

Abstract
Orofacial clefts occur with a frequency of 1 to 2 per 1000 live births. Cleft palate, which accounts for 30% of orofacial clefts, is caused by the failure of the secondary palatal processes—medially directed, oral projections of the paired embryonic maxillary processes—to fuse. Both gene mutations and environmental effects contribute to the complex etiology of this disorder. Although much progress has been made in identifying genes whose mutations are associated with cleft palate, little is known about the mechanisms by which the environment adversely influences gene expression during secondary palate development. An increasing body of evidence, however, implicates epigenetic processes as playing a role in adversely influencing orofacial development. Epigenetics refers to inherited changes in phenotype or gene expression caused by processes other than changes in the underlying DNA sequence. Such processes include, but are not limited to, DNA methylation, microRNA effects, and histone modifications that alter chromatin conformation. In this review, we describe our current understanding of the possible role epigenetics may play during development of the secondary palate. Specifically, we present the salient features of the embryonic palatal methylome and profile the expression of numerous microRNAs that regulate protein-encoding genes crucial to normal orofacial ontogeny.
Keywords: cleft palate, CpG islands, DMR, methylation promoter arrays, methylome, microRNAs, secondary palate
Introduction
Development of the secondary palate serves as a paradigm for embryonic development in general, inasmuch as virtually all of the molecular processes and signaling pathways associated with normal palatal ontogeny are mirrored in the embryogenesis of multiple other systems. The secondary palate (roof of the mouth) separates the oral cavity from the nasal cavity. A defect in the development of the secondary palate causes cleft palate (CP1), which, if left untreated, can lead to feeding and speech impediments, hearing loss (and ear infections), and breathing difficulties (MacLean et al. 2009; Nackashi et al. 2002; Wehby and Cassell 2010). Orofacial clefts occur with a frequency of 1 to 2 per 1000 live births and represent half of all craniofacial anomalies (Stanier and Moore 2004). Besides CP, orofacial clefts also include cleft lip with or without cleft palate (CL/P1), a disorder attributed primarily to defects in the formation of the upper lip. CP constitutes approximately 30% of orofacial clefts and occurs either as an isolated defect (nonsyndromic) or as a component of a syndrome, of which there are more than 200 that include CP as part of their phenotype.2 Some common syndromes that manifest CP include Apert's (Ibrahimi et al. 2005), Stickler's (Johnson et al. 2011), and Treacher Collins (Trainor 2010), whereas CL/P is primarily associated with Van der Woude's (Rizos and Spyropoulos 2004) and Patau's (Patau et al. 1960) syndromes. Population-based studies, as well as our current understanding of the molecular underpinnings of orofacial development, suggest that gene mutations responsible for CL/P and CP are distinct (Fogh-Anderson 1942; Juriloff and Harris 2008; Murray 2002; Spritz 2001).
Both gene mutations and environmental effects underlie the complex etiology of CP (Beaty et al. 2011; Fraser and Calnan 1961; Greene and Pisano 2010). Although some progress has been made in identifying genes that, when mutated, cause CP, very little progress has been made in elucidating how environmental factors contribute to this defect. Environmental influences can adversely affect development of the secondary palate by means of genetic variants or epigenetic alterations. Single-nucleotide polymorphisms (SNPs1) in specific genes can confer an increased risk to adverse developmental outcomes subsequent to exposure to certain environmental hazards. For instance, infants carrying a specific transforming growth factor alpha (TGF 1) polymorphism and whose mothers smoked cigarettes during their pregnancy exhibit a significantly increased risk for CP (Beaty et al. 1997; Hwang et al. 1995; Shaw et al. 1996). Although SNPs are easily identifiable because they involve a change in DNA sequence, epigenetic alterations can trigger gene expression changes without altering DNA sequence. Consequently, epigenetic changes cannot be identified by techniques such as genome-wide association studies that scan DNA for nucleotide changes. Epigenetic mechanisms include, but are not limited to, DNA methylation, microRNA (miRNA1) function, and histone modification. In this review, we focus on the current state of epigenetics as it relates specifically to (murine) secondary palate development and to processes that contribute to CP.
Murine Secondary Palate Development
Morphogenesis and Signaling Pathways
Orofacial development primarily begins with the migration of neural crest cells derived from the neuroectoderm of rhombomeres 1 to 3 (Köntges and Lumsden 1996) into the first two branchial arches, followed by the diversification of neural crest cell fates (Jheon and Schneider 2009; LaBonne and Bronner-Fraser 1999). Growth of the secondary palate depends on the survival of, synthesis of extracellular matrix (ECM1) by, and active proliferation of mesenchymal cells derived from the cranial neural crest and mesodermal cells of the first branchial arch. The palatal processes (shelves) originate as bilateral extensions of the oral aspect of the maxillary processes and eventually fuse to give rise to the secondary palate. In mice, these events occur during gestational days (GDs1) 12 to 14 and during the 7th week of gestation in humans (Figure 1). Elevation of the paired palatal processes and their subsequent adhesion and fusion are highly conserved in mammals. Processes that affect the size of the palatal processes, their reorientation to a position above the tongue, or their fusion to each other can contribute to CP. Developmental processes that underlie these morphogenetic events include cell proliferation, ECM metabolism, epithelial mesenchymal transition (EMT1), apoptosis, cell migration, and the activity of a diverse array of signal transduction pathways. Mutation analyses in mice indicate that many of the genes responsible for development of the secondary palate encode transcription factors, growth and signaling molecules and their receptors, and ECM components (Carinci et al. 2007; Cobourne 2004; Gritli-Linde 2007; Jugessur et al. 2009; Juriloff and Harris 2008; Lidral and Moreno 2005; Murray and Schutte 2004; Murthy and Bhaskar 2009; Rice 2005; Stanier and Moore 2004; Vieira 2008; Yu et al. 2009). Several of the genes that play essential roles in palate development are members of key signal transduction pathways, including the wingless-type MMTV integration site (WNT)-, TGF;#57442;-, platelet derived growth factor (PDGF)-, fibroblast growth factor–, and Sonic hedgehog (SHH)–signaling systems (Greene and Pisano 2010; Gritli-Linde 2007; Jugessur et al. 2009; Murray and Schutte 2004; Yu et al. 2009).
Figure 1.
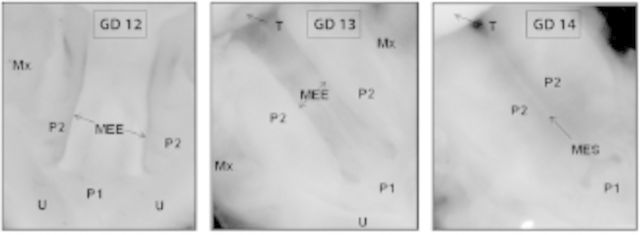
Key stages in the development of the mammalian secondary palate. Panels depict ventral views of the oral cavity with the tongue removed. Formation of the secondary palate occurs between gestational day (GD) 12 and GD 14 in mice (shown) and gestational weeks 6.5 to 10 in humans, in which the developmental steps depicted are similar. During this process, the pair of secondary palatal shelves (P2) grow toward each other, make contact along their respective medial edge epithelia (MEE; GD 12 and GD 13) to form the medial edge seam (MES; GD 14), effectively separating the oral from the nasal cavities. Mx, maxilla; P1, primary palate; P2, secondary palate; T, tongue; U, upper lip.
In this review, we describe results from our published and ongoing studies that examine two types of epigenetic mechanisms—changes in DNA methylation and regulation of gene expression by miRNAs—during development of the murine secondary palate.
Epigenetics and the Secondary Palate
Epigenetics is the study of inherited changes in phenotype or gene expression by means of mechanisms that do not alter the DNA sequence. The most actively investigated epigenetic mechanisms are DNA methylation of C in CG (often referred to as CpG) dinucleotides, the action of miRNAs, and chromatin remodeling involving histone modifications. Aberrant DNA methylation of susceptible genes can result in gene silencing, changes in miRNA levels can affect transcription of target genes, and alterations in histone methylation can affect chromatin conformation (closed and open conformations repress and facilitate gene transcription, respectively). Such epigenetic alterations may adversely affect key biochemical pathways and developmental processes that are temporally programmed and requisite for normal palatal development. These epigenetic mechanisms are not mutually exclusive. For instance, a decrease in miRNA levels could be due to DNA methylation of regulatory regions/CpG islands of miRNA-encoding genes (Chen et al. 2012; Vrba et al. 2010). Methylation-induced gene silencing can occur either by impeding the binding of transcription factors and/or the methyl group serving as a substrate for methyl-CpG binding proteins (Bogdanović and Veenstra 2009). The latter may, in turn, recruit chromatin remodeling proteins that affect various histone modifications to establish a repressive (condensed) chromatin environment that results in dramatically altered levels of gene expression (Deaton and Bird 2011). Gender differences are also observed in clefting phenotypes (Derijcke et al. 1996; James 2000; Tolarova 1990). It is noteworthy that isolated CP occurs twice as frequently in females as in males, and paternal, but not maternal, age is strongly associated with CP (Bille et al. 2005). At least one gene, TBX2, which is located on the X chromosome, has been found to be mutated in patients with CP (Andreou et al. 2007). Although it is not clear if these effects are all sex chromosome–based, it is possible that they could be due to differential methylation of specific autosomal genes in males and females. Interestingly, several sexually dimorphic genes have been observed to occur in liver, adipose tissue, muscle, and brain, which implies that gene expression differences between sexes are prevalent in many somatic tissues (Yang et al. 2006).
