrite scavenger metalloporphyrin MnTMPyP against I-R-related mitochondrial dysfunction and acute hepatotoxicity was investigated [140]. MnTMPyP pretreatment markedly suppressed the I-R-related elevation of serum transaminase levels, histological damage, iNOS expression, and oxidative modifications of key mitochondrial proteins. MnTMPyP treatment also restored the activities of some essential mitochondria enzymes including ALDH2, thiolase, and ATP synthase inhibited under I-R condition. Based on our recent data [35], the activities of these mitochondrial enzymes in the I-R-mediated acute liver injury model could have been inhibited by protein nitration and then restored in the presence of a peroxynitrite scavenger. For this reason, the levels of nitrated proteins should be also evaluated in the future translational studies when testing the benefit of a protective agent.
Mitochondria are important for energy supply, antioxidant defense, apoptosis, and intermediary metabolism (including ammonia, urea, heme, and so forth) and fatty acid oxidation to provide an alternative energy source of ketone bodies when glucose supply is limited [210–212]. In this regard, it would be of particular importance to characterize the nitrated mitochondrial proteins to gain new mechanistic insights in mitochondrial dysfunction and disease progression in acute and chronic liver diseases in the absence or presence of a protective agent. In addition, the proteomics approach of characterizing nitrated proteins can be very useful in the evaluation of many antioxidants other than NAC such as S-adenosylmethionine [213], curcumin [214, 215], and green tea extracts, which decreased nitroxidative stress-mediated development of NAFLD in obese (ob/ob) mice [216]. Furthermore, this approach can be used in identifying new disease biomarkers in various organs and various disease stages in experimental models or clinical specimens. For instance, the levels of nitrated proteins in plasma, platelets, and liver tissue were positively correlated with the severity of hepatic cirrhosis [132] and diet-induced obesity [217]. In fact, an excellent review article for identifying potential biomarkers from its discovery to its application in clinical studies has been recently reported [218]. The initial stage is known as the discovery phase with identification of biomarker candidates. Various systems biology approaches including bioinformatics, genomics, proteomics, and metabolomic analysis should be used simultaneously to identify potential biomarkers in experimental animal models or human specimens with a specific disease of interest. The potential biomarkers with relative long half-lives and stabilities have to be evaluated or verified in a large number of patient samples with certain disease states for clinical validation purpose.
6. Concluding Remarks
Many reports suggest that PTMs, including Tyr nitration, seem to play an important role, at least partially, in various pathophysiological conditions including AFLD and NAFLD. The causal pathologic role of nitrated proteins, however, was questioned due to their relatively rapid turn-over, degradation, and removal of damaged mitochondria by autophagy. It can be argued that the persistence of the insult-mediated protein nitration over extended periods of time and/or consequent imbalance between protein nitration and turnover can alter protein function. In addition, some of the nitrated proteins may disrupt normal signaling pathways (e.g., insulin signaling pathway), contributing to initiation of a cascade of deleterious events. Alternatively, other nitrated proteins may stimulate hepatic macrophage Kupffer cells with elevated cytokines and chemokines, which promote infiltration of neutrophils and inflammatory tissue injury. When these signaling pathways are initiated, even the removal of nitrative stress or nitrated proteins may not stop the ongoing destructive cascades. These events may explain the fact that certain diseases, albeit depending on disease stages, may not be fully treated or reversed even with the FDA-approved therapeutic drugs.
Nitrated proteins may not only play a role in mediating acute liver injury including DILI but also serve as a biomarker for some hepatic diseases. Due to the many forms of protein PTM, it is still challenging to dissect the unique role of nitrated proteins in promoting certain disease states. Further, many studies relied on positive correlation between protein nitration and injury development without detailed mechanistic studies. Thus, to investigate the role of nitrated proteins, a systematic and comprehensive approach should include identification, biochemical characterization, evaluation of overall consequence, and potential prevention or reversal of nitrated proteins and subsequent tissue injuries in the absence or presence of beneficial agents. This systematic approach would serve as a platform for the development of therapeutic agents against AFLD and NAFLD. However, extra caution should be taken into consideration regarding the functional consequence of any nitrated protein since nitration can also activate certain proteins (e.g., microsomal GST-1), as discussed above. It is also important to use sensitive and advanced methods to cover most peptide areas of a target protein to avoid or minimize the potential overlook in identifying nitrated peptides or proteins.
Protein nitration seems to occur in many subcellular compartments including mitochondria. Because of low levels of glutathione and antioxidant enzymes in mitochondria than in cytosol, mitochondrial lipids, DNA, and proteins can be more susceptible to oxidative and nitrative modifications under increased nitrative stress, leading to mitochondrial dysfunction and tissue injury (Figure 1). In fact, many studies dealt with experimental disease models and clinical specimens from patients showed that mitochondrial proteins are abundantly nitrated and/or oxidatively modified, often accompanied with altered functions. Thus, future studies should lead to better understanding of the underlying mechanism(s) of mitochondrial dysfunction through nitration of many additional mitochondrial proteins and their functional implications in disease development and progression. In addition, the occurrence of protein nitration in different liver cells including hepatocytes, Kupffer cells, endothelial cells, and hepatic stellate cells and their functional implications in various liver diseases are poorly understood although these areas should be characterized further. Furthermore, future translational research should also include the development of mitochondria-directed antioxidants or peroxynitrite scavengers to prevent protein nitration and ultimately treat AFLD, NAFLD, and DILI as well as various diseases in other organs.
Acknowledgments
This research was supported by the Intramural Program Fund at the National Institute on Alcohol Abuse and Alcoholism. The authors thank Dr. Klaus Gawrisch for his support.
