

Abstract
The centrosome is essential for cytotoxic T lymphocyte (CTL) function, contacting the plasma membrane and directing cytotoxic granules for secretion at the immunological synapse. Centrosome docking at the plasma membrane also occurs during cilia formation. The primary cilium, formed in nonhematopoietic cells, is essential for vertebrate Hedgehog (Hh) signaling. Lymphocytes do not form primary cilia, but we found and describe here that Hh signaling played an important role in CTL killing. T cell receptor activation, which “prearms” CTLs with cytotoxic granules, also initiated Hh signaling. Hh pathway activation occurred intracellularly and triggered Rac1 synthesis. These events “prearmed” CTLs for action by promoting the actin remodeling required for centrosome polarization and granule release. Thus, Hh signaling plays a role in CTL function, and the immunological synapse may represent a modified cilium.
Cytotoxic T lymphocytes (CTLs) recognize tumor and virally infected cells via their T cell receptor (TCR). Recognition triggers a cascade of intracellular signaling that leads to the formation of the immunological synapse and polarization of the centrosome to contact the plasma membrane (1) at the central supramolecular activation complex (cSMAC) (2) where TCRs cluster within the synapse (1, 3). Cytotoxic granules move toward the docked centrosome and deliver their contents precisely at the point of TCR-mediated recognition, which focuses secretion toward the target cell to be destroyed. Docking of the centrosome also occurs during cilia formation, when the mother centriole contacts the plasma membrane, forming the basal body from which the cilium extends. Although lymphocytes are one of very few cell types that do not form primary cilia (4), morphological and functional similarities can be drawn between the immunological synapse and cilia. Endocytosis and exocytosis are focused at the point of centrosome docking in both cases (5); ciliary intraflagellar transport (IFT) proteins are found in T cells (6), and both structures form important signaling platforms (1, 2, 7, 8).
In Hedgehog (Hh) signaling, binding of exogenous Sonic, Indian, or Desert Hh (Shh, Ihh, or Dhh) to the transmembrane receptor Patched (Ptch) regulates translocation of Smoothened (Smo) to primary cilia (9, 10). The ciliary localization of Smo is required to initiate transduction of Gli-mediated transcription of target genes, including Gli1, which serves as a reporter of Hh signaling (7, 8). We asked whether proteins of the Hh pathway are expressed in T cells and whether TCR activation triggered Hh signaling. Naïve CD8 T cells and CTLs derived after 4 to 5 days of in vitro TCR activation were isolated from OT-I TCR transgenic mice. TCR cross-linking was triggered in both naïve CD8 T cell and CTL populations using plate-bound antibody against CD3 (Fig. 1A). In naïve CD8 T cells, Gli1 mRNA was not detected, but expression was induced upon TCR cross-linking, peaking at 12 hours. Controls lacking antibody against CD3 showed no Gli1 expression. CTLs had low Gli1 mRNA levels that increased 180-fold after TCR ligation (Fig. 1A). In addition, the genes encoding Ptch1 and 2 receptors, the signal transducer Smo, and the ligand Ihh were all expressed in both naïve CD8 T cells and CTLs (fig. S1A), and protein expression of Ptch, Gli1, and Ihh increased after TCR activation of naïve CD8 cells (Fig. 1B) and CTLs (fig. S1B). Neither Shh nor Dhh were detected in CD8 T cells before or after 24-hour TCR activation or in EL4 and P815 target cell lines (fig. S1, C and D). When TCR signaling was severely impaired by deletion of the upstream tyrosine kinase Lck (11), induction of Gli1 was also diminished in naïve CD8 T cells (Fig. 1, C and D). Thus, CD8 T cells express Hh pathway components and require TCR signaling to trigger Hh signaling.
Fig. 1. TCR activation triggers Hh signaling and expression of Hh components in CD8 T cells.