Indications that gene methylation might contribute to the etiology of CP first began to appear in the mid-1990s. Studies by Rogers and colleagues (1994) indicated a high frequency of CP when the DNA demethylating agent 5-aza-2’-deoxycytidine was administered in high doses to pregnant female mice on GD 10. A similar observation was made in the rat, although there were significant differences in the developmental responses in both species (Branch et al. 1999). Bulut and colleagues (1999) noticed that congenital abnormalities, including CP, arose when 5-azacytidine (a nucleoside analog of 5-aza-2’-deoxycytidine) was administered to pregnant mice on the 11th and 14th days of pregnancy, identifying a window of susceptibility to the demethylating effects of this compound during early embryogenesis. These studies established a direct link between gene methylation and secondary palate formation. Differences in the rate of secondary palate morphogenesis have been observed in the congenic mouse pair B10/B10.A, which are genetically identical except for a 3-18 cM region on chromosome 17 that harbors the Igf2r imprinted locus (Melnick et al. 1998). The B10.A embryonic palates are slower growing and contain higher levels of Igf2r transcripts than their B10 counterparts. When exposed to equivalent amounts of corticosteroids on embryonic day 12, the B10.A strain manifested a higher frequency of CP compared with B10, and this was attributed to higher Igf2r expression resulting from a relaxation of methylation that permitted a switch from monoallelic to biallelic expression (Melnick et al. 1998). Overexpression of Igf2, an imprinted gene, in mouse embryos is frequently associated with CP (Wise and Pravatcheva 1997). Indeed, overexpression of IGF2 is observed in 10-20% of Beckwith-Wiedemann syndrome patients who present with CP in addition to several other abnormalities (Romanelli et al. 2011). Aberrant secondary palate development is occasionally observed in fragile X syndrome, in which the FMR1 gene is transcriptionally silenced (Bastaki et al. 2004; Hagerman and Hagerman 2002). Silencing of FMR1 occurs through a process termed methylation mosaicism, which involves both the amplification of CGG repeats and methylation of the CG dinucleotides contained therein (Giampietro et al. 1996). Kuriyama and colleagues (2008) were the first to observe changes in the methylation status of CpG islands, as well as in global DNA methylation, in the secondary palates of mice born to mothers exposed to all-trans retinoic acid at doses that induced CP. Using restriction landmark genome sequencing, they identified six genes that were differentially expressed, presumably due to differential gene methylation, between control and all-trans retinoic acid–treated mice. Studies on A/J mice treated with teratogens such as dilantin, corticosteroid, and 6-aminonicotinamaide have identified the Nat2 gene (expressing N-acetyltransferase 2) as a possible culprit in such teratogen-induced orofacial clefting (Erickson 2010). The substrate for Nat2, p-aminobenzoylglutamate, is a breakdown product of folate metabolism (which generates methyl groups for various biochemical reactions), thereby linking DNA methylation to clefting induced by these teratogens. Except for the aforementioned reports, the epigenetic components underlying secondary palate development and their contribution to CP have only recently begun to attract significant research attention. Specifically, no information exists about the methylome of the embryonic secondary palate. Identifying genes whose promoters undergo differential methylation during development of the palate will be a significant step toward identification of genes that are uniquely susceptible to environmental perturbation. Furthermore, little is known regarding the role miRNAs play during development of the palate. We have, therefore, recently begun to define global promoter methylation profiles during development of the murine secondary palate as a prelude to identifying differentially methylated genes and expressed miRNAs in this tissue.
Role of DNA Methylation in Murine Secondary Palate Development
Basic Features and Current Concepts
DNA methylation involves the transfer of a methyl group from S-adenosyl methionine to carbon-5 of a cytosine ring resulting in 5-methylcytosine, often referred to as the fifth base. Cytosine methylation typically occurs in CpG dinucleotide sequences (Jones and Takai 2001) but also occurs in non-CpG residues (CpA, CpC, and CpT) (Barrès et al. 2009; Laurent et al. 2010). More recently, 5-hydroxy methylcytosine (Kriaucionis and Heintz 2009; Tahiliani et al. 2009), a product formed by the oxidation of 5- methylcytosine by ten eleven translocation proteins, has been garnering considerable attention because of its critical role in DNA methylation reprogramming in the mammalian zygote (Iqbal et al. 2011; Wossidlo et al. 2011). Analysis of embryonic stem cells indicates strong enrichment of 5-hydroxy methylcytosine at transcriptional start sites associated with bivalent chromatin of developmentally regulated genes (Pastor et al. 2011).
CpG methylation, however, remains the best-studied and predominant type of DNA methylation (Bird 1980). CpG residues tend to concentrate in genomic regions known as CpG islands, which have a G/C content of 55% or greater and are most often located in the promoter regions of many genes. Methylation of CpG islands in gene promoters correlates with transcriptional silencing (Antequera 2003; Caiafa and Zampieri 2005) by means of either inhibition of transcription factor binding (Geiman and Robertson 2002) or interaction of methyl-CpG binding proteins associated with transcriptional repression (Deaton and Bird 2011; Meehan et al. 1992). Promoter-associated CpG islands of housekeeping genes tend to remain unmethylated, thus facilitating active gene transcription. On the other hand, for tissue-specific genes, these islands can exhibit differential methylation such that the genes are variably expressed in different tissues. Nearly 50% of tissue-specific genes possess CpG islands (Suzuki et al. 2001), and CpG island methylation patterns for each tissue or cell type are unique (Shiota et al. 2002). Indeed, differential methylation of promoter-associated CpG islands has long been thought to be the mechanism of tissue-specific gene regulation. Recent studies, however, that demonstrate that regions outside CpG islands, termed CpG island shores, harbor differentially methylated regions (DMRs1) that dictate tissue specificity have prompted reexamination of this paradigm (Doi et al. 2009; Irizarry et al. 2009). It is also becoming increasingly apparent that CpG residues critically involved in gene regulation have a propensity to be located in CpG-sparse regions (Eckhardt et al. 2006; Nagae et al. 2011; Schmidl et al. 2009; Sun et al. 2011; Yuen et al. 2011).
Tissue-specific DNA methylation patterns are precisely programmed during embryogenesis (Kafri et al. 1992; Monk et al. 1987). Failure to establish correct methylation patterns can lead to embryonic lethality (Li et al. 1992) or can result in developmental craniofacial malformations (Abu-Amero et al. 2008; Gonzales et al. 2005; Kakutani et al. 1996; Matsuda and Yasutomi 1992; Ohgane et al. 2001) including CP (Bliek et al. 2008; Kuriyama et al. 2008; Loenarz et al. 2010). Previous studies in our laboratory, utilizing messenger RNA (mRNA1)–based microarray analysis, have demonstrated the expression of a number of methyltransferases and methyl-CpG binding proteins in GD 9.5 murine embryonic craniofacial tissue (Mukhopadhyay et al. 2006a). This provides evidence that DNA methylation is a dynamic process occurring during secondary palate development.