Abbreviations
- 3-NT:
3-NitroTyr
- AFLD:
Alcoholic fatty liver disease
- AGE:
Advanced glycation end products
- ALDH2:
Aldehyde dehydrogenase-2
- ALS:
Amyotrophic lateral sclerosis
- CPS-1:
Carbamoyl phosphatesynthase-1
- CYP2B6:
Cytochrome P450 2B6
- CYP2E1:
Ethanol-inducible cytochrome P450 2E1
- DILI:
Drug induced liver injury
- ER:
Endoplasmic reticulum
- ETC:
Electron transport chain
- GR:
Glutathione reductase
- GS:
Glutamine synthetase
- GSH:
Glutathione
- GPX:
Glutathione peroxidase
- GST-1:
Glutathione S-transferase-1
- HFD:
High fat diet
- Hsp90:
The 90-kDa heat-shock protein
- I-R:
Ischemia-reperfusion
- IRβ:
Insulin receptor-β
- IRS-1:
Insulin receptor substrate
- iNOS:
Induced nitric oxide synthase
- LC:
Liquid chromatography
- LPS:
Lipopolysaccharide
- MS:
Mass spectrum
- NAC:
N-Acetyl cysteine
- NAFLD:
Nonalcoholic fatty liver disease
- NO:
Nitric oxide
- PTM:
Posttranslational modification
- RNS:
Reactive nitrogen species
- ROS:
Reactive oxygen species
- PP2A:
Protein phosphatase type 2A
- PPAR-α:
Peroxisome proliferator-activated receptor-α
- SOD1:
Cu/ZN superoxide dismutase
- SOD2:
Mn superoxide dismutase
- Thiolase:
3-Ketoacyl-CoA thiolase
- Trp:
Tryptophan
- Tyr:
Tyrosine.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Nathan C. Nitric oxide as a secretory product of mammalian cells. The FASEB Journal. 1992;6(12):3051–3064. [PubMed] [Google Scholar]
- 2.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347(6295):768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 3.Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(22):9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koeck T, Fu X, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. Rapid and selective oxygen-regulated protein tyrosine denitration and nitration in mitochondria. Journal of Biological Chemistry. 2004;279(26):27257–27262. doi: 10.1074/jbc.M401586200. [DOI] [PubMed] [Google Scholar]
- 6.Saxena I, Shekhawat GS. Nitric oxide (NO) in alleviation of heavy metal induced phytotoxicity and its role in protein nitration. Nitric Oxide. 2013;32:13–20. doi: 10.1016/j.niox.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Martínez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radical Biology and Medicine. 2011;51(1):17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Song BJ, Abdelmegeed MA, Henderson LE, et al. Increased nitroxidative stress promotes mitochondrial dysfunction in alcoholic and nonalcoholic fatty liver disease. Oxidative Medicine and Cellular Longevity. 2013;2013:14 pages. doi: 10.1155/2013/781050.781050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calcerrada P, Peluffo G, Radi R. Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications. Current Pharmaceutical Design. 2011;17(35):3905–3932. doi: 10.2174/138161211798357719. [DOI] [PubMed] [Google Scholar]
- 10.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls: the cytotoxic potential of superoxide and nitric oxide. Journal of Biological Chemistry. 1991;266(7):4244–4250. [PubMed] [Google Scholar]
- 11.Lancaster JR., Jr. Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chemical Research in Toxicology. 2006;19(9):1160–1174. doi: 10.1021/tx060061w. [DOI] [PubMed] [Google Scholar]
- 12.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annual Review of Pharmacology and Toxicology. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 13.Purohit V, Gao B, Song B-J. Molecular mechanisms of alcoholic fatty liver. Alcoholism: Clinical and Experimental Research. 2009;33(2):191–205. doi: 10.1111/j.1530-0277.2008.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen JI, Chen X, Nagy LE. Redox signaling and the innate immune system in alcoholic liver disease. Antioxidants and Redox Signaling. 2011;15(2):523–534. doi: 10.1089/ars.2010.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKim SE, Gäbele E, Isayama F, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125(6):1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 16.Venkatraman A, Shiva S, Wigley A, et al. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40(3):565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 17.Purohit V, Rapaka R, Kwon OS, Song BJ. Roles of alcohol and tobacco exposure in the development of hepatocellular carcinoma. Life Sciences. 2013;92(1):3–9. doi: 10.1016/j.lfs.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bansal S, Liu C-P, Sepuri NBV, et al. Mitochondria-targeted cytochrome P450 2E1 induces oxidative damage and augments alcohol-mediated oxidative stress. Journal of Biological Chemistry. 2010;285(32):24609–24619. doi: 10.1074/jbc.M110.121822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. Journal of Clinical Investigation. 2000;105(8):1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuriel T, Hansler A, Gross SS. Protein nitrotryptophan: formation, significance and identification. Journal of Proteomics. 2011;74(11):2300–2312. doi: 10.1016/j.jprot.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kansanen E, Jyrkkänen H-K, Levonen A-L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radical Biology and Medicine. 2012;52(6):973–982. doi: 10.1016/j.freeradbiomed.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 22.Patel A, Liebner F, Netscher T, Mereiter K, Rosenau T. Vitamin E chemistry. Nitration of non-α-tocopherols: products and mechanistic considerations. Journal of Organic Chemistry. 2007;72(17):6504–6512. doi: 10.1021/jo0706832. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura M, Nagayoshi R, Ijiri K, Nakashima-Matsushita N, Takeuchi T, Matsuyama T. Nitration and chlorination of folic acid by peroxynitrite and hypochlorous acid, and the selective binding of 10-nitro-folate to folate receptor β . Biochemical and Biophysical Research Communications. 2002;297(5):1238–1244. doi: 10.1016/s0006-291x(02)02359-8. [DOI] [PubMed] [Google Scholar]
- 24.Berlett BS, Levine RL, Stadtman ER. Carbon dioxide stimulates peroxynitrite-mediated nitration of tyrosine residues and inhibits oxidation of methionine residues of glutamine synthetase: both modifications mimic effects of adenylylation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):2784–2789. doi: 10.1073/pnas.95.6.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knapp LT, Kanterewicz BI, Hayes EL, Klann E. Peroxynitrite-induced tyrosine nitration and inhibition of protein kinase C. Biochemical and Biophysical Research Communications. 2001;286(4):764–770. doi: 10.1006/bbrc.2001.5448. [DOI] [PubMed] [Google Scholar]
- 26.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Archives of Biochemistry and Biophysics. 1998;356(1):1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 27.Souza JM, Daikhin E, Yudkoff M, Raman CS, Ischiropoulos H. Factors determining the selectivity of protein tyrosine nitration. Archives of Biochemistry and Biophysics. 1999;371(2):169–178. doi: 10.1006/abbi.1999.1480. [DOI] [PubMed] [Google Scholar]
