

Abstract
We hypothesized that endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species during type I-diabetes impairs carotid ACE2-angiotensin-(1–7)-Mas axis functionality, which accounts for the impaired carotid flow in diabetic rats. We also hypothesized that angiotensin-(1–7) chronic treatment of diabetic rats restores carotid ACE2-angiotensin-(1–7)-Mas axis functionality and carotid flow. Relaxant curves for angiotensin II or angiotensin-(1–7) were obtained in carotid from streptozotocin-induced diabetic rats. Superoxide or hydrogen peroxide levels were measured by flow cytometry in carotid endothelial cells. Carotid flow was also determined. We found that endothelial AT1-activated NAD(P)H oxidase-driven generation of superoxide and hydrogen peroxide in diabetic rat carotid impairs ACE2-angiotensin-(1–7)-Mas axis functionality, which reduces carotid flow. In this mechanism, hydrogen peroxide derived from superoxide dismutation inhibits ACE2 activity in generating angiotensin-(1–7) seemingly by activating I Cl,SWELL, while superoxide inhibits the nitrergic Mas-mediated vasorelaxation evoked by angiotensin-(1–7). Angiotensin-(1–7) treatment of diabetic rats restored carotid ACE2-angiotensin-(1–7)-Mas axis functionality by triggering a positive feedback played by endothelial Mas receptors, that blunts endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species. Mas-mediated antioxidant effects also restored diabetic rat carotid flow, pointing to the contribution of ACE2-angiotensin-(1–7)-Mas axis in maintaining carotid flow.
1. Introduction
Vascular dysfunction triggered by type I-diabetes has been extensively described as an important risk factor for the development of carotid atherosclerosis in the genesis of cerebrovascular diseases, such as stroke [1–4]. The major mechanisms underlying diabetic vascular dysfunction result from changes in the functionality of the main systems involved in the control of arterial tonus [5–9], such as renin-angiotensin system (RAS). Indeed, the upregulation of angiotensin converting enzyme- (ACE-) angiotensin II-AT1 axis from vascular RAS seems to play a crucial role in the pathogenesis of diabetic vascular dysfunction and complications. Yousif et al. [5, 6] and Pernomian et al. [8, 9] showed that type I-diabetes enhances the vasocontractile response evoked by angiotensin II [5, 6, 8, 9], which damages vascular function and contributes with atherogenesis by affecting both the vascular tone and the progression of vascular inflammation [10]. Moreover, angiotensin converting enzyme (ACE) inhibitors or AT1 antagonists attenuate carotid atherosclerosis during diabetes by improving vascular function [11–13].
Despite the aggressive effects assigned to ACE-angiotensin II-AT1 axis on vascular function during diabetic conditions, there is another important axis from RAS, namely, ACE2-angiotensin-(1–7)-Mas axis, that triggers opposite effects to those produced by the former [14, 15]. In this alternative axis, ACE homologue (ACE2) hydrolyzes angiotensin II into angiotensin-(1–7) [16, 17], which is the endogenous ligand of Mas receptors [18]. In vessels, such as rat carotid, the activation of Mas receptors evokes a nitrergic relaxation [19] that has been correlated with vasoprotective effects in diabetic conditions [20]. This perspective has pointed the vascular ACE2-angiotensin-(1–7)-Mas axis as a potential therapeutic target to attenuate diabetic endothelial dysfunction and the subsequent vascular complications. Nevertheless, the vasoprotective therapeutic efficacy of drugs aimed at the activation of ACE2-Angiotensin-(1–7)-Mas axis depends on the integrity of the functionality of this axis during the disease. To the best of our knowledge, there are no evidences concerning the consequences of type I-diabetes on the functionality of vascular ACE2-angiotensin-(1–7)-Mas axis. However, our previous findings suggest an indirect evidence concerning these consequences: in rat carotid, type I-diabetes shifts the Gaussian-like shape of angiotensin II-evoked contraction curve into a sigmoidal shape, due to endothelial AT1-activated NAD(P)H oxidase-driven generation of superoxide (O2 −), hydrogen peroxide (H2O2), and hydroxyl radical (∙OH) [8]. In turn, the Gaussian-like shape of angiotensin II-induced contraction curve in rat carotid results from the relaxation triggered by micromolar concentrations of angiotensin II, which is mediated by Mas receptors [19] probably upon the hydrolysis of angiotensin II into angiotensin-(1–7). Thus, our previous findings [8] allow us to hypothesize that endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species during type I-diabetes impairs the functionality of ACE2-angiotensin-(1–7)-Mas axis in rat carotid by inhibiting both the hydrolysis of angiotensin II into angiotensin-(1–7) and the nitrergic signaling pathway underlying Mas receptors activation. Considering the vasoprotective effects assigned to vascular ACE2-angiotensin-(1–7)-Mas axis [20], we also hypothesize that the impairment of the functionality of this axis would contribute to damage carotid blood flow and resistance in type I-diabetic rats. Furthermore, since the activation of Mas receptors inhibits NAD(P)H oxidase-driven generation of reactive oxygen species in endothelial cells [21], we expect that the chronic treatment of type I-diabetic rats with angiotensin-(1–7) would restore the functionality of carotid ACE2-angiotensin-(1–7)-Mas axis and carotid blood flow and resistance by restoring both the local hydrolysis of angiotensin II into angiotensin-(1–7) and the nitrergic signaling pathway underlying Mas receptors activation.