The Murine Secondary Palate Methylome
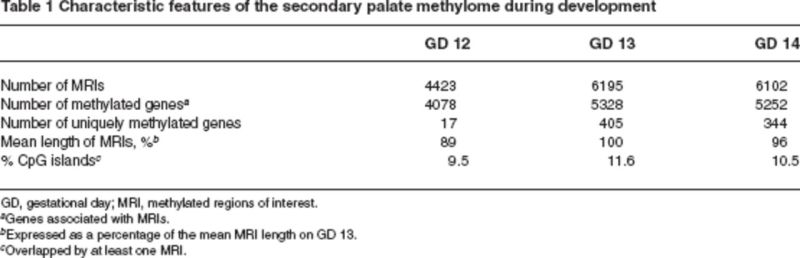
To characterize the methylome in the developing murine secondary palate, we determined global promoter methylation profiles in the secondary palate on GDs 12, 13, and 14 (representing the gestational period during which the palate forms) (Figure 1). A two-step experimental strategy was employed: first, genomic DNA fragments, enriched for methylated CpG residues, were isolated from the secondary palate and converted into hybridization probes; second, these probes were hybridized to murine promoter microarrays (Seelan et al., unpublished data) (Figure 2). Regions containing at least six contiguously methylated probes within a 900–base pair region were identified and designated as methylated regions of interest. Based on this criterion, approximately 5600 genes were found to be methylated, with a majority showing little evidence of differential methylation during palate development (GDs 12–14) (Table 1). Methylated regions of interest were observed to occur predominantly in intragenic regions (i.e., gene bodies that include 3’ untranslated regions, 5’ untranslated regions, exons, and introns) rather than in promoters and upstream regions of genes. This finding is consistent with studies of the human methylome, wherein methylated regions of interest were found predominantly in gene bodies (approximately 50%), whereas only approximately 6% occurred at the 5’ end of genes (Rauch et al. 2009). In contrast with genes methylated in promoters, which are often associated with decreased gene expression, intragenic methylation tends to be positively correlated with transcription (Rauch et al. 2009). Although the significance of intragenic methylation is yet to be elucidated, it has been suggested that it may repress expression of antisense transcripts, inhibit transcription from alternate mRNA start sites, modify transcription efficiency, and/or bring about conformational changes in histones (Ball et al. 2009; Shenker and Flanagan 2012; Suzuki and Bird 2008).
Figure 2.
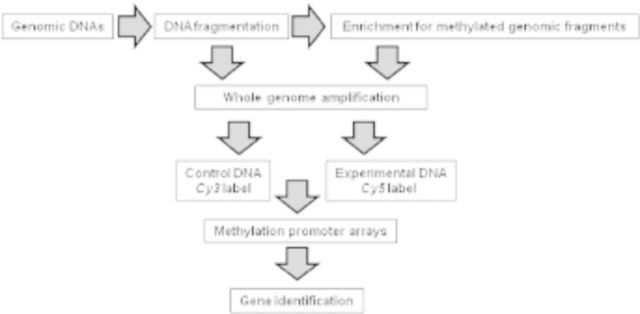
A schematic outline to identify differentially methylated genes using promoter microarrays. Genomic DNAs from secondary palates (gestation days 12–14) were isolated and fragmented by sonication. Sonicated DNA was incubated with MBD2b, a methyl-CpG binding protein (Active Motif, Inc., Carlsbad, CA), for enrichment of methylated genomic fragments. The enriched DNA fragments from all three gestational days were amplified by whole genome amplification. Genomic DNAs not subjected to methylation enrichment were used as controls. Control DNA, which was labeled with Cy3, and experimental DNA, which was labeled with Cy5, were hybridized to NimbleGen 2.1 M mouse promoter microarrays (Roche NimbleGen, Madison, WI). Nine microarrays representing three biologic replicates on each of gestation days 12, 13, and 14 were probed. After hybridization and washing, the microarray chips were scanned and the accrued data were analyzed using biostatistical methods (Seelan et al., unpublished data).
Table 1.
|
When gene ontology analysis using the Database for Annotation, Visualization and Integrated Discovery was performed to identify signaling pathways affected by genes methylated during palate development (Seelan et al., unpublished data), several categories associated with palatogenesis were readily apparent. These include apoptosis, cell adhesion, signaling systems mediated by Wnt, Hedgehog, and Notch, and the biosynthesis of proteoglycans necessary for ECM formation (see “Introduction”). Thus, DNA methylation appears to be critical for several processes that underlie palate morphogenesis. The secondary palate methylome that we have broadly characterized is currently being evaluated by pyrosequencing techniques to determine methylation levels of individual CpG residues localized within the methylated regions of interest of specific genes. These analyses are expected to provide robust evidence of differential methylation, if any, in genes during palate development from GDs 12 to 14. The secondary palate methylome should be a valuable reference for examining the role environment plays in the etiology of CP.
The Sox4 Paradigm
Sox4 is widely expressed in the murine embryo (Dy et al. 2008) and regulates multiple aspects of neural crest cell development (Hong and Saint-Jeannet 2005). Although its role in palatal development or CP is not known, Sox4 has been implicated as a candidate gene for human CL with CP (Juriloff and Harris 2008). Sox4 is a transcription factor belonging to the SoxC family and is functionally associated with the TGFß and Wnt-ß-catenin signaling pathways (Scharer et al. 2009). Significant downregulation of Sox4 gene expression occurs in developing palatal tissue between GD 12 and GD 13 and remains steady thereafter from GD 13 to GD 14 (Mukhopadhyay et al. 2006a). This expression pattern could be due to critical CpG residues in the upstream regulatory region of Sox4 becoming increasingly methylated from GD 12 to GD 13.
To test this premise, the CpG methylation profile of the upstream region of Sox4 was determined by bisulfite sequencing of genomic DNAs obtained from GDs 12, 13, and 14 secondary palatal tissue (Greene and Pisano 2010). Six polymerase chain reaction amplicons from bisulfite-modified DNA were generated corresponding to approximately 2 kb of the Sox4 upstream region (relative to the ATG start codon) (Figure 3) and were sequenced to determine the methylation levels of nearly all CpG residues. Comparison of percentage methylation levels of these amplicons from the three gestational days revealed significant methylation differences in amplicons 3 and 4 only from GD 12 to GD 13, but not between GD 13 and GD 14 (Figure 3). None of the four remaining amplicons displayed any significant change in methylation levels on any GD, and methylation levels of all amplicons on GD 13 were almost identical to their counterparts on GD 14 (Figure 3). Interestingly, the two amplicons that exhibited differential methylation between GD 12 and GD 13 were located in regions outside the CpG island of the Sox4 gene (which spans amplicon 1), consistent with their presence in CpG island shores (Irizarry et al. 2009). The two amplicons, therefore, represent putative Sox4 DMRs. Notably, the CpG island of the Sox4 promoter remained unmethylated on all three gestational days, with methylation levels ranging 0–1.5% (Figure 3). The distal DMR (amplicon 4) contained CpG residues that showed significant increase (>30% change) in methylation levels from GD 12 to GD 13 (data not shown), which correlated with a decrease in Sox4 mRNA levels during these developmental time points; no significant change in methylation levels was observed in these CpG residues between GD 13 and GD 14. On the other hand, the proximal DMR (amplicon 3) contained CpG residues that showed a significant decrease in methylation from GD 12 to GD 13, which was inconsistent with decreasing Sox4 mRNA levels detected in the secondary palate (Mukhopadhyay et al. 2006a). Efforts are now underway to assess the functional relevance of these CpG residues in Sox4 expression in the palate. Thus, detailed CpG profiling of the upstream sequence of Sox4, a candidate gene likely involved in murine palatogenesis, reveals that the period of dramatic change in DNA methylation occurs between GD 12 and GD 13, that CpG islands are predominantly unmethylated and exhibit no differential methylation during palatogenesis, and that DMRs likely associated with palate development are localized in CpG-sparse shore regions. Sox4 could be one of the few genes that exhibit differential methylation during secondary palate development.
Figure 3.
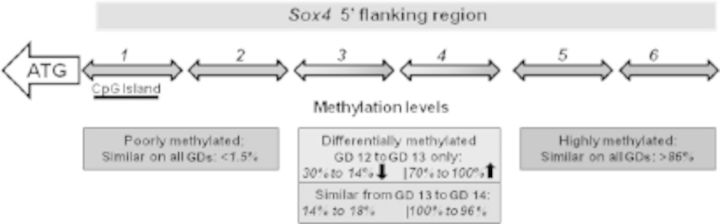
CpG methylation profiling of murine Sox4 upstream region. Six polymerase chain reaction amplicons (numbered 1-6) representing the Sox4 upstream region (relative to the ATG start site) were generated from genomic DNAs isolated from gestational days (GDs) 12 to 14 secondary palate for CpG methylation profiling. CpG profiling was undertaken by amplifying bisulfite-modified DNA (Clark et al. 1994) and sequencing the cloned amplicons. Methylation levels of each CpG residue were inferred by sequencing approximately 10 clones. Amplicons 1 and 2 are poorly methylated (0-1.5%) and amplicons 5 and 6 are heavily methylated (86-96%) on all three GDs. Amplicons 3 and 4 are differentially methylated from GD 12 to GD 13 (upper box) but not from GD 13 to GD 14 (lower box). Note that methylation decreases from GD 12 to GD 13 in amplicon 3 and increases from GD 13 to GD 14 in amplicon 4. Changes in methylation levels in amplicon 4 are consistent with Sox4 messenger RNA expression (see text). No significant changes were observed in methylation levels in amplicons 3 and 4 from GD 13 to GD 14.