- 28.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 29.Bregere C, Rebrin I, Sohal RS. Detection and characterization of in vivo nitration and oxidation of tryptophan residues in proteins. Methods in Enzymology. 2008;441:339–349. doi: 10.1016/S0076-6879(08)01219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamakura F, Kawasaki H. Post-translational modifications of superoxide dismutase. Biochimica et Biophysica Acta. 2010;1804(2):318–325. doi: 10.1016/j.bbapap.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Deeb RS, Nuriel T, Cheung C, et al. Characterization of a cellular denitrase activity that reverses nitration of cyclooxygenase. The American Journal of Physiology—Heart and Circulatory Physiology. 2013;305(5):H687–H698. doi: 10.1152/ajpheart.00876.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenacre S, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radical Research. 2001;34(6):541–581. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- 33.Takakusa H, Mohar I, Kavanagh TJ, Kelly EJ, Kaspera R, Nelson SD. Protein tyrosine nitration of mitochondrial carbamoyl phosphate synthetase 1 and its functional consequences. Biochemical and Biophysical Research Communications. 2012;420(1):54–60. doi: 10.1016/j.bbrc.2012.02.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Görg B, Wettstein M, Metzger S, Schliess F, Häussinger D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology. 2005;41(5):1065–1073. doi: 10.1002/hep.20662. [DOI] [PubMed] [Google Scholar]
- 35.Abdelmegeed MA, Jang S, Banerjee A, Hardwick JP, Song BJ. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radical Biology and Medicine. 2013;60:211–222. doi: 10.1016/j.freeradbiomed.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larosche I, Lettéron P, Berson A, et al. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. Journal of Pharmacology and Experimental Therapeutics. 2010;332(3):886–897. doi: 10.1124/jpet.109.160879. [DOI] [PubMed] [Google Scholar]
- 37.Moon K-H, Hood BL, Kim B-J, et al. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44(5):1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 38.Moon K-H, Upreti VV, Yu L-R, et al. Mechanism of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated mitochondrial dysfunction in rat liver. Proteomics. 2008;8(18):3906–3918. doi: 10.1002/pmic.200800215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin H-L, Myshkin E, Waskell L, Hollenberg PF. Peroxynitrite inactivation of human cytochrome P450s 2B6 and 2E1: heme modification and site-specific nitrotyrosine formation. Chemical Research in Toxicology. 2007;20(11):1612–1622. doi: 10.1021/tx700220e. [DOI] [PubMed] [Google Scholar]
- 40.Abdelmegeed MA, Moon K-H, Chen C, Gonzalez FJ, Song B-J. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochemical Pharmacology. 2010;79(1):57–66. doi: 10.1016/j.bcp.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji Y, Neverova I, van Eyk JE, Bennett BM. Nitration of tyrosine 92 mediates the activation of rat microsomal glutathione S-transferase by peroxynitrite. Journal of Biological Chemistry. 2006;281(4):1986–1991. doi: 10.1074/jbc.M509480200. [DOI] [PubMed] [Google Scholar]
- 42.Savvides SN, Scheiwein M, Böhme CC, et al. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. Journal of Biological Chemistry. 2002;277(4):2779–2784. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 43.Francescutti D, Baldwin J, Lee L, Mutus B. Peroxynitrite modification of glutathione reductase: modeling studies and kinetic evidence suggest the modification of tyrosines at the glutathione disulfide binding site. Protein Engineering. 1996;9(2):189–194. doi: 10.1093/protein/9.2.189. [DOI] [PubMed] [Google Scholar]
- 44.Fujimoto M, Shimizu N, Kunii K, Martyn JAJ, Ueki K, Kaneki M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes. 2005;54(5):1340–1348. doi: 10.2337/diabetes.54.5.1340. [DOI] [PubMed] [Google Scholar]
- 45.Reinehr R, Görg B, Höngen A, Häussinger D. CD95-tyrosine nitration inhibits hyperosmotic and CD95 ligand-induced CD95 activation in rat hepatocytes. Journal of Biological Chemistry. 2004;279(11):10364–10373. doi: 10.1074/jbc.M311997200. [DOI] [PubMed] [Google Scholar]
- 46.Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. The American Journal of Physiology—Heart and Circulatory Physiology. 2004;286(1):H22–H29. doi: 10.1152/ajpheart.00766.2003. [DOI] [PubMed] [Google Scholar]
- 47.Jian-Hong Zhu J-HZ, Zhang X, Roneker CA, et al. Role of copper,zinc-superoxide dismutase in catalyzing nitrotyrosine formation in murine liver. Free Radical Biology and Medicine. 2008;45(5):611–618. doi: 10.1016/j.freeradbiomed.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franco MC, Ye Y, Refakis CA, et al. Nitration of Hsp90 induces cell death. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(12):E1102–E1111. doi: 10.1073/pnas.1215177110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu F, Wilson JX. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovascular Research. 2009;81(1):38–45. doi: 10.1093/cvr/cvn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Souza JM, Peluffo G, Radi R. Protein tyrosine nitration—functional alteration or just a biomarker? Free Radical Biology and Medicine. 2008;45(4):357–366. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Abello N, Kerstjens HAM, Postma DS, Bischoff R. Protein tyrosine nitration: selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. Journal of Proteome Research. 2009;8(7):3222–3238. doi: 10.1021/pr900039c. [DOI] [PubMed] [Google Scholar]
- 52.Grune T, Blasig IE, Sitte N, Roloff B, Haseloff R, Davies KJA. Peroxynitrite increases the degradation of aconitase and other cellular proteins by proteasome. Journal of Biological Chemistry. 1998;273(18):10857–10862. doi: 10.1074/jbc.273.18.10857. [DOI] [PubMed] [Google Scholar]
- 53.Souza JM, Choi I, Chen Q, et al. Proteolytic degradation of tyrosine nitrated proteins. Archives of Biochemistry and Biophysics. 2000;380(2):360–366. doi: 10.1006/abbi.2000.1940. [DOI] [PubMed] [Google Scholar]
- 54.Schildknecht S, Gerding HR, Karreman C, et al. Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. Journal of Neurochemistry. 2013;125(4):491–511. doi: 10.1111/jnc.12226. [DOI] [PubMed] [Google Scholar]
- 55.Chavarria C, Souza JM. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Archives of Biochemistry and Biophysics. 2013;533(1-2):25–32. doi: 10.1016/j.abb.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 56.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79(1):13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 57.Ciechanover A, Schwartz AL. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):2727–2730. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH. Rodent models of alcoholic liver disease: of mice and men. Alcohol. 2012;46(8):715–725. doi: 10.1016/j.alcohol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nature Protocols. 2013;8(3):627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J-F, Greenberg SS, Spitzer JJ. Chronic alcohol administration stimulates nitric oxide formation in the rat liver with or without pretreatment by lipopolysaccharide. Alcoholism: Clinical and Experimental Research. 1995;19(2):387–393. doi: 10.1111/j.1530-0277.1995.tb01520.x. [DOI] [PubMed] [Google Scholar]