2. Material and Methods
The experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals. A prior approval was granted by the Animal Ethics Committee of the Faculty of Medicine from Ribeirão Preto (FMRP) from the University of São Paulo (USP) in Brazil (approval reference number: 007/2009). Male Wistar rats (Rattus norvegicus) were used in this study. These animals were kept under a 12 light/12 dark cycle (light from 06:00 to 18:00 h) and fed with regular chow and water ad libitum.
2.1. Experimental Design and Animal Groups
Type I-diabetes was induced by a single intraperitoneal injection of streptozotocin (STZ, 55 mg/kg) dissolved in citrate buffer (0.09 mol/L, pH 4.5) (day 0) in eight-week-old male Wistar rats (350–400 g). The control group was composed by age-matched normoglycaemic rats that underwent to citrate buffer injection. Fasting glucose levels were determined from rat tail blood samples prior to and 48 h (day 2) after STZ or vehicle injection, by using a one-touch glucometer (LifeScan Inc., Milpitas, CA, USA). Diabetic rats presented glycaemia higher than 300 mg/dL (Table 1). Six weeks after STZ or vehicle injection (day 42), the animals underwent to body weight measurement (Table 2), then they were sacrificed, and the experiments were performed [8, 9]. In some protocols, STZ- or vehicle-treated rats (eight-weeks old) were chronically treated with intraperitoneal daily injections of the selective ACE2 inhibitor DX600 (5 μg/kg/day) or with daily intraperitoneal injections of the selective Mas receptors agonist angiotensin-(1–7) (576 μg/kg/day), combined or not with the selective Mas receptors antagonist A779 (1 mg/kg/day), for six weeks [22]. All these treatments started at the time of STZ or vehicle injection.
Table 1.
Fasting blood glucose levels from control or diabetic rats.
| Rats | Glycaemia (mg·dL−1) | |
|---|---|---|
| Day 0 | Day 2 | |
| Nontreated Control | 68.7 ± 3.11 | 73.0 ± 6.35 |
| Nontreated Diabetic | 75.4 ± 5.17 | 392.6 ± 11.47∗,# |
| DX600-treated control | 70.5 ± 4.20 | 71.8 ± 5.23 |
| DX600-treated diabetic | 69.9 ± 6.31 | 402.7 ± 15.25∗,# |
| Angiotensin-(1–7)-treated control | 73.1 ± 5.19 | 67.9 ± 5.24 |
| Angiotensin-(1–7)-treated diabetic | 71.2 ± 4.98 | 385.9 ± 13.26∗,# |
| A779-treated control | 72.1 ± 4.57 | 76.3 ± 7.93 |
| A779-treated diabetic | 74.2 ± 7.21 | 406.0 ± 14.69∗,# |
| Angiotensin-(1–7) + A779-treated control | 67.5 ± 6.52 | 69.3 ± 6.45 |
| Angiotensin-(1–7) + A779-treated diabetic | 71.2 ± 7.09 | 409.1 ± 17.33∗,# |
The values are significantly different (P < 0.01; n = 11) from the respective control rats at the same day (∗) or the same rats at day 0 (#).
Table 2.
Body weight from control or diabetic rats.
| Rats | Body weight (g) | |
|---|---|---|
| Day 0 | Day 42 | |
| Nontreated control | 374.9 ± 7.5 | 714.1 ± 10.2# |
| Nontreated diabetic | 365.2 ± 6.9 | 286.3 ± 9.1∗,# |
| DX600-treated control | 361.5 ± 5.11 | 721.2 ± 11.4# |
| DX600-treated diabetic | 378.0 ± 4.9 | 267.5 ± 10.3∗,# |
| Angiotensin-(1–7)-treated control | 373.8 ± 6.2 | 733.9 ± 8.7# |
| Angiotensin-(1–7)-treated diabetic | 354.2 ± 8.2 | 275.3 ± 11.6∗,# |
| A779-treated control | 369.1 ± 5.7 | 729.4 ± 7.5# |
| A779-treated diabetic | 372.4 ± 9.1 | 281.6 ± 13.2∗,# |
| Angiotensin-(1–7) + A779-treated control | 368.4 ± 7.2 | 719.1 ± 9.5# |
| Angiotensin-(1–7) + A779-treated diabetic | 355.7 ± 6.9 | 272.3 ± 11.4∗,# |
The values are significantly different (P < 0.01; n = 11) from the respective control rats at the same day (∗) or the same rats at day 0 (#).