MicroRNAs
MicroRNAs and Embryogenesis
MicroRNAs are a unique class of gene silencers discovered nearly two decades ago that belong to the largest family of small, noncoding RNAs in plant and animal systems (Lee et al. 1993; reviewed in Wang et al. 2007; Wightman et al. 1993; Zhang et al. 2007). MicroRNAs, existing either in polycistronic clusters or within the introns of protein-coding genes, are transcribed as long primary miRNAs (Bartel 2009; Chuang and Jones 2007). Processing of miRNAs into double-stranded, stem-loop precursor miRNAs occurs in the nucleus by means of a protein complex comprised of a type 3 ribonuclease (Drosha) (Lee et al. 2004). Following transport to the cytoplasm, these precursor miRNAs are processed further by Dicer, another type 3 ribonuclease, into mature, functional miRNAs (Lund et al. 2004). The mature miRNAs are subsequently incorporated in RNA-induced silencing complexes (Lee et al. 2002), which can silence numerous, downstream gene targets by binding to specific sequences in the 3’ untranslated regions or within the coding regions (Tay et al. 2008). Although such binding usually silences expression of target genes by means of either inhibition of translation or mRNA destabilization (Bartel 2009; Pan and Chegini 2008), recent studies also document miRNAs as translation stimulators (Vasudevan et al. 2007). Experimental findings reveal that complete complementarity of binding between miRNAs and gene targets, although rare in animals, can result in degradation of the target (Meister 2007), whereas partial complementarity triggers translational repression (Kim and Nam 2006). Current estimation indicates that miRNAs account for approximately 1% of predicted genes in higher eukaryotic genomes and that approximately 30% of protein coding genes are regulated by miRNAs (Ross et al. 2007).
Over the past several years, numerous studies have established miRNAs as critical regulators of proliferation, differentiation, and apoptosis—cellular events indispensable for proper embryonic development (reviewed in Conrad et al. 2006; Lee et al. 2006; Mineno et al. 2006). During organogenesis, expression of avian (Darnell et al. 2006), zebrafish (Giraldez et al. 2005), and murine (Takada et al. 2006) miRNAs are tightly regulated in a spatiotemporal manner (Saetrom et al. 2007; Wienholds et al. 2005). Loss of the miRNA-processing enzyme Dicer1 in mice leads to embryolethality (Bernstein et al. 2003; Yang et al. 2005). Inactivation of Dicer in Caenorhabditis elegans and in zebrafish results in heterochronic phenotypes (Grishok et al. 2001) and overall growth arrest during embryogenesis (Wienholds et al. 2003), respectively. The indispensability of miRNAs to stem cell proliferation and differentiation during embryogenesis has been further demonstrated by the failure of differentiation of embryoid body-like structures formed by Dicer1-deficient embryonic stem cells and by the rescue of the differentiation defect in Dicer1-reconstructed clones (Bernstein et al. 2003; Kanellopoulou et al. 2005). The importance of the maintenance and regulation of endogenous miRNA levels during mammalian neurogenesis and neurodevelopment has also been reported (Giraldez et al. 2005; Hosako et al. 2009). Monoallelic deletion of murine dgcr8, a gene encoding an RNA-binding protein required for miRNA biogenesis, resulted in abnormal postnatal development of prefrontal cortical circuitry, which eventually led to cognitive deficits (Schofield et al. 2011). Cardiomyocyte-specific deletion of dgcr8 revealed a fully penetrant cardiomyopathy phenotype and premature lethality, which highlights the vital roles of miRNAs in maintaining cardiac function (Rao et al. 2009). These studies reinforce the centrality of miRNAs in orchestrating normal embryogenesis.
MicroRNA Gene Expression Signature of the Developing Orofacial Tissue
The physiologic roles of miRNAs in orofacial (including secondary palate) development have only recently begun to be examined. In zebrafish, miRNA 140 has been reported to inhibit PDGF receptor #57441;–mediated attraction of cranial neural crest cells to the oral ectoderm (Eberhart et al. 2008), a process necessary for normal morphogenesis of the secondary palate. Lately, an intriguing hint that polymorphisms in human miRNA-coding genes might underlie the etiology of CP was presented by the association of an SNP in the human gene encoding miR-140 with isolated CP in a small cohort from western China (Li et al. 2010). An array of morphogens, growth factors, and signaling mediators, such as members of the TGFß family, bone morphogenetic proteins (BMPs1), Wnts, Shh, epidermal growth factor, mitogen-activated protein kinases, cyclic adenosine monophosphate (cAMP), and retinoic acid, as well as a variety of downstream transcription factors, such as Sma and MAD related family proteins (Smads), cAMP response element-binding protein (CREB), CREB binding protein, E1A binding protein p300, Sloan-Kettering Institute protein (Ski), Ski-related novel protein N, nuclear receptor corepressors, and histone deacetylases, have been reported to interact and coordinate the dynamic process of orofacial ontogenesis (Brunet et al. 1995; Gehris and Greene 1992; Lan et al. 2006; Mendelsohn et al. 1994; Mukhopadhyay et al. 2006a, 2006b; Warner et al. 2002; Weston et al. 1998). Many of the aforementioned signaling mediators and transcriptional regulators have been shown to be targeted by an ever-growing number of miRNAs (Kawasaki and Taira 2003; Kennell et al. 2008; Li and Carthew 2005; Lin et al. 2009; Martello et al. 2007; Zhao et al. 2008). For instance, chondrocyte-specific miRNA 140 was shown to regulate endochondral bone development by BMP signaling, and loss of miR-140 expression resulted in orofacial deformities (Nakamura et al. 2011). In another study, TGFß-mediated EMT, a process critical to normal development of the secondary palate (Sun et al. 1998), was shown to be associated with marked downregulation of miR-205 and all five members of the miRNA 200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) (Gregory et al. 2008). Remarkably, induced expression of the miR-200 family alone was sufficient to prevent TGFß-induced EMT.
Interestingly, miRNAs can also function as effectors of key signaling mediators (such as, TGFßs, BMPs, and Wnts, among others), governing diverse cellular events essential for orofacial development. For example, miR-335, regulated by Wnt signaling, orchestrates mesenchymal stem cell proliferation, migration, and differentiation (Tomé et al. 2011). Regulation of cellular differentiation by BMP signaling, by modulating the expression of members of the miRNA-17-92 cluster, has also been demonstrated (Wang et al. 2010). Furthermore, recent studies reported TGFß-induced myofibroblast differentiation and collagen expression by means of miR-21 and miR-29, respectively (Maurer et al. 2010; Yao et al. 2011).
A recent study defined unique signatures of hundreds of miRNAs expressed within developing orofacial tissue and identified several developmentally regulated clusters of miRNAs that target genes that encode proteins associated with cellular proliferation, adhesion, differentiation, apoptosis, and EMT, all processes central to normal orofacial morphogenesis (Mukhopadhyay et al. 2010). Findings from this study revealed that out of 588 murine miRNA genes examined, 68, 72, and 66 miRNAs were found to be expressed in the developing orofacial region on GD 12, GD 13, and GD 14, respectively. Among these groups, 57 were common to all three days of gestation. A number of differentially expressed miRNAs (such as miR-20a, miR-20b, miR-193, miR-193b, miR-140, members of the let-7 family of miRNAs, miR-152, miR-122, and miR-206, among others) were identified by target analyses as potentially vital modulators of downstream gene targets that encode proteins that execute pivotal functions in orofacial development. Target analyses with multiple miRNA target prediction software programs (e.g., miRDB: http://mirdb.org/miRDB/; PicTar: http://pictar.mdc-berlin.de; TargetScan: www.targetscan.org; and, miRanda: www.microrna.org/microrna/home.do), demonstrated that within the growing orofacial tissue various developmentally regulated miRNAs could potentially target an array of cytoskeletal proteins, growth and differentiation factors, signal transduction modulators and effectors, and transcription factors, all documented to play crucial roles in orofacial ontogeny (Figure 4).
Figure 4.
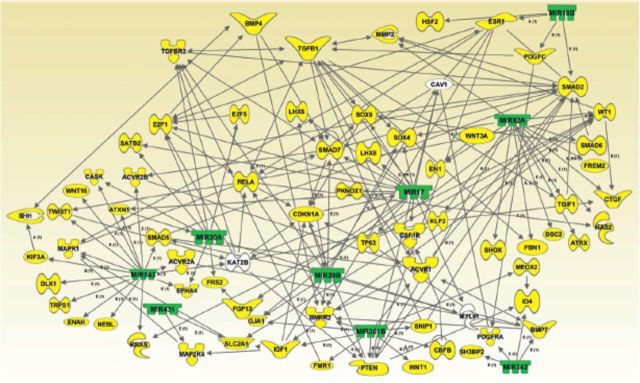
Computational gene interaction predictions: gene network with microRNAs demonstrating diminished expression in developing orofacial tissue on gestational day (GD) 13 versus GD 12. A network with selected genes encoding microRNAs (green) with decreased expression on GD 13 versus GD 12 developing orofacial tisue and their known or predicted target genes (yellow) critical for orofacial ontogenesis was constructed with ingenuity pathway analysis software and the miRDB (http://mirdb.org/miRDB/) database. Solid lines specify direct relationships, whereas dotted lines indicate indirect interactions (Mukhopadhyay et al. 2010).