- 61.Kessova IG, Ho Y-S, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology. 2003;38(5):1136–1145. doi: 10.1053/jhep.2003.50450. [DOI] [PubMed] [Google Scholar]
- 62.Kim B-J, Hood BL, Aragon RA, et al. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6(4):1250–1260. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Seminars in Liver Disease. 2010;30(4):391–401. doi: 10.1055/s-0030-1267539. [DOI] [PubMed] [Google Scholar]
- 64.Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxidants and Redox Signaling. 2011;15(2):421–424. doi: 10.1089/ars.2011.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.French SW, Benson NC, Sun PS. Centrilobular liver necrosis induced by hypoxia in chronic ethanol-fed rats. Hepatology. 1984;4(5):912–917. doi: 10.1002/hep.1840040521. [DOI] [PubMed] [Google Scholar]
- 66.Tsukamoto H, Xi XP. Incomplete compensation of enhanced hepatic oxygen consumption in rats with alcoholic centrilobular liver necrosis. Hepatology. 1989;9(2):302–306. doi: 10.1002/hep.1840090223. [DOI] [PubMed] [Google Scholar]
- 67.Song B-J, Moon K-H, Olsson NU, Salem N., Jr. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. Journal of Hepatology. 2008;49(2):262–273. doi: 10.1016/j.jhep.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki T, Murakami T, Iino R, et al. F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of ε subunit in response to proton motive force and ADP/ATP balance. Journal of Biological Chemistry. 2003;278(47):46840–46846. doi: 10.1074/jbc.M307165200. [DOI] [PubMed] [Google Scholar]
- 69.Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biological Chemistry. 2002;383(3-4):401–409. doi: 10.1515/BC.2002.044. [DOI] [PubMed] [Google Scholar]
- 70.Venkatraman A, Landar A, Davis AJ, et al. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. Journal of Biological Chemistry. 2004;279(21):22092–22101. doi: 10.1074/jbc.M402245200. [DOI] [PubMed] [Google Scholar]
- 71.Mansouri A, Tarhuni A, Larosche I, et al. MnSOD overexpression prevents liver mitochondrial DNA depletion after an alcohol binge but worsens this effect after prolonged alcohol consumption in mice. Digestive Diseases. 2010;28(6):756–775. doi: 10.1159/000324284. [DOI] [PubMed] [Google Scholar]
- 72.Zelickson BR, Benavides GA, Johnson MS, et al. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochimica et Biophysica Acta. 2011;1807(12):1573–1582. doi: 10.1016/j.bbabio.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38(1):141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 74.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58(2):395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 75.Albano E, Clot P, Morimoto M, Tomasi A, Ingelman-Sundberg M, French SW. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology. 1996;23(1):155–163. doi: 10.1002/hep.510230121. [DOI] [PubMed] [Google Scholar]
- 76.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiological Reviews. 1997;77(2):517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 77.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radical Biology and Medicine. 2008;44(5):723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abdelmegeed MA, Banerjee A, Jang S, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radical Biology and Medicine. 2013;65:1238–1245. doi: 10.1016/j.freeradbiomed.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morgan K, French SW, Morgan TR. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology. 2002;36(1):122–134. doi: 10.1053/jhep.2002.33720. [DOI] [PubMed] [Google Scholar]
- 80.Butura A, Nilsson K, Morgan K, et al. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. Journal of Hepatology. 2009;50(3):572–583. doi: 10.1016/j.jhep.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 81.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radical Biology and Medicine. 2010;49(9):1406–1416. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knockaert L, Fromenty B, Robin M-A. Mechanisms of mitochondrial targeting of cytochrome P450 2E1: physiopathological role in liver injury and obesity. FEBS Journal. 2011;278(22):4252–4260. doi: 10.1111/j.1742-4658.2011.08357.x. [DOI] [PubMed] [Google Scholar]
- 83.Raza H, Prabu SK, Robin M-A, Avadhani NG. Elevated mitochondrial cytochrome P450 2E1 and glutathione S-transferase A4-4 in streptozotocin-induced diabetic rats: tissue-specific variations and roles in oxidative stress. Diabetes. 2004;53(1):185–194. doi: 10.2337/diabetes.53.1.185. [DOI] [PubMed] [Google Scholar]
- 84.Neve EPA, Ingelman-Sundberg M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. Journal of Biological Chemistry. 2000;275(22):17130–17135. doi: 10.1074/jbc.M000957200. [DOI] [PubMed] [Google Scholar]
- 85.Robin M-A, Sauvage I, Grandperret T, Descatoire V, Pessayre D, Fromenty B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Letters. 2005;579(30):6895–6902. doi: 10.1016/j.febslet.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 86.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Laboratory Investigation. 2004;84(3):282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 87.Hollander D. The intestinal permeability barrier. A hypothesis as to its regulation and involvement in Crohn’s disease. Scandinavian Journal of Gastroenterology. 1992;27(9):721–726. doi: 10.3109/00365529209011172. [DOI] [PubMed] [Google Scholar]
- 88.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol- induced liver damage. The American Journal of Gastroenterology. 1999;94(1):200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 89.Keshavarzian A, Choudhary S, Holmes EW, et al. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. Journal of Pharmacology and Experimental Therapeutics. 2001;299(2):442–448. [PubMed] [Google Scholar]
- 90.Keshavarzian A, Fields J. Alcohol: “ice-breaker” yes, “gut barrier-breaker,” maybe. The American Journal of Gastroenterology. 2000;95(5):1124–1125. doi: 10.1111/j.1572-0241.2000.01998.x. [DOI] [PubMed] [Google Scholar]
- 91.Keshavarzian A, Fields J. Alcoholic liver disease: is it an “extraintestinal” complication of alcohol-induced intestinal injury? Journal of Laboratory and Clinical Medicine. 2003;142(5):285–287. doi: 10.1016/S0022-2143(03)00140-9. [DOI] [PubMed] [Google Scholar]
- 92.Robinson GM, Orrego H, Israel Y. Low-molecular-weight polyethylene glycol as a probe of gastrointestinal permeability after alcohol ingestion. Digestive Diseases and Sciences. 1981;26(11):971–977. doi: 10.1007/BF01314757. [DOI] [PubMed] [Google Scholar]