2.2. Ex Vivo Arterial Reactivity Studies
The functionality of vascular ACE2-angiotensin-(1–7)-Mas axis was studied by functional assays of angiotensin II or angiotensin-(1–7) cumulative concentration-response relaxant curves, obtained in ex vivo arterial reactivity studies in carotid rings from control or diabetic rats.
2.2.1. Carotid Rings Preparation
Rats were sacrificed by abdominal aortic exsanguination and common carotid arteries were isolated. Carotid rings (4 mm) were placed in 5.0 mL of Krebs-Henseleit bicarbonate buffer (composition in mmol/L: NaCl 118.4; KCl 4.7; CaCl2 1.9; KH2PO4 1.2; MgSO4·7H2O 1.2; NaHCO3 25; C6H12O6 11.6) in organ bath chambers, gassed with 95% O2 and 5% CO2 and maintained at 37°C and pH 7.4. The rings were connected to an isometric force transducer (Letica Scientific Instruments, Barcelona, Spain) to measure changes in the isometric tension [8, 9, 23, 24]. After 60 min of stabilization at a resting tension of 1 g, carotid rings viability was tested with the appropriate molar concentration of phenylephrine that produces 50% of the maximum contraction response (EC50) in each experimental group (phenylephrine EC50 = 0.1 μmol/L for control rat carotid or phenylephrine EC50 = 10 nmol/L for diabetic rat carotid), as previously determined by cumulative concentration-response curves for phenylephrine. The endothelial integrity was verified with the appropriate molar concentration of acetylcholine that produces 100% of the maximum relaxation response (EC100) in each experimental group (acetylcholine EC100 = 1.0 μmol/L for control rat carotid or acetylcholine EC100 = 100 μmol/L for diabetic rat carotid), over phenylephrine-induced precontraction [8, 9]. For studies with endothelium-intact vessels, the ring was discarded if the maximum relaxation induced by acetylcholine did not reach 80–100%. When necessary, the endothelium was mechanically removed by gentle rubbing the vessel with a thin wire. Endothelium was deemed absent when the relaxation response to acetylcholine did not occur [8, 9, 23, 24].
2.2.2. Experimental Protocols
(1) Cumulative Concentration Response Relaxant Curves for Angiotensin II. The functionality of vascular ACE2 (i.e., the activity of vascular ACE2 in generating angiotensin-(1–7)) was studied by relaxant curves induced by angiotensin II in rat carotid. This approach was supported by two evidences: (1) the angiotensin 1–7 Mas receptor antagonist, A779, partially inhibits the relaxation evoked by angiotensin II in rat carotid [19], suggesting that angiotensin II is converted into angiotensin-(1–7) in this bed; and (2) the unique enzyme that is able to convert angiotensin II into angiotensin-(1–7) in vascular tissues is ACE2 [17, 25].
Relaxation cumulative concentration-response curves for angiotensin II (10−9–10−4 mol/L) were obtained in endothelium-intact or endothelium-denuded carotid rings from control or diabetic rats, over the precontraction induced by the appropriate molar concentration of phenylephrine that produces 80% of the maximum contraction response (EC80) in each experimental group (phenylephrine EC80 = 1.0 μmol/L for control rat carotid or phenylephrine EC80 = 0.1 μmol/L for diabetic rat carotid). In this study, we have chosen phenylephrine EC80 instead of EC50 due to the higher magnitude of the precontraction evoked by the former concentration (too much closer to the maximum contraction). This avoids significant changes on the precontraction value upon pharmacological intervention. In functional assays, this is an important approach, since it avoids significant interferences from the precontraction magnitude variation on the relaxant response magnitude. Finally, EC80 can be safely used to induce a precontraction because it is as accurate for the homogeneous distribution of date as the EC50, since both of them can be found at the central linear region of sigmoidal curves. The relaxant curves for angiotensin II were obtained in carotid rings from nontreated control or diabetic rats, in the absence or presence of DX600 (10 μmol/L, 30 min) [26], A779 (5.0 μmol/L, 30 min) [19] or the selective AT2 antagonist, PD123,319 (0.5 μmol/L, 30 min) [19]. As posteriorly described in the Results section, this protocol confirms that ACE2 converts angiotensin II into angiotensin-(1–7), which in turn partially mediates the relaxation evoked by angiotensin II by activating Mas receptors. Thus, this protocol validates the functional studies of angiotensin II-evoked relaxant curves in rat carotid as a method to study vascular ACE2 functionality.