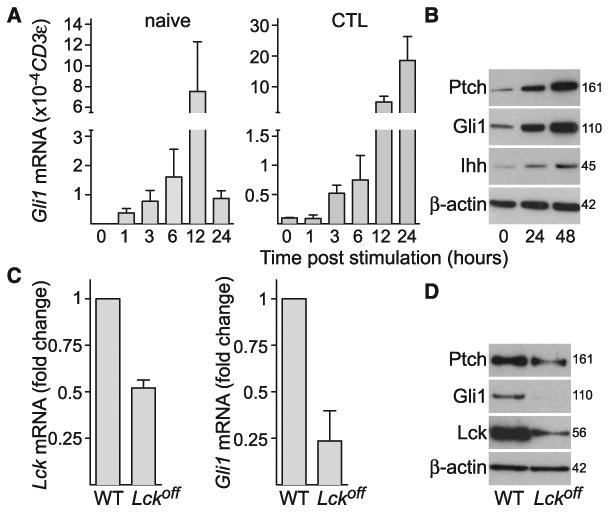
(A) Quantitative polymerase chain reaction (qPCR) showing mRNA levels of Gli1 in naïve CD8 T cells (left) and CTLs (right) at times shown after TCR cross-linking with plate-bound antibody against CD3 relative to CD3ε as a reference gene; n = 3 (naïve) or 2 (CTLs); data are means ± SD. Similar results were obtained using the gene for TATA box–binding protein (Tbp) as a reference gene (not shown). Cells plated without antibody against CD3 showed no Gli1 induction over 12 hours. (B) Immunoblot analysis of protein expression of Ptch, Gli1, Ihh, and actin at 0, 24, and 48 hours after TCR stimulation in naïve CD8 T cells; n = 3. Molecular masses are shown in kilodaltons. Similar results were also obtained from CD8 T cells derived from C57BL/6 and BALB/c mice (not shown). (C and D) Naïve CD8 T cells were purified from spleens of wild-type (WT) or Lckoff mice and stimulated for 12 hours with plate-bound antibody against CD3. (C) Graphs showing mRNA levels of Lck (left) and Gli1 (right) in Lckoff CD8 T cells relative to WT control; n = 2, data are means ± SD. (D) Immunoblot analysis of Ptch, Gli1, Lck, and actin in Lckoff and WT control CD8 T cells after 12 hours of TCR stimulation; n = 3. Molecular masses are shown in kilodaltons.
Because only T cells were present in these assays, CD8 T cells must have both synthesized and responded to Hh proteins to activate this signaling pathway. This is unusual, as Hh signaling is usually paracrine, with one cell type producing Hh and another responding to this cue. We noted that Ihh was detected as a 45-kD protein, which indicated that it was not fully processed into the secreted form (12, 13). This raised the possibility that Ihh might bind Ptch intracellularly. We used recombinant Ihh protein to ask whether CTLs responded to exogenous Ihh. Although cross-linking of TCRs triggered Gli1 expression, stimulating CTLs with extracellular Ihh alone did not. Furthermore, exogenous Ihh did not enhance Gli1 expression in response to TCR activation (fig. S1E). Thus, Ihh encounters its receptor, Ptch, intracellularly in CTLs.
We next asked where Ptch, Ihh, and Smo localize in CTLs using antibodies to detect endogenous Ihh and endogenous Smo and Ptch1 fused with enhanced yellow fluorescent protein (Ptch1-EYFP) to localize the receptor (Fig. 2). In CTLs, Ptch1 was found on intracellular vesicles (Fig. 2A). Ihh colocalized with Ptch1 on a subset of these vesicles, which polarized toward the immunological synapse upon recognition of a target cell (Fig. 2, B and C). This is consistent with the idea that Ihh-mediated signaling via Ptch1 takes place intracellularly, and we confirmed the interaction between Ihh and Ptch1 on intracellular vesicles using a proximity ligation assay (fig. S2A). Ihh was also seen near sites of actin accumulation (fig. S2, B and C), which raised the possibility that Ihh may also influence actin, as shown for other Hh components (14, 15). Endogenous Smo was also associated with intracellular vesicles in CTLs (Fig. 2D) that are predominantly Lamp1-positive (fig. S2D) and distinct from the Ihh-positive compartment (fig. S2E). Live imaging of Smo fused with enhanced green fluorescent protein (Smo-EGFP) expressed in CTLs showed that, upon recognition of a target cell via TCR, the intracellular pool of Smo polarized to the immunological synapse (Fig. 2E and movie S1), analogous to the Hh-triggered translocation of Smo into the cilium (9, 10, 16, 17).