- 93.Tang Y, Banan A, Forsyth CB, et al. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcoholism: Clinical and Experimental Research. 2008;32(2):355–364. doi: 10.1111/j.1530-0277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 94.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. Journal of Pharmacololgy and Experimental Therapeutics. 2003;305(3):880–886. doi: 10.1124/jpet.102.047852. [DOI] [PubMed] [Google Scholar]
- 95.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Preservation of intestinal structural integrity by zinc is independent of metallothionein in alcohol-intoxicated mice. The American Journal of Pathology. 2004;164(6):1959–1966. doi: 10.1016/S0002-9440(10)63756-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. Journal of Hepatology. 1987;4(1):8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 97.Tang Y, Forsyth CB, Farhadi A, et al. Nitric oxide-mediated intestinal injury is required for alcohol-induced gut leakiness and liver damage. Alcoholism: Clinical and Experimental Research. 2009;33(7):1220–1230. doi: 10.1111/j.1530-0277.2009.00946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Forsyth CB, Voigt RM, Shaikh M, et al. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2013;305(2):G185–G195. doi: 10.1152/ajpgi.00354.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Upreti VV, Moon K-H, Yu L-R, et al. Increased oxidative-modifications of cytosolic proteins in 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-exposed rat liver. Proteomics. 2011;11(2):202–211. doi: 10.1002/pmic.201000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smathers RL, Galligan JJ, Stewart BJ, Petersen DR. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chemico-Biological Interactions. 2011;192(1-2):107–112. doi: 10.1016/j.cbi.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51(5):1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 102.Wieckowska A, McCullough AJ, Feldstein AE. Noninvasive diagnosis and monitoring of nonalcoholic steatohepatitis: present and future. Hepatology. 2007;46(2):582–589. doi: 10.1002/hep.21768. [DOI] [PubMed] [Google Scholar]
- 103.Aubert J, Begriche K, Knockaert L, Robin MA, Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clinics and Research in Hepatology and Gastroenterology. 2011;35(10):630–637. doi: 10.1016/j.clinre.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 104.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Seminars in Liver Disease. 2008;28(4):370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 105.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. Journal of Hepatology. 2012;57(4):860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ha S-K, Chae C. Inducible nitric oxide distribution in the fatty liver of a mouse with high fat diet-induced obesity. Experimental Animals. 2010;59(5):595–604. doi: 10.1538/expanim.59.595. [DOI] [PubMed] [Google Scholar]
- 107.Chalasani N, Christopher Gorski J, Asghar MS, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37(3):544–550. doi: 10.1053/jhep.2003.50095. [DOI] [PubMed] [Google Scholar]
- 108.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27(1):128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 109.Fujita K, Nozaki Y, Yoneda M, et al. Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcoholism: Clinical and Experimental Research. 2010;34(supplement 1):S18–S24. doi: 10.1111/j.1530-0277.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 110.Seth RK, Das S, Kumar A, et al. CYP2E1-dependent and leptin-mediated hepatic CD57 expression on CD8+ T cells aid progression of environment-linked nonalcoholic steatohepatitis. Toxicology and Applied Pharmacology. 2014;274(1):42–54. doi: 10.1016/j.taap.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0083481.e83481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang W, Zhao C, Zhou J, Zhen Z, Wang Y, Shen C. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0076538.e76538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Solís Herruzo JA, García Ruiz I, Pérez Carreras M, Muñoz Yagüe MT. Non-alcoholic fatty liver disease. From insulin resistance to mitochondrial dysfunction. Revista Espanola de Enfermedades Digestivas. 2006;98(11):844–874. doi: 10.4321/s1130-01082006001100006. [DOI] [PubMed] [Google Scholar]
- 114.Kathirvel E, Morgan K, French SW, Morgan TR. Overexpression of liver-specific cytochrome P4502E1 impairs hepatic insulin signaling in a transgenic mouse model of nonalcoholic fatty liver disease. European Journal of Gastroenterology and Hepatology. 2009;21(9):973–983. doi: 10.1097/MEG.0b013e328328f461. [DOI] [PubMed] [Google Scholar]
- 115.Kathirvel E, Chen P, Morgan K, French SW, Morgan TR. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. Journal of Gastroenterology and Hepatology. 2010;25(6):1136–1143. doi: 10.1111/j.1440-1746.2009.06196.x. [DOI] [PubMed] [Google Scholar]
- 116.Menghini R, Casagrande V, Menini S, et al. TIMP3 overexpression in macrophages protects from insulin resistance, adipose inflammation, and nonalcoholic fatty liver disease in mice. Diabetes. 2012;61(2):454–462. doi: 10.2337/db11-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chatterjee S, Ganini D, Tokar EJ, et al. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. Journal of Hepatology. 2013;58(4):778–784. doi: 10.1016/j.jhep.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li X, Li H, Lu N, Feng Y, Huang Y, Gao Z. Iron increases liver injury through oxidative/nitrative stress in diabetic rats: involvement of nitrotyrosination of glucokinase. Biochimie. 2012;94(12):2620–7262. doi: 10.1016/j.biochi.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 119.Solís-Muñoz P, Solís-Herruzo JA, Fernández-Moreira D, et al. Melatonin improves mitochondrial respiratory chain activity and liver morphology in ob/ob mice. Journal of Pineal Research. 2011;51(1):113–123. doi: 10.1111/j.1600-079X.2011.00868.x. [DOI] [PubMed] [Google Scholar]
- 120.Charbonneau A, Marette A. Inducible nitric oxide synthase induction underlies lipid-induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes. 2010;59(4):861–871. doi: 10.2337/db09-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miyagi M, Sakaguchi H, Darrow RM, et al. Evidence that light modulates protein nitration in rat retina. Molecular & Cellular Proteomics. 2002;1(4):293–303. doi: 10.1074/mcp.m100034-mcp200. [DOI] [PubMed] [Google Scholar]
- 122.Chen H-JC, Chang C-M, Lin W-P, Cheng D-L, Leong M-I. H2O2/nitrite-induced post-translational modifications of human hemoglobin determined by mass spectrometry: redox regulation of tyrosine nitration and 3-nitrotyrosine reduction by antioxidants. ChemBioChem. 2008;9(2):312–323. doi: 10.1002/cbic.200700541. [DOI] [PubMed] [Google Scholar]
- 123.Chen H-JC, Chang C-M, Chen Y-M. Hemoprotein-mediated reduction of nitrated DNA bases in the presence of reducing agents. Free Radical Biology and Medicine. 2003;34(2):254–268. doi: 10.1016/s0891-5849(02)01246-7. [DOI] [PubMed] [Google Scholar]