To verify if the reactive oxygen species derived from AT1-activated NAD(P)H oxidase impair the functionality of ACE2 in diabetic rat carotid, angiotensin II-induced relaxant curves were obtained in the absence or presence of the selective AT1 antagonist, losartan (1.0 μmol/L, 30 min) [27], the selective NAD(P)H oxidase inhibitor, apocynin (0.1 mmol/L, 30 min) [8, 23], the selective O2 − scavenger, tiron (0.1 mmol/L, 30 min) [8, 23], or the selective H2O2 scavenger, PEG-catalase (250 U/mL, 30 min) [8, 23]. Also, we investigated the hypothesis that the reactive oxygen species-mediated activation of volume-sensitive Cl− current (I Cl,SWELL) impairs ACE2 functionality in diabetic rat carotid, by obtaining angiotensin II-evoked relaxant curves in the presence of the selective inhibitor of I Cl,SWELL, DCPIB (10 μmol/L, 10 min) [28, 29]. This hypothesis was suggested based on the following evidences: (1) the cleavage of angiotensin II by ACE2 is reduced by increasing the physiological extracellular levels of Cl− [30]; and (2) H2O2 generated from NAD(P)H oxidase-derived O2 − can activate I Cl,SWELL in vascular smooth muscle cells, leading to the efflux of Cl− and thus increasing Cl− extracellular levels [31, 32].
In order to verify if the Mas-mediated effects against AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species restore the functionality of ACE2 in diabetic rat carotid, the relaxation curves for angiotensin II were obtained in carotid rings from control or diabetic rats that were chronically treated with angiotensin-(1–7). This protocol was not repeated in carotid rings from rats treated with angiotensin-(1–7) combined with A779 because angiotensin II-evoked relaxation is partially mediated by Mas receptors [19].
(2) Cumulative Concentration-Response Relaxant Curves for Angiotensin-(1–7). The functionality of vascular Mas receptors (i.e., nitrergic vasorelaxant response evoked by angiotensin-(1–7) upon Mas receptors activation) was studied by relaxation cumulative concentration-response curves for angiotensin-(1–7) (10−11–10−5 mol/L), that were obtained in endothelium-intact or endothelium-denuded carotid rings from control or diabetic rats. In these protocols, carotid rings were also precontracted with phenylephrine EC80 (1.0 μmol/L for control rat carotid or 0.1 μmol/L for diabetic rat carotid), due to the reasons previously described.
The mediators of angiotensin-(1–7)-induced relaxation were characterized by obtaining these curves in carotid rings from nontreated control or diabetic rats, in the absence or presence of A779 (5.0 μmol/L, 30 min), PD123,319 (0.5 μmol/L, 30 min), the selective nitric oxide (NO) scavenger hydroxocobalamin (0.1 mmol/L, 30 min) [33], the nonselective NOS inhibitor L-NNA (0.1 mmol/L, 30 min), the selective neuronal NOS (nNOS) inhibitor L-NPA (50 nmol/L, 30 min), or the selective inducible NOS (iNOS) inhibitor 1400 W (10 nmol/L, 30 min) [9].
To verify if the reactive oxygen species derived from AT1-activated NAD(P)H oxidase impair the functionality of angiotensin-(1–7)-Mas receptors in diabetic rat carotid, angiotensin-(1–7)-induced relaxant curves were obtained in the absence or presence of losartan (1.0 μmol/L, 30 min), apocynin (0.1 mmol/L), tiron (0.1 mmol/L), or PEG-catalase (250 U/mL). We also investigated if the reactive oxygen species-activated I Cl,SWELL impairs the functionality of angiotensin-(1–7)-Mas receptors in diabetic rat carotid, by obtaining angiotensin II-evoked relaxant curves in the presence of DCPIB (10 μmol/L, 10 min).
In order to verify if the Mas-mediated effects against AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species restore the functionality of angiotensin-(1–7)-Mas receptors in diabetic rat carotid, the relaxation curves for angiotensin II were obtained in carotid rings from control or diabetic rats that were chronically treated with angiotensin-(1–7). This protocol was not repeated in carotid rings from rats treated with angiotensin-(1–7) combined with A779 because angiotensin-(1–7)-evoked relaxation is mediated by Mas receptors [19].
2.3. Flow Cytometry in Endothelial Cells
To confirm the functional evidences that the Mas-mediated effects induced by the chronic treatment with angiotensin-(1–7) on ACE2-angiotensin-(1–7)-Mas axis functionality in diabetic rat carotid involve antioxidant actions against endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species (O2 − and H2O2), flow cytometry assays were performed in carotid-derived endothelial cells, loaded with selective probes for reactive oxygen species.