To investigate cause–consequence relationships between developmentally regulated miRNAs and their putative gene targets, ingenuity pathway analysis of miRNA gene expression profiling data was performed to assign these miRNAs to specific cellular pathways (Mukhopadhyay et al. 2010). One representative gene network is noteworthy for connecting eight differentially expressed miRNAs (miR-20a, miR-20b, miR-22, miR-206, miR-362, miR-106a, miR-152, and miR-140) and associating these with important physiologic events, such as cellular proliferation, growth of epithelial cells, and cell survival (Figure 5). The importance of this network is further emphasized by the presence of several direct and indirect gene targets (of the differentially expressed miRNAs) reported to be expressed and to execute (or likely to execute) crucial roles during orofacial ontogenesis. Examples of such target genes include CREBBP (Warner et al. 2004), CBP/p300 (Warner et al. 2006), Cdkn1a/p21 (Yu et al. 2008), and Bcl2, Cux1, Foxc1, and Mybl1 (Mukhopadhyay et al. 2006a, 2006b). Furthermore, a range of signal transduction pathways, most of which are engaged in normal orofacial development, can be linked directly or indirectly to various miRNAs and genes present in the network (not shown). Examples of such relevant pathways include the TGFß-, BMP-, Wnt-, retinoic acid-, janus tyrosine kinase/signal transducer and activator of transcription, vascular endothelial growth factor, phosphoinositide 3-kinase/protein kinase B, and calcium-signal transduction pathways (not shown). Such gene interaction maps firmly underscore the paramount importance of a range of developmentally regulated miRNAs in orchestrating orofacial ontogenesis by modulating expression of crucial, downstream gene targets.
Figure 5.

Computational gene interaction predictions: selected microRNA gene network in developing orofacial tissue. A network with selected genes encoding microRNAs was constructed with ingenuity pathway analysis software. Several differentially regulated microRNA genes from the study were used to construct a gene association map (Figure 4) for predicting various cellular and molecular events operative within the developing mouse orofacial tissue. Solid lines specify direct relationships, whereas dotted lines indicate indirect interactions (Mukhopadhyay et al. 2010).
Finally, the importance of the miRNAs differentially expressed within the embryonic orofacial region in governing expression of key genes indispensable for orofacial morphogenesis is convincingly highlighted by two miRNA–mRNA gene networks developed with ingenuity pathway analysis and the miRDB online database (Mukhopadhyay et al. 2010). It is noteworthy that these networks that display either diminished (Figure 4) or enhanced (not shown) expression within the developing orofacial tissue directly link potential gene targets reported to be crucial for development of the orofacial region. Examples of some of these gene targets are TGFßs and BMPs (Brunet et al. 1995; Zhang et al. 2002), Meox-2 (Jin and Ding 2006), PDGF (Choi et al. 2009), matrix metalloproteinases (Blavier et al. 2001; de Oliveira Demarchi et al. 2010), Cdkn1a/p21 (Yu et al. 2008), Satb2 (Leoyklang et al. 2007), and Cask (Laverty and Wilson 1998). Collectively, these studies emphasize that differentially expressed miRNAs, which regulate crosstalk among various signaling cascades, orchestrate differentiation and morphogenesis of developing orofacial tissue such as the secondary palate. Because individual miRNAs may target multiple downstream genes, complex, cooperatively interacting regulatory combinations are likely to exist.
Concluding Remarks
Epigenetic studies that pertain to secondary palate development are still in a nascent stage and represent a fertile area for intensive investigation. Identification and functional validation of genes whose expression is driven by differential methylation of promoter regions and determination of the functional role of miRNAs during palatogenesis will provide deep insights into the mechanistic basis by which environmental influences impact secondary palate development. Studies that examine chromatin conformation states at the genome-wide level during secondary palate development are currently lacking. Such studies have the potential to identify “poised” developmental genes (Pastor et al. 2011) that are primed for expression during secondary palate development and therefore likely to play crucial roles in the etiology of CP. A pertinent observation here is that neural tube defects and orofacial clefts, including CP, are often seen when pregnant mothers are exposed to valproic acid (Ornoy 2009), an antiepileptic drug and a known human teratogen. The teratogenicity of this drug is likely due to its ability to inhibit histone deacetylases, whose function is to reduce the acetylation of histones, thereby altering chromatin conformation states that affect gene transcription (Phiel et al. 2001). The implication of this finding is that inappropriate chromatin conformation induced in the early embryo/fetus can impair craniofacial development, including secondary palate/orofacial tissue development. A comparatively larger body of literature exists for CL/P, which occurs much more frequently than CP, but extensive epigenetic studies are likewise lacking for understanding the etiology of this disorder. However, a growing body of evidence clearly suggests that epigenetic processes contribute to the etiology of this order (Bliek et al. 2008; Plamondon et al. 2011; Spritz 2001).
Acknowledgments
This research was supported in part by grants from the National Institute of Health (grants HD053509 and DE018215); the Cleft Palate Foundation; and the Centers of Biomedical Research Excellence program of the National Institute of General Medical Sciences (P20 RR017702). The authors are indebted to Dr. Guy N. Brock and Ms. Savitri N. Appana, School of Public Health, University of Louisville, for their help in the biostatistic analysis of the microarray data.
Biography
Ratnam S. Seelan, PhD, and Partha Mukhopadhyay, PhD, are assistant professors in the Department of Molecular, Cellular and Craniofacial Biology, Birth Defects Center; M. Michele Pisano, PhD, is a professor in the Department of Molecular, Cellular and Craniofacial Biology and director of the Birth Defects Center and NIH Center of Biomedical Research Excellence (COBRE); and Robert M. Greene, PhD, is professor and chair of the Department of Molecular, Cellular Craniofacial Biology and director of the Birth Defects Center and NIH Center of Biomedical Research Excellence (COBRE), all at ULSD, University of Louisville, Kentucky.
Footnotes
Abbreviations that appear ≥3x throughout this article: BMP, bone morphogenetic protein; cAMP, cyclic adenosine monophosphate; CP, cleft palate; CL/P, cleft lip with or without cleft palate; DMR, differentially methylated region; ECM, extracellular matrix; EMT, epithelial mesenchymal transition; GD, gestational day; miRNA, microRNA; mRNA, messenger RNA; PDGF, platelet-derived growth factor; SHH, Sonic hedgehog; SNP, single-nucleotide polymorphism; TGF, transforming growth factor; WNT, Wingless-type MMTV integration site.
Data from Online Mendelian Inheritence in Man, www.ncbi.nlm.nih.gov/omim (this and other websites cited in this article were accessed on November 5, 2012).