- 124.Kamisaki Y, Wada K, Bian K, et al. An activity in rat tissues that modifies nitrotyrosine-containing proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(20):11584–11589. doi: 10.1073/pnas.95.20.11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Irie Y, Saeki M, Kamisaki Y, Martin E, Murad F. Histone H1.2 is a substrate for denitrase, an activity that reduces nitrotyrosine immunoreactivity in proteins. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):5634–5639. doi: 10.1073/pnas.1131756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuo W-N, Kanadia RN, Shanbhag VP, Toro R. Denitration of peroxynitrite-treated proteins by “protein nitratases” from rat brain and heart. Molecular and Cellular Biochemistry. 1999;201(1-2):11–16. doi: 10.1023/a:1007024126947. [DOI] [PubMed] [Google Scholar]
- 127.Görg B, Qvartskhava N, Voss P, Grune T, Häussinger D, Schliess F. Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Letters. 2007;581(1):84–90. doi: 10.1016/j.febslet.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 128.Smallwood HS, Lourette NM, Boschek CB, et al. Identification of a denitrase activity against calmodulin in activated macrophages using high-field liquid chromatography—FTICR mass spectrometry. Biochemistry. 2007;46(37):10498–10505. doi: 10.1021/bi7009713. [DOI] [PubMed] [Google Scholar]
- 129.Kang M, Akbarali HI. Denitration of L-type calcium channel. FEBS Letters. 2008;582(20):3033–3036. doi: 10.1016/j.febslet.2008.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Godoy LC, Muñoz-Pinedo C, Castro L, et al. Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2653–2658. doi: 10.1073/pnas.0809279106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eccleston HB, Andringa KK, Betancourt AM, et al. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxidants and Redox Signaling. 2011;15(2):447–459. doi: 10.1089/ars.2010.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Annie-Jeyachristy S, Geetha A, Surendran R, et al. Level of nitrated proteins in the plasma, platelets and liver of patients with liver cirrhosis. Redox Report. 2009;14(6):259–266. doi: 10.1179/135100009X12525712409616. [DOI] [PubMed] [Google Scholar]
- 133.Spruss A, Kanuri G, Uebel K, Bischoff SC, Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxidants and Redox Signaling. 2011;14(11):2121–2135. doi: 10.1089/ars.2010.3263. [DOI] [PubMed] [Google Scholar]
- 134.Abdelmegeed MA, Yoo S-H, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song B-J. PPARα expression protects male mice from high fat-induced nonalcoholic fatty liver. Journal of Nutrition. 2011;141(4):603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wagnerberger S, Spruss A, Kanuri G, et al. Toll-like receptors 1-9 are elevated in livers with fructose-induced hepatic steatosis. The British Journal of Nutrition. 2012;107(12):1727–1738. doi: 10.1017/S0007114511004983. [DOI] [PubMed] [Google Scholar]
- 136.Locatelli I, Sutti S, Vacchiano M, Bozzola C, Albano E. NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clinical Science. 2013;124(4):279–287. doi: 10.1042/CS20120289. [DOI] [PubMed] [Google Scholar]
- 137.Banerjee A, Abdelmegeed MA, Jang S, Song BJ. Zidovudine (AZT) and hepatic lipid accumulation: implication of inflammation, oxidative and endoplasmic reticulum stress mediators. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0076850.e76850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 139.Yoo SH, Park O, Henderson LE, Abdelmegeed MA, Moon K-H, Song B-J. Lack of PPARα exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicology Letters. 2011;202(1):23–29. doi: 10.1016/j.toxlet.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Moon K-H, Hood BL, Mukhopadhyay P, et al. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135(4):1344–1357. doi: 10.1053/j.gastro.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hinson JA, Pike SL, Pumford NR, Mayeux PR. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chemical Research in Toxicology. 1998;11(6):604–607. doi: 10.1021/tx9800349. [DOI] [PubMed] [Google Scholar]
- 142.Knight TR, Ho Y-S, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. Journal of Pharmacology and Experimental Therapeutics. 2002;303(2):468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 143.Cover C, Mansouri A, Knight TR, et al. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. Journal of Pharmacology and Experimental Therapeutics. 2005;315(2):879–887. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- 144.Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Letters. 1996;385(1-2):63–66. doi: 10.1016/0014-5793(96)00347-x. [DOI] [PubMed] [Google Scholar]
- 145.Berlett BS, Friguet B, Yim MB, Chock PB, Stadtman ER. Peroxynitrite-mediated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylylation: relevance to signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(5):1776–1780. doi: 10.1073/pnas.93.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Agarwal R, MacMillan-Crow LA, Rafferty TM, et al. Acetaminophen-induced hepatotoxicity in mice occurs with inhibition of activity and nitration of mitochondrial manganese superoxide dismutase. Journal of Pharmacology and Experimental Therapeutics. 2011;337(1):110–116. doi: 10.1124/jpet.110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ni H-M, Bockus A, Boggess N, Jaeschke H, Ding W-X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55(1):222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kitam VO, Maksymchuk OV, Chashchyn MO. The possible mechanisms of CYP2E1 interactions with HSP90 and the influence of ethanol on them. BMC Structural Biology. 2012;12, article 33 doi: 10.1186/1472-6807-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dey A, Cederbaum AI. Geldanamycin, an inhibitor of Hsp90 increases cytochrome P450 2E1 mediated toxicity in HepG2 cells through sustained activation of the p38MAPK pathway. Archives of Biochemistry and Biophysics. 2007;461(2):275–286. doi: 10.1016/j.abb.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dey A, Cederbaum AI. Geldanamycin, an inhibitor of Hsp90, potentiates cytochrome P4502E1-mediated toxicity in HepG2 cells. Journal of Pharmacology and Experimental Therapeutics. 2006;317(3):1391–1399. doi: 10.1124/jpet.106.101808. [DOI] [PubMed] [Google Scholar]
- 151.Vadseth C, Souza JM, Thomson L, et al. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. Journal of Biological Chemistry. 2004;279(10):8820–8826. doi: 10.1074/jbc.M306101200. [DOI] [PubMed] [Google Scholar]