Carotid arteries were isolated after abdominal aortic exsanguination. Thus, the arteries were longitudinally sectioned and endothelial cells were mechanically isolated by gentle friction with a plastic stem in plates containing Hanks' solution (composition in mmol/L: CaCl2 1.6; MgSO4 1.0; NaCl 145.0; KCl 5.0; NaH2PO4 0.5; dextrose 10.0; HEPES 10.0) at pH 7.4. The cell suspensions were centrifuged at 1.375 hg for 5 min, and the pellets were resuspended in 0.5 mL of Hanks's solution in a humidified incubator at 37°C until use [8, 9, 23, 24, 34]. Each n comprised a pool of six carotid arteries. The cell viability was previously determined by trypan blue staining (2%) and counting in a Neubauer chamber (Weber Scientific International, Germany).
The endothelial cells were loaded with the nonselective fluorescent dye for reactive oxygen species, dihydroethidium (DHE, 2.5 μmol/L, 20 min, 37°C) [8, 23] or with the selective fluorescent dye for H2O2, 5(6)-carboxy-2′,7′-dichlorofluorescein diacetate (CDCF-DA, 1 μmol/L, 20 min, 25°C) [9, 35].
The basal levels of O2 − from endothelial cells isolated from carotid arteries of nontreated control or diabetic rats were measured in the absence or presence of tiron (0.1 mmol/L, 30 min), apocynin (0.1 mmol/L, 30 min), losartan (1.0 μmol/L, 30 min), or DCPIB (10 μmol/L, 10 min), added to the cellular samples during DHE loading. The basal levels of O2 − were also measured in endothelial cells from carotid arteries of control or diabetic rats chronically treated with angiotensin-(1–7), combined or not with A779.
The basal levels of H2O2 from endothelial cells isolated from carotid arteries of nontreated control or diabetic rats were measured in the absence or presence of PEG-catalase (3,000 U/mL, 30 min) [36], tiron (0.1 mmol/L, 30 min), apocynin (0.1 mmol/L, 30 min), losartan (1.0 μmol/L, 30 min), or DCPIB (10 μmol/L, 10 min), added to the cellular samples during CDCF-DA loading. The basal levels of H2O2 were also measured in endothelial cells from carotid arteries of control or diabetic rats chronically treated with angiotensin-(1–7), combined or not with A779.
2.4. Immunohistochemical Assays
The expression of ACE2 and Mas receptors, as well as the levels of angiotensin-(1–7), was assessed by immunohistochemical assays. These data were correlated with the functionality of ACE2-angiotensin-(1–7)-Mas axis in nontreated control or diabetic rat carotid arteries. The effects of the chronic treatment with angiotensin-(1–7) on ACE2 and Mas receptors expression in control or diabetic rat carotid were also studied. In turn, the expression of angiotensin-(1–7) was not assessed in carotid arteries from control or diabetic rats that were chronically treated with angiotensin-(1–7), since this treatment tends to increase the vascular levels of the heptapeptide. Furthermore, to confirm the functional evidences concerning the generation of angiotensin-(1–7) by ACE2 in rat carotid and the impairment of ACE2 activity in generating angiotensin-(1–7) in diabetic rat carotid, angiotensin-(1–7) levels were also assessed in carotid arteries from nontreated or DX600-treated control or diabetic rats.
As we aimed to evaluate the expression of ACE2, angiotensin-(1–7) and Mas receptors in each vascular layer, mainly in the endothelium, we have chosen immunohistochemistry instead of western blotting (WB) assays, since it is not viable to isolate endothelial cells from rat carotid to perform WB (each n of endothelial cells sample would comprise more than 20 carotid arteries to yield a sufficient protein level). In addition, angiotensin-(1–7) levels could not be evaluated by western blotting assays since the molecular weight of angiotensin-(1–7) is lesser than 2.0 kDa, what precludes the use of the available polyacrylamide or tricine gels and molecular weight markers.
After abdominal aortic exsanguination, carotid arteries were isolated and included in paraffin. Ion paraffin-included carotid rings were cut into 3 μm sections and mounted on poly-L-lysine-coated slides, which were rinsed with phosphate-buffered saline (PBS) and immersed in 3% H2O2 for 20 min to block endogenous peroxidase. Nonspecific protein binding was blocked with normal serum for 30 min [8, 9]. The sections were incubated with the primary antibody against ACE2 (1 : 100) [37], Mas receptors (1 : 250) [38], or angiotensin-(1–7) (1 : 100) [39] for 2 h at 25°C. Following washes in PBS, biotinylated pan-specific universal secondary antibody (1 : 300) was applied for 30 min. The slides were incubated with avidin-biotin peroxidase complex for 30 min. They were then counterstained by haematoxylin, dehydrated, and mounted with Permount. As negative controls, all specimens were incubated with an isotope-matched control antibody under identical conditions. The immunolabeling was considered positive when distinct red nuclear or cytoplasmic staining was homogenously present [8, 9].