Fig. 2. Ihh, Ptch1, and Smo are localized on intracellular vesicles that polarize toward the immunological synapse.
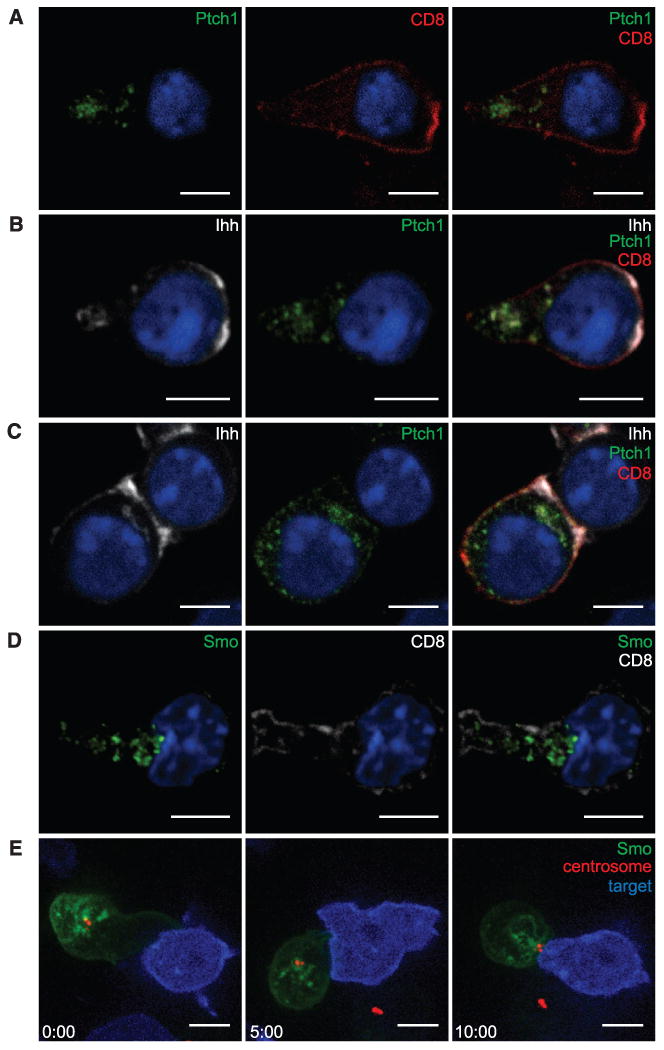
OT-I CTLs transduced with Ptch1-EYFP and labeled with antibodies to EYFP (green), CD8 (red), and Ihh (white). (A and B) show OT-1 CTLs alone and (C) conjugated with EL4 target cells. (D) Endogenous Smo (green) and CD8 (white) in OT-I CTLs. Single x-y confocal sections are shown. Nuclei are stained with Hoechst (blue). (A: n > 75;B and C: n > 35;D: n > 85; two to four independent experiments each). (E) Individual frames of a movie (movie S1) showing OT-I CTLs nucleofected with Smo-EGFP and PACT-RFP (centrosome marker, red), forming a synapse with EL4 target cells (blue). Time after initial contact is shown in minutes (n = 33). Scale bars: 5 μm.