References
- Abu-Amero S, Monk D, Frost J, Preece M, Stanier P, Moore GE. 2008. The genetic aetiology of Silver-Russell syndrome. J Med Genet 45:193–199 [DOI] [PubMed] [Google Scholar]
- Andreou AM, Pauws E, Jones MC, Singh MK, Bussen M, Doudney K, Moore GE, Kispert A, Brosens JJ, Stanier P. 2007. TBX22 missense mutations found in patients with X-linked cleft palate affect DNA binding, sumoylation, and transcriptional repression. Am J Hum Genet 81:700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F. 2003. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci 60:1647–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotech 27:361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrès R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A, Zierath JR. 2009. Non-CpG methylation of the PGC-1 #57441; promoter through DNMT3B controls mitochondrial density. Cell Metab 10:189–198 [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136:215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastaki LA, Hegazy F, Al-Heneidi MM, Turki N, Azab AS, Naguib KK. 2004. Fragile X syndrome: A clinico-genetic study of mentally retarded patients in Kuwait. East Mediterr Health J 10:116–124 [PubMed] [Google Scholar]
- Beaty TH, Maestri NE, Hetmanski JB, Wyszynski DF, Vanderkolk CA, Simpson JC, McIntosh I, Smith EA, Zeiger JS, Raymond GV, Panny SR, Tifft CJ, Lewanda AF, Cristion CA, Wulfsberg EA. 1997. Testing for interaction between maternal smoking and TGFA genotype among oral cleft cases born in Maryland 1992–1996. Cleft Palate Craniofac J 34:447–454 [DOI] [PubMed] [Google Scholar]
- Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, Wu T, Patel PJ, Jin SC, Zhang TX, Schwender H, Wu-Chou YH, Chen PK, Chong SS, Cheah F, Yeow V, Ye X, Wang H, Huang S, Jabs EW, Shi B, Wilcox AJ, Lie RT, Jee SH, Christensen K, Doheny KF, Pugh EW, Ling H, Scott AF. 2011. Evidence for gene–environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol 35:469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. 2003. Dicer is essential for mouse development. Nat Genet 35:215–217 [DOI] [PubMed] [Google Scholar]
- Bille C, Skytthe A, Vach W, Knudsen LB, Andersen AM, Murray JC, Christensen K. 2005. Parent's age and the risk of oral clefts. Epidemiology 16:311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP. 1980. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res 8:1499–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. 2001. TGF-#57442;3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell 12:1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliek BJ, Steegers-Theunissen RP, Blok LJ, Santegoets LA, Lindemans J, Oostra BA, Steegers EA, de Klein A. 2008. Genome-wide pathway analysis of folate-responsive genes to unravel the pathogenesis of orofacial clefting in man. Birth Defects Res A Clin Mol Teratol 82:627–635 [DOI] [PubMed] [Google Scholar]
- Bogdanović O, Veenstra GJ. 2009. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 118:549–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch S, Chernoff N, Brownie C, Francis BM. 1999. 5-AZA-2'-deoxycytidine-induced dysmorphogenesis in the rat. Teratog Carcinog Mutagen 19:329–338 [PubMed] [Google Scholar]
- Brunet CL, Sharpe PM, Ferguson MW. 1995. Inhibition of TGF-#57442;3 (but not TGF-#57442;1 or TGF-#57442;2) activity prevents normal mouse embryonic palate fusion. Int J Dev Biol 39:345–355 [PubMed] [Google Scholar]
- Bulut HE, Ozdemir O, Başimoglu-Koca Y, Korkmaz M, Atalay A. 1999. Effects of a DNA demethylating agent—5-azacytidine—on testicular morphology during mouse embryo development. Okajimas Folia Anat Jpn 76:47–53 [DOI] [PubMed] [Google Scholar]
- Caiafa P, Zampieri M. 2005. DNA methylation and chromatin structure: The puzzling CpG islands. J Cell Biochem 94:257–265 [DOI] [PubMed] [Google Scholar]
- Carinci F, Scapoli L, Palmieri A, Zollino I, Pezzetti F. 2007. Human genetic factors in nonsyndromic cleft lip and palate: An update. Int J Pediatr Otorhinolaryngol 71:1509–1519 [DOI] [PubMed] [Google Scholar]
- Chen X, Hu H, Guan X, Xiong G, Wang Y, Wang K, Li J, Xu X, Yang K, Bai Y. 2012. CpG island methylation status of miRNAs in esophageal squamous cell carcinoma. Int J Cancer 130:1607–1613 [DOI] [PubMed] [Google Scholar]
- Choi SJ, Marazita ML, Hart PS, Sulima PP, Field LL, McHenry TG, Govil M, Cooper ME, Letra A, Menezes R, Narayanan S, Mansilla MA, Granjeiro JM, Vieira AR, Lidral AC, Murray JC, Hart TC. 2009. The PDGF-C regulatory region SNP rs28999109 decreases promoter transcriptional activity and is associated with CL/P. Eur J Hum Genet 17:774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Jones PA. 2007. Epigenetics and microRNAs. Pediatr Res 61:24R–29R [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Paul CL, Frommer M. 1994. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 22:2990–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne MT. 2004. The complex genetics of cleft lip and palate. Eur J Orthod 26:7–16 [DOI] [PubMed] [Google Scholar]
- Conrad R, Barrier M, Ford LP. 2006. Role of miRNA and miRNA processing factors in development and disease. Birth Defects Res C Embryo Today 78:107–117 [DOI] [PubMed] [Google Scholar]
- Darnell DK, Kaur S, Stanislaw S, Konieczka JH, Yatskievych TA, Antin PB. 2006. MicroRNA expression during chick embryo development. Dev Dyn 235:3156–3165 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25:1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Demarchi AC, Zambuzzi WF, Paiva KB, da Silva-Valenzuela MG, Nunes FD, de Cássia Sávio Figueira R, Sasahara RM, Demasi MA, Winnischofer SM, Sogayar MC, Granjeiro JM. 2010. Development of secondary palate requires strict regulation of ECM remodeling: Sequential distribution of RECK, MMP-2, MMP-3, and MMP-9. Cell Tissue Res 340:61–69 [DOI] [PubMed] [Google Scholar]
- Derijcke A, Eerens A, Carels C. 1996. The incidence of oral clefts: A review. Br J Oral Maxillofac Surg 34:488–494 [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. 2009. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41:1350–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy P, Penzo-Méndez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. 2008. The three SoxC proteins—Sox4, Sox11 and Sox12—exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res 36:3101–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. 2008. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet 40:290–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38:1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP. 2010. Genes, environment, and orofacial clefting: N-acetyltransferase and folic acid. J Craniofac Surg 21: 1384–1387 [DOI] [PubMed] [Google Scholar]
- Fogh-Anderson P. 1942. Inheritance of Harelip and Cleft Palate. Copenhagen: Arnold Busck [Google Scholar]
- Fraser GR, Calnan JS. 1961. Cleft lip and palate: Seasonal incidence, birth weight, birth rank, sex, site, associated malformations and parental age. A statistical survey. Arch Dis Child 36:420–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehris AL, Greene RM. 1992. Regulation of murine embryonic epithelial cell differentiation by transforming growth factors beta. Differentiation 49:167–173 [DOI] [PubMed] [Google Scholar]
- Geiman TM, Robertson KD. 2002. Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem 87:117–125 [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Haas BR, Lipper E, Gutman A, Zellers NJ, La Trenta GS, Brooks SS, Matalon R, Kaul R, Ding XH, Brown WT. 1996. Fragile X syndrome in two siblings with major congenital malformations. Am J Med Genet 63:396–400 [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. 2005. MicroRNAs regulate brain morphogenesis in zebrafish. Science 308:833–838 [DOI] [PubMed] [Google Scholar]
- Gonzales B, Henning D, So RB, Dixon J, Dixon MJ, Valdez BC. 2005. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum Mol Genet 14:2035–2043 [DOI] [PubMed] [Google Scholar]
- Greene RM, Pisano MM. 2010. Palate morphogenesis: Current understanding and future directions. Birth Defects Res C Embryo Today 90:133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. 2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601 [DOI] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. 2001. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106:23–34 [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. 2007. Molecular control of secondary palate development. Dev Biol 301:309–326 [DOI] [PubMed] [Google Scholar]
- Harrington RJ, Harrington PJ. 2002. Fragile X Syndrome: Diagnosis, Treatment, and Research. Baltimore: Johns Hopkins University Press [Google Scholar]
- Hong CS, Saint-Jeannet JP. 2005. Sox proteins and neural crest development. Semin Cell Dev Biol 16:694–703 [DOI] [PubMed] [Google Scholar]
- Hosako H, Martin GS, Barrier M, Chen YA, Ivanov IV, Mirkes PE. 2009. Gene and microRNA expression in p53-deficient day 8.5 mouse embryos. Birth Defects Res A Clin Mol Teratol 85:546–555 [DOI] [PubMed] [Google Scholar]
- Hwang SJ, Beaty TH, Panny SR, Street NA, Joseph JM, Gordon S, McIntosh I, Francomano CA. 1995. Association study of transforming growth factor alpha (TGF#57441;) TaqI polymorphism and oral clefts: indication of gene-environment interaction in a population-based sample of infants with birth defects. Am J Epidemiol 141:629–636 [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Chiu ES, McCarthy JG, Mohammadi M. 2005. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg 115:264–270 [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, Szabó PE. 2011. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A 108:3642–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41:178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James WH. 2000. Are orofacial clefts a consequence of maternal hormone imbalance? Evidence from sex ratios of sibs of probands. Teratol 62:342–345 [DOI] [PubMed] [Google Scholar]