2.5. Carotid Blood Flow Measurement
In order to verify if the functionality of local ACE2-angiotensin-(1–7)-Mas axis contributes to the control of carotid blood flow and resistance and if the Mas-mediated effects restore carotid blood flow and resistance in diabetic rats, these experiments were performed in control or diabetic rats that were chronically treated or not with DX600, A779 or with angiotensin-(1–7), combined or not with A779.
Rats were anaesthetized with ketamine (50 mg/kg) and xylazine (10 mg/kg), by intraperitoneal injection. The left and right common carotid arteries were exposed. A noninvasive transit-time flow probe (model 1.5RB; Transonic Systems, Inc., Ithaca, NY, USA) was placed around each carotid artery and connected to a flow meter (model T-206; Transonic system, USA). Basal mean blood pressure was determined in anaesthetized rats after femoral artery cannulation [9, 40]. Blood pressure and carotid flow were simultaneously measured after the slow infusion of normal saline (0.9%, 1 mL), that was used as volume expander, in order to prevent the eventual interferences related to the reduced tissue perfusion volume in diabetic rats, whose body weight is reduced (Table 2).
2.6. Data Analysis
Data were expressed as the mean ± S.E.M., and the differences between the mean values were assessed by one-way ANOVA and Bonferroni post-hoc. The significance level considered was 0.05. We chose one-way ANOVA as the statistical test since there was only one independent variable in the experimental design (i.e., the treatment factor), which was divided into two categorical (nominal) variables: (1) the in vivo treatment of rats with STZ, citrate buffer, DX600, angiotensin-(1–7), and/or A779; and (2) the in vitro pretreatment of carotid rings or carotid endothelial cells with DX600, A779, PD123,319, losartan, hydroxocobalamin, apocynin, tiron, PEG-catalase, or DCPIB. Endothelial removal is also considered as an in vitro pretreatment variable, since the removal of endothelial modulation on vascular responses can be equally reached by mechanical or chemical induction.
In the functional studies, relaxant responses were recorded as reductions in the muscular tone evoked by the preconstrictor agent and expressed as grams of tension (absolute relaxation values) on negative scale. The concentration-response curves were fitted using a nonlinear interactive fitting program (GraphPad Prism 5.00; GraphPad Software Inc., San Diego, CA). The maximum effect (E max) of AngII or Ang-(1–7) was obtained from the nonlinear regression of the agonist curve.
In the flow cytometry analysis, the median values of the fluorescence intensity (FI) emitted by endothelial cells were determined using DIVA software and expressed in fluorescence units (U).
The percentage of the stained area was determined by ImageJ Program (1.46r, Wayne Rasband, NIH, USA) in immunohistochemical assays, in which it was delimited an area of 900 μm2 for the media or an area of 45 μm2 for the endothelium from carotid sections.
In the in vivo experiments, the mean carotid blood flow (F) was recorded by 10 min in a computational acquisition system (Dataq, USA), which provided an actual volume flow measurement in a resolution of 0.05 mL/min. Mean blood pressure (BP) baseline values were calculated as the average of the 10 min recording, by using the acquisition system (Dataq, USA). F and BP were used to calculate the carotid resistance (R) by applying the formulae R = BP/F.
2.7. Drugs, Chemical Reagents, and Other Materials
They are STZ, angiotensin II, angiotensin-(1–7), losartan, PD123,319, hydroxocobalamin, L-NNA, 1400 W, apocynin, tiron, PEG-catalase, CDCF-DA, and DCPIB (Sigma, St. Louis, Mo., USA); DX600 (Anaspec Inc., Fremont, CA, USA); A779 (Bachem California Inc., Torrance, CA, USA); L-NPA (Tocris, Avonmouth, UK); DHE (Invitrogen, Carlsbad, CA, USA); ketamine (União Química, Jabaquara, SP, Brazil); xylazine (Calier Laboratory, Jubatuba, MG, Brazil); rabbit polyclonal anti-ACE2 antibody (Abcam, Cambridge, MA, USA); rabbit anti-angiotensin-(1–7) antibody (Phoenix Pharmaceuticals Inc., Burlingame, CA, USA); rabbit polyclonal anti-angiotensin-(1–7)-Mas receptor antibody (Alomone Labs, Jerusalem, Israel); biotinylated universal secondary antibody, avidin-biotin peroxidase complex (Vectastain Elite ABC kit, Universal, Vector Laboratories Inc. U.S. Headquarters, Burlingame, CA, USA).