To determine whether TCR-triggered Hh signaling affects CTL-mediated killing, we made use of a genetic model in which Smo is conditionally deleted. Although Hh signaling is required for T cell development (18–21), T cells from mice in which exon 1 of Smo is inducibly deleted in adult hemopoietic cells under Mx1-Cre control develop normally (22, 23). CTLs generated from Smo-deleted mice showed greatly reduced (1.5%) levels of Smo mRNA relative to controls and greatly reduced Smo protein levels (Fig. 3, A and B). Gli1 mRNA up-regulation in response to TCR ligation was also reduced: CTLs from control mice showed a threefold increase in levels of Gli1 mRNA, whereas CTLs from Smo-deleted mice gave only a 1.5-fold increase (Fig. 3A). TCR signaling was not altered in Smo-deleted CTLs (Fig. 3C and fig. S3). However, when we assessed Smo-deleted CTLs for their cytotoxic effector function, we found reduced levels of target cell killing compared with control CTLs (Fig. 3D), which suggested that Hh signaling contributes to CTL killing. Because centrosome polarization to the plasma membrane is a key step in CTL-mediated killing, we asked whether Hh signaling affected centrosome docking at the cSMAC during conjugate formation between CTLs and target cells. Smo-deleted CTLs showed a ∼50% reduction in centrosomal docking at the cSMAC (Fig. 3E), which was consistent with the reduction in Smo protein levels and CTL killing.
Fig. 3. Hh signaling is required for CTL killing and centrosome polarization to the immunological synapse.
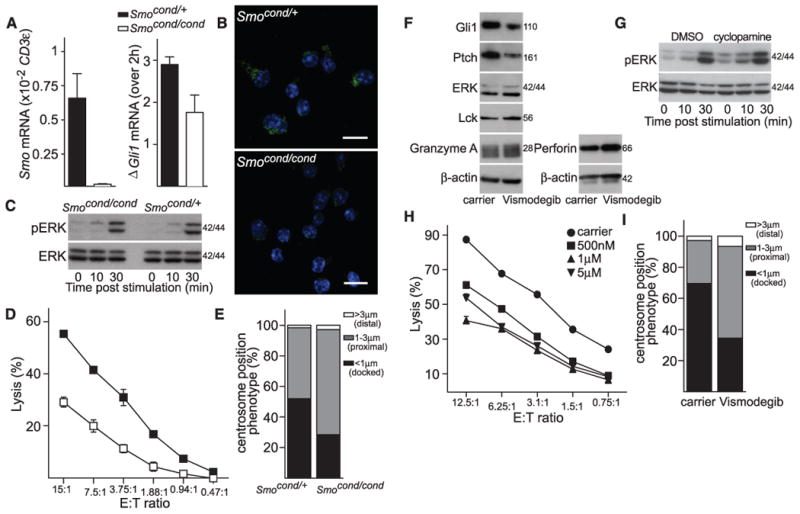
(A) qPCR analysis of Smo mRNA expression in Mx1-Cre Smocond/cond and control Mx1-Cre Smocond/+ CTLs relative to CD3ε as a reference gene (left) (n = 4, data are means ± SD); or Gli1 mRNA levels 2 hours after TCR activation with plate-bound antibody against CD3 for Mx1-Cre Smocond/+ and Mx1-Cre Smocond/cond CTLs relative to unstimulated (right); n = 3, data are means ± SD. (B) Representative staining of endogenous Smo (green) expression in Mx1-Cre Smocond/+ and Mx1-Cre Smocond/cond CTLs. Nuclei stained with Hoechst (blue); n > 100. Scale bars: 5 μm. (C) Mx1-Cre Smocond/+ and Mx1-Cre Smocond/cond CTLs were stimulated with plate-bound CD3-specific antibody for times indicated and blotted for protein expression of phosphorylated ERK (pERK) and ERK; n = 2. Molecular masses are shown in kilodaltons. (D) Percentage lysis of P815 target cells by Mx1-Cre Smocond/+ and Mx1-Cre Smocond/cond CTLs at effector:target (E:T) ratios shown (n = 6). (E) Centrosome position relative to clustered Lck was classified as <1 μm (docked), 1 to 3 μm (proximal) or >3 μm (distal) as percentage of conjugates of Mx1-Cre Smocond/+ (n = 129) and Mx1-Cre Smocond/cond (n = 121) with P815 targets as depicted in fig. S4 (n = 3), data are means ± SD. (F) Immunoblots of cell lysates from OT-I CTLs