- 152.Giasson BI, Duda JE, Murray IVJ, et al. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 153.Ahsan H. 3-Nitrotyrosine: a biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Human Immunology. 2013;74(10):1392–1399. doi: 10.1016/j.humimm.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 154.Ohmori H, Kanayama N. Immunogenicity of an inflammation-associated product, tyrosine nitrated self-proteins. Autoimmunity Reviews. 2005;4(4):224–229. doi: 10.1016/j.autrev.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 155.Birnboim HC, Lemay A-M, Lam DKY, Goldstein R, Webb JR. Cutting edge: MHC class ii-restricted peptides containing the inflammation-associated marker 3-nitrotyrosine evade central tolerance and elicit a robust cell-mediated immune response. Journal of Immunology. 2003;171(2):528–532. doi: 10.4049/jimmunol.171.2.528. [DOI] [PubMed] [Google Scholar]
- 156.Thomson L, Christie J, Vadseth C, et al. Identification of immunoglobulins that recognize 3-nitrotyrosine in patients with acute lung injury after major trauma. The American Journal of Respiratory Cell and Molecular Biology. 2007;36(2):152–157. doi: 10.1165/rcmb.2006-0288SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nagaraj S, Gupta K, Pisarev V, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Medicine. 2007;13(7):828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hardy LL, Wick DA, Webb JR. Conversion of tyrosine to the inflammation-associated analog 3°-nitrotyrosine at either TCR- or MHC-contact positions can profoundly affect recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells. Journal of Immunology. 2008;180(9):5956–5962. doi: 10.4049/jimmunol.180.9.5956. [DOI] [PubMed] [Google Scholar]
- 159.Monroe RK, Halvorsen SW. Environmental toxicants inhibit neuronal Jak tyrosine kinase by mitochondrial disruption. NeuroToxicology. 2009;30(4):589–598. doi: 10.1016/j.neuro.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 160.Kurdi M, Booz GW. JAK redux: a second look at the regulation and role of JAKs in the heart. The American Journal of Physiology—Heart and Circulatory Physiology. 2009;297(5):H1545–H1556. doi: 10.1152/ajpheart.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gole MD, Souza JM, Choi I, et al. Plasma proteins modified by tyrosine nitration in acute respiratory distress syndrome. The American Journal of Physiology—Lung Cellular and Molecular Physiology. 2000;278(5):L961–L967. doi: 10.1152/ajplung.2000.278.5.L961. [DOI] [PubMed] [Google Scholar]
- 162.Mantena SK, Vaughn DP, Jr., Andringa KK, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochemical Journal. 2009;417(1):183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fromenty B, Pessayre D. Impaired mitochondrial function in microvesicular steatosis: effects of drugs, ethanol, hormones and cytokines. Journal of Hepatology. 1997;26(supplement 2):43–53. doi: 10.1016/s0168-8278(97)80496-5. [DOI] [PubMed] [Google Scholar]
- 165.Pessayre D, Mansouri A, Fromenty B. Nonalcoholic steatosis and steatohepatitis V. mitochondrial dysfunction in steatohepatitis. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2002;282(2):G193–G199. doi: 10.1152/ajpgi.00426.2001. [DOI] [PubMed] [Google Scholar]
- 166.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends in Biochemical Sciences. 2010;35(9):505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Song B-J, Moon K-H, Upreti VV, Eddington ND, Lee IJ. Mechanisms of mdma (ecstasy)-induced oxidative stress, mitochondrial dysfunction, and organ damage. Current Pharmaceutical Biotechnology. 2010;11(5):434–443. doi: 10.2174/138920110791591436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wallace KB. Mitochondrial off targets of drug therapy. Trends in Pharmacological Sciences. 2008;29(7):361–366. doi: 10.1016/j.tips.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 169.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radical Biology and Medicine. 1999;27(7-8):891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 170.Giulivi C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radical Biology and Medicine. 2003;34(4):397–408. doi: 10.1016/s0891-5849(02)01298-4. [DOI] [PubMed] [Google Scholar]
- 171.Janssen-Heininger YMW, Mossman BT, Heintz NH, et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radical Biology and Medicine. 2008;45(1):1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ford E, Hughes MN, Wardman P. Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radical Biology and Medicine. 2002;32(12):1314–1323. doi: 10.1016/s0891-5849(02)00850-x. [DOI] [PubMed] [Google Scholar]
- 173.Fernández-Checa JC, Kaplowitz N, García-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Seminars in Liver Disease. 1998;18(4):389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 174.Davies KJA, Shringarpure R. Preferential degradation of oxidized proteins by the 20S proteasome may be inhibited in aging and in inflammatory neuromuscular diseases. Neurology. 2006;66(2, supplement 1):S93–S96. doi: 10.1212/01.wnl.0000192308.43151.63. [DOI] [PubMed] [Google Scholar]
- 175.Pickering AM, Davies KJ. Differential roles of proteasome and immunoproteasome regulators Pa28alphabeta, Pa28gamma and Pa200 in the degradation of oxidized proteins. Archives of Biochemistry and Biophysics. 2012;523(2):181–190. doi: 10.1016/j.abb.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pickering AM, Davies KJ. Degradation of damaged proteins: the main function of the 20S proteasome. Progress in Molecular Biology and Translational Science. 2012;109:227–248. doi: 10.1016/B978-0-12-397863-9.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Caldwell SH, de Freitas LAR, Park SH, et al. Intramitochondrial crystalline inclusions in nonalcoholic steatohepatitis. Hepatology. 2009;49(6):1888–1895. doi: 10.1002/hep.22851. [DOI] [PubMed] [Google Scholar]
- 178.Campello S, Scorrano L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Reports. 2010;11(9):678–684. doi: 10.1038/embor.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Song B-J, Abdelmegeed MA, Yoo S-H, et al. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. Journal of Proteomics. 2011;74(12):2691–2702. doi: 10.1016/j.jprot.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochimica et Biophysica Acta. 2011;1807(6):620–625. doi: 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wan J, Bae M-A, Song B-J. Acetoaminophen-induced accumulation of 8-oxodeoxyguanosine through reduction of Ogg1 DNA repair enzyme in C6 glioma cells. Experimental and Molecular Medicine. 2004;36(1):71–77. doi: 10.1038/emm.2004.10. [DOI] [PubMed] [Google Scholar]
- 182.Monsalve M, Borniquel S, Valle I, Lamas S. Mitochondrial dysfunction in human pathologies. Frontiers in Bioscience. 2007;12(3):1131–1153. doi: 10.2741/2132. [DOI] [PubMed] [Google Scholar]
- 183.Mansouri A, Fromenty B, Berson A, et al. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. Journal of Hepatology. 1997;27(1):96–102. doi: 10.1016/s0168-8278(97)80286-3. [DOI] [PubMed] [Google Scholar]
- 184.Mansouri A, Gaou I, de Kerguenec C, et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology. 1999;117(1):181–190. doi: 10.1016/s0016-5085(99)70566-4. [DOI] [PubMed] [Google Scholar]