3. Results
3.1. Angiotensin II-Induced Relaxation
Phenylephrine EC80 evoked a precontraction of 0.29 ± 0.021 g (n = 9) in endothelium-intact carotid rings from nontreated control rats. In these vessels, angiotensin II produced a biphasic response over phenylephrine-induced precontraction, which was characterized by a residual contraction (0.15 ± 0.019 g, n = 9) evoked by nanomolar concentrations of angiotensin II, followed by a relaxant response (E max = −0.36 ± 0.028 g, n = 9) induced by micromolar concentrations of angiotensin II (Figures 1(a) and 1(c)). Endothelium removal did not alter the magnitude of phenylephrine-induced precontraction (0.34 ± 0.032 g, n = 9) or the relaxant response evoked by angiotensin II (P > 0.05, one-way ANOVA, Bonferroni post-hoc). In endothelium-denuded carotid rings from nontreated control rat, this relaxant response was completely inhibited by DX600 but partially inhibited by A779 (Figures 1(c) and 1(g)). Losartan inhibited the residual contraction evoked by angiotensin II and thus reduced the maximum relaxation triggered by angiotensin II in endothelium-intact carotid rings from nontreated control rat (Figures 3(a) and 3(c)). The magnitude of phenylephrine-induced precontraction in control rat carotid rings was not altered by DX600 (0.35 ± 0.031 g, n = 9), A779 (0.32 ± 0.025 g, n = 9), or losartan (0.29 ± 0.023 g, n = 9) (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
Figure 1.
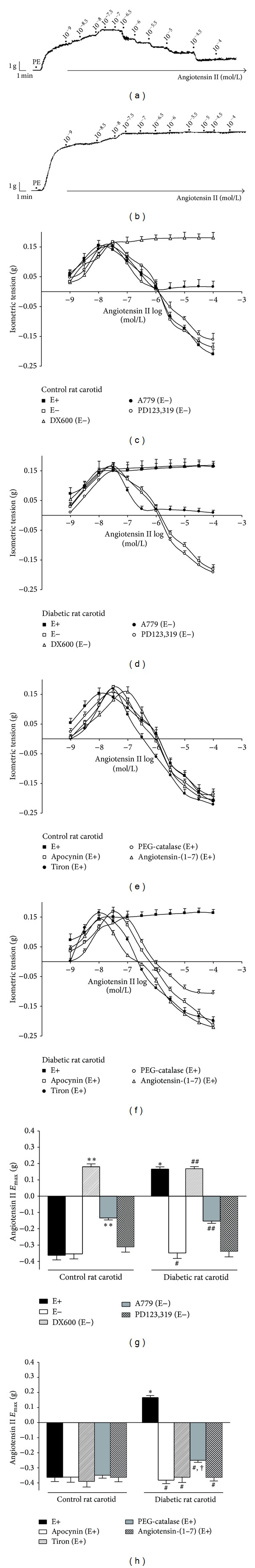
Concentration-response curves for angiotensin II in endothelium-intact (E+) or -denuded (E−) carotid rings from control or diabetic rats, over the precontraction induced by phenylephrine (PE). Representative traces from angiotensin II-evoked relaxation in E+ control (a) or diabetic (b) rat carotid. Effect of the in vitro pretreatment with DX600, A779, or PD123,319 in carotid rings from control (c) or diabetic (d) rats. Effect of the in vitro pretreatment with apocynin, tiron, PEG-catalase, or the chronic (in vivo) treatment with angiotensin-(1–7) in carotid rings from control (e) or diabetic (f) rats. Angiotensin II E max in carotid arteries from control or diabetic rats before or after the in vitro pretreatment with DX600, A779, PD123,319 (g), apocynin, tiron, or PEG-catalase or the chronic treatment (in vivo) with angiotensin-(1–7) (h). The values are significantly different (P < 0.01; n = 9) from nonpretreated E+ (∗) or E− (∗∗) carotid rings from nontreated control rats, from nonpretreated E+ (#) or E− (##) carotid rings from nontreated diabetic rats, or from PEG-catalase pretreated E+ (†) carotid rings from nontreated control rats.
Figure 3.