- Jheon AH, Schneider RA. 2009. The cells that fill the bill: Neural crest and the evolution of craniofacial development. J Dent Res 88:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JZ, Ding J. 2006. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development 133:3341–3347 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Moonis G, Green GE, Carmody R, Burbank HN. 2011. Syndromes of the first and second branchial arches, part 1: Embryology and characteristic defects. AJNR Am J Neuroradiol 32:14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Takai D. 2001. The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070 [DOI] [PubMed] [Google Scholar]
- Jugessur A, Farlie PG, Kilpatrick N. 2009. The genetics of isolated orofacial clefts: From genotypes to subphenotypes. Oral Dis 15:437–453 [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. 2008. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol 82:63–77 [DOI] [PubMed] [Google Scholar]
- Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, Cedar H, Razin A. 1992. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev 6:705–714 [DOI] [PubMed] [Google Scholar]
- Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ. 1996. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci U S A 93:12406–12411 (Erratum: Proc Natl Acad Sci U S A 98:7647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. 2005. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19:489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Taira K. 2003. Functional analysis of microRNAs during the retinoic acid-induced neuronal differentiation of human NT2 cells. Nucleic Acids Res 2003:243–244 [DOI] [PubMed] [Google Scholar]
- Kennell JA, Gerin I, MacDougald OA, Cadigan KM. 2008. The microRNA miR-8 is a conserved negative regulator of Wnt signaling. Proc Natl Acad Sci U S A 105:15417–15422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Nam JW. 2006. Genomics of microRNA. Trends Genet 22:165–173 [DOI] [PubMed] [Google Scholar]
- Köntges G, Lumsden A. 1996. Rhombencephalic neural crest segmentation is preserved throughout craniofacial ontogeny. Development 122:3229–3242 [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. 2009. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama M, Udagawa A, Yoshimoto S, Ichinose M, Sato K, Yamazaki K, Matsuno Y, Shiota K, Mori C. 2008. DNA methylation changes during cleft palate formation induced by retinoic acid in mice. Cleft Palate Craniofac J 45:545–551 [DOI] [PubMed] [Google Scholar]
- LaBonne C, Bronner-Fraser M. 1999. Molecular mechanisms of neural crest formation. Annu Rev Cell Dev Biol 15:81–112 [DOI] [PubMed] [Google Scholar]
- Lan Y, Ryan RC, Zhang Z, Bullard SA, Bush JO, Maltby KM, Lidral AC, Jiang R. 2006. Expression of Wnt9b and activation of canonical Wnt signaling during midfacial morphogenesis in mice. Dev Dyn 235:1448–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, Wei CL. 2010. Dynamic changes in the human methylome during differentiation. Genome Res 20:320–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty HG, Wilson JB. 1998. Murine CASK is disrupted in a sex-linked cleft palate mouse mutant. Genomics 53:29–41 [DOI] [PubMed] [Google Scholar]
- Lee CT, Risom T, Strauss WM. 2006. MicroRNAs in mammalian development. Birth Defects Res C Embryo Today 78:129–139 [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854 [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. 2002. MicroRNA maturation: Stepwise processing and subcellular localization. Embo J 21:4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. 2004. MicroRNA genes are transcribed by RNA polymerase II. Embo J 23:4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V. 2007. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat 28:732–738 [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926 [DOI] [PubMed] [Google Scholar]
- Li L, Meng T, Jia Z, Zhu G, Shi B. 2010. Single nucleotide polymorphism associated with nonsyndromic cleft palate influences the processing of miR-140. Am J Med Genet A 152A:856–862 [DOI] [PubMed] [Google Scholar]
- Li X, Carthew RW. 2005. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell 123:1267–1277 [DOI] [PubMed] [Google Scholar]
- Lidral AC, Moreno LM. 2005. Progress toward discerning the genetics of cleft lip. Curr Opin Pediatr 17:731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EA, Kong L, Bai XH, Luan Y, Liu CJ. 2009. miR-199a, a bone morphogenic protein 2-responsive microRNA, regulates chondrogenesis via direct targeting to Smad1. J Biol Chem 284:11326–11335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, Ratcliffe PJ, Schofield CJ. 2010. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an N#57515;-dimethyl lysine demethylase. Hum Mol Genet 19:217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. 2004. Nuclear export of microRNA precursors. Science 303:95–98 [DOI] [PubMed] [Google Scholar]
- MacLean JE, Hayward P, Fitzgerald DA, Waters K. 2009. Cleft lip and/or palate and breathing during sleep. Sleep Med Rev 13:345–354 [DOI] [PubMed] [Google Scholar]
- Martello G, Zacchigna L, Inui M, Montagner M, Adorno M, Mamidi A, Morsut L, Soligo S, Tran U, Dupont S, Cordenonsi M, Wessely O, Piccolo S. 2007. MicroRNA control of nodal signalling. Nature 449:183–188 [DOI] [PubMed] [Google Scholar]
- Matsuda M, Yasutomi M. 1992. Inhibition of cephalic neural tube closure by 5-azacytidine in neurulating rat embryos in vitro. Anat Embryol (Berl) 185:217–223 [DOI] [PubMed] [Google Scholar]
- Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH, Gay S, Distler O. 2010. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 62:1733–1743 [DOI] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, Bird AP. 1992. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res 20:5085–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G. 2007. miRNAs get an early start on translational silencing. Cell 131:25–28 [DOI] [PubMed] [Google Scholar]
- Melnick M, Chen H, Buckley S, Warburton D, Jaskoll T. 1998. Insulin-like growth factor II receptor, transforming growth factor-#57442;, and Cdk4 expression and the developmental epigenetics of mouse palate morphogenesis and dysmorphogenesis. Dev Dyn 211:11–25 [DOI] [PubMed] [Google Scholar]
- Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M. 1994. Function of the retinoic acid receptors (RARs) during development. (II) Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120:2749–2771 [DOI] [PubMed] [Google Scholar]
- Mineno J, Okamoto S, Ando T, Sato M, Chono H, Izu H, Takayama M, Asada K, Mirochnitchenko O, Inouye M, Kato I. 2006. The expression profile of microRNAs in mouse embryos. Nucleic Acids Res 34:1765–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, Boubelik M, Lehnert S. 1987. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99:371–382 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Brock G, Pihur V, Webb C, Pisano MM, Greene RM. 2010. Developmental microRNA expression profiling of murine embryonic orofacial tissue. Birth Defects Res A Clin Mol Teratol 88:511–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Greene RM, Pisano MM. 2006a. Expression profiling of TGFß superfamily genes in developing orofacial tissue. Birth Defects Res A Clin Mol Teratol 76:528–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Singh S, Greene RM, Pisano MM. 2006b. Molecular fingerprinting of BMP2- and BMP4-treated embryonic maxillary mesenchymal cells. Orthodontics and Craniofacial Res 9:93–110 [DOI] [PubMed] [Google Scholar]
- Murray JC. 2002. Gene/environment causes of cleft lip and/or palate. Clin Genet 61:248–256 [DOI] [PubMed] [Google Scholar]
- Murray JC, Schutte BC. 2004. Cleft palate: Players, pathways, and pursuits. J Clin Invest 113:1676–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy J, Bhaskar L. 2009. Current concepts in genetics of nonsyndromic clefts. Indian J Plast Surg 42:68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackashi JA, Dedlow RE, Dixon-Wood V. 2002. Health care for children with cleft lip and palate: Comprehensive services, and infant feeding. In: Wyszynski DF, ed. Cleft Lip and Palate from Origin to Treatment. New York: Oxford University Press; p 127–158 [Google Scholar]
- Nagae G, Isagawa T, Shiraki N, Fujita T, Yamamoto S, Tsutsumi S, Nonaka A, Yoshiba S, Matsusaka K, Midorikawa Y, Ishikawa S, Soejima H, Fukayama M, Suemori H, Nakatsuji N, Kume S, Aburatani H. 2011. Tissue-specific demethylation in CpG-poor promoters during cellular differentiation. Hum Mol Genet 20:2710–2721 [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. 2011. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate BMP signaling. Mol Cell Biol 31:3019–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgane J, Wakayama T, Kogo Y, Senda S, Hattori N, Tanaka S, Yanagimachi R, Shiota K. 2001. DNA methylation variation in cloned mice. Genesis 30:45–50 [DOI] [PubMed] [Google Scholar]
- Ornoy A. 2009. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod Toxicol 28:1–10 [DOI] [PubMed] [Google Scholar]
- Pan Q, Chegini N. 2008. MicroRNA signature and regulatory functions in the endometrium during normal and disease states. Semin Reprod Med 26:479–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. 2011. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473:394–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP. 1960. Multiple congenital anomaly caused by an extra autosome. Lancet 275:790–793 [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. 2001. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276:36734–36741 [DOI] [PubMed] [Google Scholar]