treated with 5 μM vismodegib for 36 hours, probed with antibodies against Gli1, Ptch, ERK, Lck, granzyme A, perforin, and β-actin; n = 2. (G) OT-I CTLs treated with 10 μM cyclopamine for 24 hours, stimulated with plate-bound CD3-specific antibody for times indicated and blotted for protein expression of pERK and ERK; n = 2. Molecular masses are shown in kilodaltons. (H) Percentage lysis of EL4 target cells by OT-I CTLs treated with vismodegib at concentrations stated; n = 5. The x axis shows varying CTL effector:target (E:T) ratios. (I) OT-I CTLs treated with vismodegib (5 μM) were labeled with antibodies against Lck, γ-tubulin, and CD8. Quantification of centrosome polarization after treatment is shown (n > 60).
We also confirmed that Hh signaling is important for centrosome polarization and CTL-mediated killing using three separate inhibitors: cyclopamine and vismodegib (GDC-0449), both of which inhibit Smo (24, 25), and GANT61, which targets Gli transcription factors (26). The inhibitors reduced levels of Gli1 and Ptch protein, both targets of the Hh signaling pathway, but did not affect levels of TCR-associated kinases Lck and extracellular signal–regulated kinase (ERK) or granzyme A and perforin, two CTL proteins required for target cell lysis (Fig. 3F and fig. S4A). TCR signaling was also unaffected by these inhibitors (Fig. 3G and fig. S4B). All three inhibitors diminished CTL-mediated killing in a dose-dependent manner (Fig. 3H and fig. S4C), without impairing conjugate formation with target cells or clustering of Lck at the cSMAC in response to TCR signaling (fig. S4, E and F). Centrosome docking at the cSMAC was also reduced in conjugates formed by CTLs treated with inhibitor (Fig. 3I).
Centrosome polarization has been correlated with actin remodelling at the immunological synapse (1, 3). Carrier-treated CTLs reorganized actin into a distal ring at the immunological synapse and polarized the centrosome within 5 min of encountering the target (fig. S5A and movie S2). By contrast, in cyclopamine-treated CTLs, actin accumulated across the immunological synapse [at time (t) = 0], but failed to reorganize into the distal actin ring (fig. S5B). Centrosome polarization to the plasma membrane was also disrupted (movie S3): 92% of conjugates polarized the centrosome to the synapse in control carrier–treated CTLs compared with 59% of conjugates in cyclopamine-treated CTL. Actin clearance from the immunological synapse was also greatly reduced in CTLs from Smo-deleted mice (Fig. 4, A to C), and Smo-deleted CTLs showed a 60% reduction in actin clearance from the synapse compared with control CTLs. Thus, Hh signaling might also be required to promote actin reorganization at the immunological synapse.
Fig. 4. Hh signaling in CTLs controls Rac1 expression and actin reorganization at the immunological synapse.