- 185.Ding W-X, Li M, Yin X-M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy. 2011;7(2):248–249. doi: 10.4161/auto.7.2.14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34(1):9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 187.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 189.Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine-nitrated proteins. Methods in Molecular Biology. 2004;279:151–165. doi: 10.1385/1-59259-807-2:151. [DOI] [PubMed] [Google Scholar]
- 190.Macmillan-Crow LA, Thompson JA. Immunoprecipitation of nitrotyrosine-containing proteins. Methods in Enzymology. 1999;301:135–145. doi: 10.1016/s0076-6879(99)01076-9. [DOI] [PubMed] [Google Scholar]
- 191.Helman M, Givol D. Isolation of nitrotyrosine-containing peptides by using an insoluble-antibody column. Biochemical Journal. 1971;125(4):971–974. doi: 10.1042/bj1250971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Heijnen HFG, van Donselaar E, Slot JW, et al. Subcellular localization of tyrosine-nitrated proteins is dictated by reactive oxygen species generating enzymes and by proximity to nitric oxide synthase. Free Radical Biology and Medicine. 2006;40(11):1903–1913. doi: 10.1016/j.freeradbiomed.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 193.Dekker F F, Abello N, Wisastra R, Bischoff R. Current Protocols in Protein Science. Chapter 14: Unit 14.3. 2012. Enrichment and detection of tyrosine-nitrated proteins. [DOI] [PubMed] [Google Scholar]
- 194.Zhang Q, Qian W-J, Knyushko TV, et al. A method for selective enrichment and analysis of nitrotyrosine-containing peptides in complex proteome samples. Journal of Proteome Research. 2007;6(6):2257–2268. doi: 10.1021/pr0606934. [DOI] [PubMed] [Google Scholar]
- 195.Tsikas D, Caidahl K. Recent methodological advances in the mass spectrometric analysis of free and protein-associated 3-nitrotyrosine in human plasma. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2005;814(1):1–9. doi: 10.1016/j.jchromb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 196.Anderon L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. Journal of Physiology. 2005;563, part 1:23–60. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhan X, Desiderio DM. The human pituitary nitroproteome: detection of nitrotyrosyl-proteins with two-dimensional western blotting, and amino acid sequence determination with mass spectrometry. Biochemical and Biophysical Research Communications. 2004;325(4):1180–1186. doi: 10.1016/j.bbrc.2004.10.169. [DOI] [PubMed] [Google Scholar]
- 198.Zheng L, Settle M, Brubaker G, et al. Localization of nitration and chlorination sites on apolipoprotein A-I catalysed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. Journal of Biological Chemistry. 2005;280(1):38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]
- 199.Butt YK-C, Lo SC-L. Detecting nitrated proteins by proteomic technologies. Methods in Enzymology. 2008;440:17–31. doi: 10.1016/S0076-6879(07)00802-6. [DOI] [PubMed] [Google Scholar]
- 200.Tsikas D, Schmidt M, Bohmer A, Zoerner AA, Gutzki FM, Jordan J. UPLC-MS/MS measurement of S-nitrosoglutathione (GSNO) in human plasma solves the S-nitrosothiol concentration enigma. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2013;927:147–157. doi: 10.1016/j.jchromb.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 201.Pho DB, Roustan C, Tot ANT, Pradel L-A. Evidence for an essential glutamyl residue in yeast hexokinase. Biochemistry. 1977;16(20):4533–4537. doi: 10.1021/bi00639a031. [DOI] [PubMed] [Google Scholar]
- 202.Zhan X, Desiderio DM. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Analytical Biochemistry. 2006;354(2):279–289. doi: 10.1016/j.ab.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 203.Song B-J, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. Journal of Biological Chemistry. 1989;264(6):3568–3572. [PubMed] [Google Scholar]
- 204.Saito C, Lemasters JJ, Jaeschke H. C-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicology and Applied Pharmacology. 2010;246(1-2):8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicological Sciences. 2002;65(2):166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 206.Li G, Chen JB, Wang C, et al. Curcumin protects against acetaminophen-induced apoptosis in hepatic injury. World Journal of Gastroenterology. 2013;19(42):7440–7446. doi: 10.3748/wjg.v19.i42.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicology and Applied Pharmacology. 2011;251(3):226–233. doi: 10.1016/j.taap.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Carvalho NR, da Rosa EF, da Silva MH, et al. New therapeutic approach: diphenyl diselenide reduces mitochondrial dysfunction in acetaminophen-induced acute liver failure. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0081961.e81961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chemical Research in Toxicology. 2010;23(7):1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Soboll S. Regulation of energy metabolism in liver. Journal of Bioenergetics and Biomembranes. 1995;27(6):571–582. doi: 10.1007/BF02111655. [DOI] [PubMed] [Google Scholar]
- 211.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacology and Therapeutics. 1995;67(1):101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 212.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Reviews Molecular Cell Biology. 2010;11(10):700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 213.Bailey SM, Robinson G, Pinner A, et al. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2006;291(5):G857–G867. doi: 10.1152/ajpgi.00044.2006. [DOI] [PubMed] [Google Scholar]
- 214.Darvesh AS, Carroll RT, Bishayee A, Geldenhuys WJ, van der Schyf CJ. Oxidative stress and Alzheimer’s disease: dietary polyphenols as potential therapeutic agents. Expert Review of Neurotherapeutics. 2010;10(5):729–745. doi: 10.1586/ern.10.42. [DOI] [PubMed] [Google Scholar]
- 215.Ha H-L, Shin H-J, Feitelson MA, Yu D-Y. Oxidative stress and antioxidants in hepatic pathogenesis. World Journal of Gastroenterology. 2010;16(48):6035–6043. doi: 10.3748/wjg.v16.i48.6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chung M-Y, Park HJ, Manautou JE, Koo SI, Bruno RS. Green tea extract protects against nonalcoholic steatohepatitis in ob/ob mice by decreasing oxidative and nitrative stress responses induced by proinflammatory enzymes. Journal of Nutritional Biochemistry. 2012;23(4):361–367. doi: 10.1016/j.jnutbio.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Musso G, Gambino R, de Michieli F, et al. Nitrosative stress predicts the presence and severity of nonalcoholic fatty liver at different stages of the development of insulin resistance and metabolic syndrome: possible role of vitamin a intake. The American Journal of Clinical Nutrition. 2007;86(3):661–671. doi: 10.1093/ajcn/86.3.661. [DOI] [PubMed] [Google Scholar]
- 218.Horvatovich PL, Bischoff R. Current technological challenges in biomarker discovery and validation. European Journal of Mass Spectrometry. 2010;16(1):101–121. doi: 10.1255/ejms.1050. [DOI] [PubMed] [Google Scholar]