- Plamondon JA, Harris MJ, Mager DL, Gagnier L, Juriloff DM. 2011. The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res A Clin Mol Teratol 91:716–727 [DOI] [PubMed] [Google Scholar]
- Rao PK, Toyama Y, Chiang HR, Gupta S, Bauer M, Medvid R, Reinhardt F, Liao R, Krieger M, Jaenisch R, Lodish HF, Blelloch R. 2009. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res 105:585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer G. 2009. A human B cell methylome at 100-base pair resolution. Proc Natl Acad U S A 106:671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP. 2005. Craniofacial anomalies: from development to molecular pathogenesis. Curr Mol Med 5:699–722 [DOI] [PubMed] [Google Scholar]
- Rizos M, Spyropoulos MN. 2004. Van der Woude syndrome: A review. Cardinal signs, epidemiology, associated features, differential diagnosis, expressivity, genetic counselling and treatment. Eur J Orthod 26:17–24 [DOI] [PubMed] [Google Scholar]
- Rogers JM, Francis BM, Sulik KK, Alles AJ, Massaro EJ, Zucker RM, Elstein KH, Rosen MB, Chernoff N. 1994. Cell death and cell cycle perturbation in the developmental toxicity of the demethylating agent, 5-aza-2'-deoxycytidine. Teratology 50:332–339 [DOI] [PubMed] [Google Scholar]
- Romanelli V, Meneses HN, Fernández L, Martínez-Glez V, Gracia-Bouthelier R, F Fraga M, Guillén E, Nevado J, Gean E, Martorell L, Marfil VE, García-Miñaur S, Lapunzina P. 2011. Beckwith-Wiedemann syndrome and uniparental disomy 11p: Fine mapping of the recombination breakpoints and evaluation of several techniques. Eur J Hum Genet 19:416–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JS, Carlson JA, Brock G. 2007. miRNA: The new gene silencer. Am J Clin Pathol 128:830–836 [DOI] [PubMed] [Google Scholar]
- Saetrom P, Snøve O, Jr, Rossi JJ. 2007. Epigenetics and microRNAs. Pediatr Res 61:17R–23R [DOI] [PubMed] [Google Scholar]
- Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS. 2009. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res 69:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, Rehli M. 2009. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res 19:1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CM, Hsu R, Barker AJ, Gertz CC, Blelloch R, Ullian EM. 2011. Monoallelic deletion of the microRNA biogenesis gene Dgcr8 produces deficits in the development of excitatory synaptic transmission in the prefrontal cortex. Neural Dev 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Wasserman CR, Lammer EJ, O’Malley CD, Murray JC, Basart AM, Tolarova MM. 1996. Orofacial clefts, parental cigarette smoking, and transforming growth factor-alpha gene variants. Am J Hum Genet 58:551–561 [PMC free article] [PubMed] [Google Scholar]
- Shenker N, Flanagan JM. 2012. Intragenic DNA methylation: Implications of this epigenetic mechanism for cancer research. Br J Cancer 106:248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota K, Kogo Y, Ohgane J, Imamura T, Urano A, Nishino K, Tanaka S, Hattori N. 2002. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes Cells 7:961–969 [DOI] [PubMed] [Google Scholar]
- Spritz RA. 2001. The genetics and epigenetics of orofacial clefts. Curr Opin Pediatr 13:556–560 [DOI] [PubMed] [Google Scholar]
- Stanier P, Moore GE. 2004. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet 13: R73–R81 [DOI] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED. 1998. TGF#57442;3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development 125:95–105 [DOI] [PubMed] [Google Scholar]
- Sun H, Wu J, Wickramasinghe P, Pal S, Gupta R, Bhattacharyya A, Agosto-Perez FJ, Showe LC, Huang TH, Davuluri RV. 2011. Genome-wide mapping of RNA Pol-II promoter usage in mouse tissues by ChIP-seq. Nucleic Acids Res 39:190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet 9:465–476 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Tsunoda T, Sese J, Taira H, Mizushima-Sugano J, Hata H, Ota T, Isogai T, Tanaka T, Nakamura Y, Suyama A, Sakaki Y, Morishita S, Okubo K, Sugano S. 2001. Identification and characterization of the potential promoter regions of 1031 kinds of human genes. Genome Res 11:677–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada S, Berezikov E, Yamashita Y, Lagos-Quintana M, Kloosterman WP, Enomoto M, Hatanaka H, Fujiwara S, Watanabe H, Soda M, Choi YL, Plasterk RH, Cuppen E, Mano H. 2006. Mouse microRNA profiles determined with a new and sensitive cloning method. Nucleic Acids Res 34:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. 2008. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455:1124–1128 [DOI] [PubMed] [Google Scholar]
- Tolarova MM. 1990. Genetics, gene carriers, and environment. In: Bader JD, ed. Risk Assessment in Dentistry. Chapel Hill: University of North Carolina Dental Ecology; p 116–148 [Google Scholar]
- Tomé M, López-Romero P, Albo C, Sepúlveda JC, Fernández-Gutiérrez B, Dopazo A, Bernad A, González MA. 2011. miR-335 orchestrates cell proliferation, migration and differentiation in human mesenchymal stem cells. Cell Death Differ 18:985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA. 2010. Craniofacial birth defects: The role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet A 152A:2984–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Tong Y, Steitz JA. 2007. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 318:1931–1934 [DOI] [PubMed] [Google Scholar]
- Vieira AR. 2008. Unraveling human cleft lip and palate research. J Dent Res 87:119–125 [DOI] [PubMed] [Google Scholar]
- Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S, Stampfer MR, Futscher BW. 2010. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS One 5:e8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, Huang Z, Black BL, Wang F, Martin JF. 2010. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a microRNA-mediated mechanism. Dev Cell 19:903–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Stricker HM, Gou D, Liu L. 2007. MicroRNA: Past and present. Front Biosci 12:2316–2329 [DOI] [PubMed] [Google Scholar]
- Warner DR, Bhattacherjee V, Yin X, Singh S, Mukhopadhyay P, Pisano MM, Greene RM. 2004. Functional interaction between Smad, CREB binding protein, and p68 RNA helicase. Biochem Biophys Res Commun 324:70–76 [DOI] [PubMed] [Google Scholar]
- Warner DR, Pisano MM, Greene RM. 2002. Expression of the nuclear coactivators CBP and p300 in developing craniofacial tissue. In Vitro Cell Dev Biol Anim 38:48–53 [DOI] [PubMed] [Google Scholar]
- Warner DR, Pisano MM, Greene RM. 2006. Functional analysis of CBP/p300 in embryonic orofacial mesenchymal cells. J Cell Biochem 99:1374–1379 [DOI] [PubMed] [Google Scholar]
- Wehby GL, Cassell CH. 2010. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral Dis 16:3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston WM, Potchinsky MB, Lafferty CM, Ma L, Greene RM. 1998. Cross-talk between signaling pathways in murine embryonic palate cells: effect of TGF#57442; and cAMP on EGF-induced DNA synthesis. In Vitro Cell Dev Biol Anim 34:74–78 [DOI] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. 2005. MicroRNA expression in zebrafish embryonic development. Science 309:310–311 [DOI] [PubMed] [Google Scholar]
- Wienholds E, Koudijs MJ, van Eeden FJ, Cuppen E, Plasterk RH. 2003. The microRNA-producing enzyme Dicer1 is essential for zebrafish development. Nat Genet 35:217–218 [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. 1993. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75:855–862 [DOI] [PubMed] [Google Scholar]
- Wise TL, Pravatcheva DD. 1997. Perinatal lethality in H19 enhancers-Igf2 transgenic mice. Mol Reprod Dev 48:194–207 [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2:241. [DOI] [PubMed] [Google Scholar]
- Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. 2005. Dicer is required for embryonic angiogenesis during mouse development. J Biol Chem 280:9330–9335 [DOI] [PubMed] [Google Scholar]
- Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ. 2006. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res 16:995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q, Cao S, Li C, Mengesha A, Kong B, Wei M. 2011. Micro-RNA-21 regulates TGF-#57442;-induced myofibroblast differentiation by targeting PDCD4 in tumor-stroma interaction. Int J Cancer 128:1783–1792 [DOI] [PubMed] [Google Scholar]
- Yu W, Serrano M, Miguel SS, Ruest LB, Svoboda KK. 2009. Cleft lip and palate genetics and application in early embryological development. Indian J Plast Surg 42:S35–S50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Li W, Lu Q, Wang L, Zhang X, Han P, Chen P, Pei Y. 2008. p21 is required for atRA-mediated growth inhibition of MEPM cells, which involves RAR. J Cell Biochem 104:2185–2192 [DOI] [PubMed] [Google Scholar]
- Yuen RK, Neumann SM, Fok AK, Penaherrera MS, McFadden DE, Robinson WP, Kobor MS. 2011. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenetics Chromatin 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wang Q, Pan X. 2007. MicroRNAs and their regulatory roles in animals and plants. J Cell Physiol 210:279–289 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. 2002. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129:4135–4146 [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Sun DG, Wang J, Liu SR, Zhang CY, Zhu MX, Ma X. 2008. Retinoic acid downregulates microRNAs to induce abnormal development of spinal cord in spina bifida rat model. Childs Nerv Syst 24:485–492 [DOI] [PubMed] [Google Scholar]