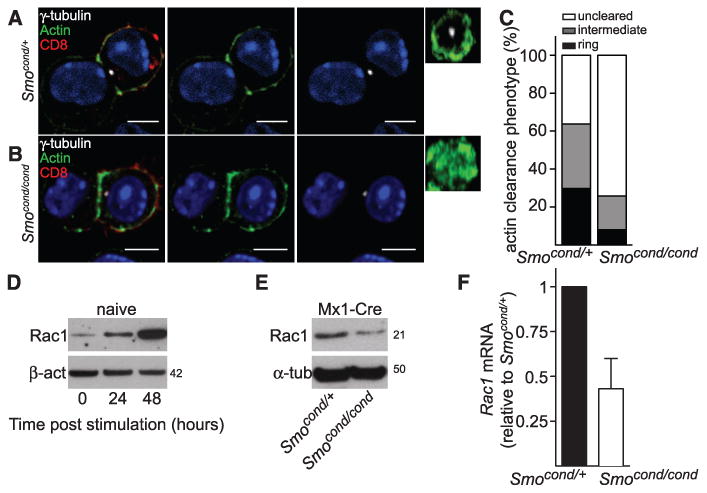
(A to C) Mx1-Cre Smocond/+(A) and Mx1-Cre Smocond/cond CTLs (B) were conjugated to P815 target cells for 15 min, then fixed and stained by using antibodies against CD8, γ-tubulin, and actin. Single x-y confocal sections and en face (y-z) constructions through the synapse are shown, which demonstrate that the actin ring does not form properly in Mx1-Cre Smocond/cond CTLs. Nuclei stained with Hoechst (blue). Scale bars: 5 μm. (C) Quantification of actin clearance at the immunological synapse, depicting the percentage of CTLs in which actin remains distributed throughout the synapse (not cleared), show an intermediate phenotype, or are cleared to form an actin ring (Smocond/+ n = 47; Mx1-Cre Smocond/cond n = 62). Immunoblot analyses of protein expression of Rac1, actin, and calnexin (D) at 0, 24, and 48 hours after TCR stimulation in naïve CD8 T cells (n = 3) and (E) Mx1-Cre Smocond/+ and Mx1-Cre Smocond/cond CTLs (n = 2). Molecular masses are shown in kilodaltons. (F) qPCR showing mRNA levels of Rac1 in Mx1-Cre Smocond/cond CTLs relative to Mx1-Cre Smocond/+ control CTLs n = 3, data are means ± SD.
Centrosome polarization in T cells is driven by a process of microtubule end-on capture-shrinkage, in which microtubules emanating from the centrosome are captured at the cortex and then undergo shrinkage (27). Both this process and actin remodeling are mediated by Rac1 (28–30). Furthermore, the Hh pathway has been implicated in Rac1-mediated actin remodelling in neurons and fibroblasts (14, 15). Therefore, we examined Rac1 expression during induction of CTLs from naïve T cells. Rac1 protein levels increased after TCR stimulation of naïve T cells (Fig. 4D). By contrast, both protein and mRNA levels of Rac1 were diminished in Smo-deleted CTLs (Fig. 4, E and F), which suggested that Rac1 levels were regulated by Hh signaling via a transcriptional effect.
These findings support a model for the regulation of CTL-mediated killing by Hh signaling (fig. S5C). Naïve T cells are small, round cells lacking both the cytotoxic granules and the highly developed cytoskeleton required for target cell killing. Upon TCR activation, these cells develop over 4 to 5 days into mature CTLs prearmed with cytotoxic granules. We now show that TCR activation also triggers Hh signaling during this time, which increases levels of Rac1 and thereby promotes centrosome polarization, actin remodeling, granule release, and target cell killing. In this way, Hh signaling prearms CTLs with the ability to rapidly polarize the cytoskeleton and deliver the cytotoxic granules within minutes when CTLs encounter targets. Our results reveal molecular parallels between primary cilia and the immunological synapse, which highlight the possible origin of the immunological synapse as a modified cilium.
Supplementary Material
Acknowledgments
We thank R. Rohatgi for the MSCV-Ptch1-EYFP construct and the Smo antibodies; S. Munro (Cambridge) for PACT (pericentrin)-RFP; M. Davidson (University of Florida) for Farnesyl-5-TagBFP2; R. Zamoyska for Lck-depleted spleens; D. Fearon, T. Crompton, J. Kaufman, M. A. de la Roche, and A. Schuldt for helpful discussions and critical reading of the manuscript; and R. Rohatgi and J. Stinchcombe for helpful discussions. We also thank the flow cytometry core facility at the Cambridge Institute for Medical Research (CIMR) for cell sorting and assistance with the calcium assay. This work was supported by a Wellcome Trust Principal Research Fellowship to G.M.G. (075880), a Wellcome Trust Strategic Award for core facilities at the CIMR (100140), NIH (R01AR05439 and R01GM095941), the Burroughs Wellcome Fund, the David and Lucile F. Packard Foundation, and the Sandler Family Supporting Foundation to J.R. The data presented in this paper are provided in the main paper and supplementary materials.
Footnotes
References and Notes
- 1.Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 2.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 3.Stinchcombe JC, Griffiths GM. Annu Rev Cell Dev Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 4.Wheatley DN. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths GM, Tsun A, Stinchcombe JC. J Cell Biol. 2010;189:399–406. doi: 10.1083/jcb.201002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finetti F, et al. Nat Cell Biol. 2009;11:1332–1339. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singla V, Reiter JF. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 8.Goetz SC, Anderson KV. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbit KC, et al. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 10.Rohatgi R, Milenkovic L, Scott MP. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 11.Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 12.Valentini RP, et al. J Biol Chem. 1997;272:8466–8473. doi: 10.1074/jbc.272.13.8466. [DOI] [PubMed] [Google Scholar]
- 13.Kornberg TB. Sci Signal. 2011;4:pe44. doi: 10.1126/scisignal.2002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki N, Kurisu J, Kengaku M. Mol Cell Neurosci. 2010;45:335–344. doi: 10.1016/j.mcn.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Polizio AH, et al. J Biol Chem. 2011;286:19589–19596. doi: 10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Zhou Z, Walsh CT, McMahon AP. Proc Natl Acad Sci USA. 2009;106:2623–2628. doi: 10.1073/pnas.0812110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milenkovic L, Scott MP, Rohatgi R. J Cell Biol. 2009;187:365–374. doi: 10.1083/jcb.200907126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Andaloussi A, et al. Nat Immunol. 2006;7:418–426. doi: 10.1038/ni1313. [DOI] [PubMed] [Google Scholar]
- 19.Outram SV, et al. Blood. 2009;113:2217–2228. doi: 10.1182/blood-2008-03-144840. [DOI] [PubMed] [Google Scholar]
- 20.Uhmann A, et al. Blood. 2007;110:1814–1823. doi: 10.1182/blood-2007-02-075648. [DOI] [PubMed] [Google Scholar]
- 21.Crompton T, Outram SV, Hager-Theodorides AL. Nat Rev Immunol. 2007;7:726–735. doi: 10.1038/nri2151. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann I, et al. Cell Stem Cell. 2009;4:559–567. doi: 10.1016/j.stem.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, et al. Cell Stem Cell. 2009;4:548–558. doi: 10.1016/j.stem.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JK, Taipale J, Cooper MK, Beachy PA. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Low JA, de Sauvage FJ. J Clin Oncol. 2010;28:5321–5326. doi: 10.1200/JCO.2010.27.9943. [DOI] [PubMed] [Google Scholar]
- 26.Lauth M, Bergström A, Shimokawa T, Toftgård R. Proc Natl Acad Sci USA. 2007;104:8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi J, et al. J Cell Biol. 2013;202:779–792. doi: 10.1083/jcb.201301004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wittmann T, Bokoch GM, Waterman-Storer CM. J Biol Chem. 2004;279:6196–6203. doi: 10.1074/jbc.M307261200. [DOI] [PubMed] [Google Scholar]
- 29.Filbert EL, Le Borgne M, Lin J, Heuser JE, Shaw AS. J Immunol. 2012;188:5421–5427. doi: 10.4049/jimmunol.1200242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimura Y, Applegate K, Davidson MW, Danuser G, Waterman CM. PLOS ONE. 2012;7:e41413. doi: 10.1371/journal.pone.0041413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.