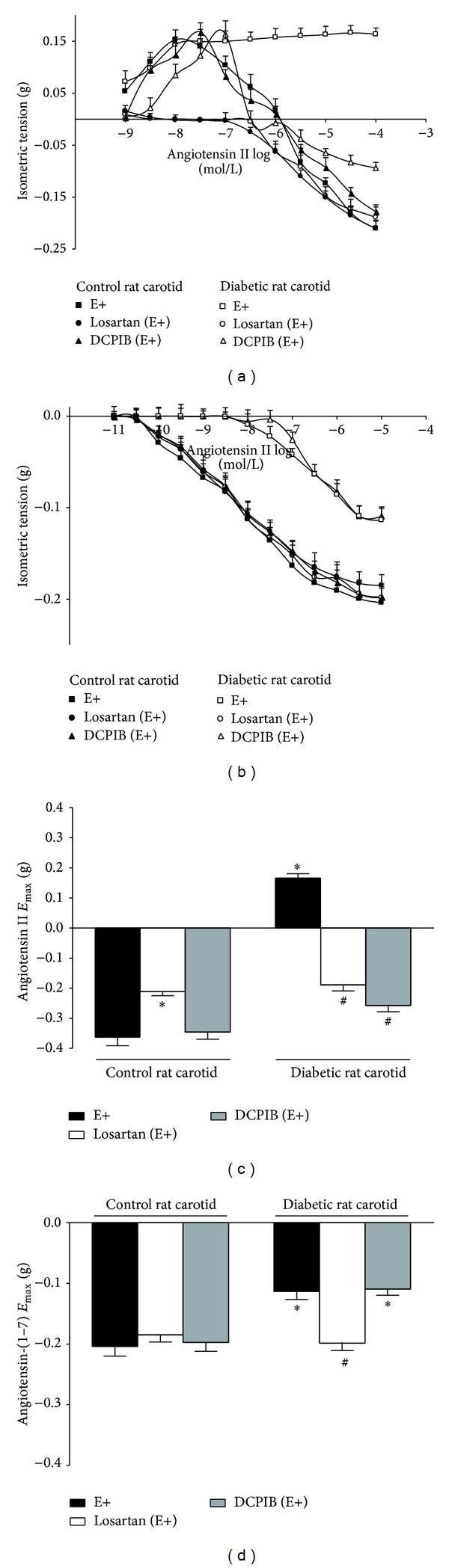
Concentration-response curves for angiotensin II or angiotensin-(1–7) in endothelium-intact (E+) carotid from control or diabetic rats after the in vitro pretreatment with losartan or DCPIB. Effect of the in vitro pretreatment with losartan or DCPIB on the relaxation induced by angiotensin II (a) or angiotensin-(1–7) (b). Angiotensin II E max (c) or angiotensin-(1–7) E max (d) before or after the in vitro pretreatment with losartan or DCPIB. The values are significantly different (P < 0.01; n = 9) from nonpretreated carotid rings (E+) from nontreated control (∗) or diabetic (#) rats.
Phenylephrine EC80 evoked a precontraction of 0.51 ± 0.033 g (n = 9) in endothelium-intact carotid rings from nontreated diabetic rats. This value was significantly higher than that one evoked in endothelium-intact carotid rings from nontreated control rats (P < 0.001, one-way ANOVA, Bonferroni post-hoc). In carotid artery from nontreated diabetic rat, nanomolar concentrations of angiotensin II evoked a persistent residual contraction (0.17 ± 0.014 g, n = 9) over phenylephrine-induced precontraction (Figures 1(b) and 1(d)). Diabetes abrogated the relaxant response induced by micromolar concentrations of angiotensin II in rat carotid, which was completely restored by endothelium removal (this result required that the characterization of the mediators of angiotensin II-induced relaxation was performed in endothelium-denuded carotid rings from diabetic rats, as well as from control rats, to compare the responses). In endothelium-denuded carotid rings from nontreated diabetic rats, the relaxant response evoked by angiotensin II was completely inhibited by DX600 but partially inhibited by A779 (Figures 1(d) and 1(g)). Losartan inhibited the residual contraction induced by angiotensin II but allowed micromolar concentrations of angiotensin II to elicit a relaxant response in endothelium-intact carotid rings from nontreated diabetic rat (Figures 3(a) and 3(c)). The magnitude of phenylephrine-induced precontraction in carotid rings from nontreated diabetic rats was not altered by endothelium removal (0.46 ± 0.041 g, n = 9) or the pretreatment with DX600 (0.49 ± 0.037 g, n = 9), A779 (0.47 ± 0.028 g, n = 9), or losartan (0.52 ± 0.045 g, n = 9), when compared to nonpretreated endothelium-intact diabetic rat carotid (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
In endothelium-denuded carotid rings from nontreated control or diabetic rats, the maximum relaxation induced by angiotensin II in the presence of A779 had the same extent (in absolute values of g of tension) as the maximum contraction evoked by angiotensin II in these arteries (Figures 1(c), 1(d), and 1(g)).
Although the relaxation induced by angiotensin II has not been altered by apocynin, tiron, PEG-catalase, and DCPIB or by the chronic treatment with angiotensin-(1–7) in endothelium-intact carotid rings from nontreated control rats, this response was completely restored by apocynin or tiron and partially restored by PEG-catalase or DCPIB in endothelium-intact carotid rings from nontreated diabetic rats. Similar to the effects of apocynin or tiron, the chronic treatment with angiotensin-(1–7) completely restored the relaxation induced by angiotensin II in endothelium-intact carotid rings from diabetic rats (Figures 1(e), 1(f), 1(h), 3(a), and 3(c)). In control rat carotid, the magnitude of phenylephrine-induced precontraction was not altered by apocinin (0.28 ± 0.019 g, n = 9), tiron (0.27 ± 0.022 g, n = 9), PEG-catalase (0.30 ± 0.023 g, n = 9), angiotensin-(1–7) (0.32 ± 0.031 g, n = 9), or DCPIB (0.31 ± 0.024 g, n = 9). Also, in diabetic rat carotid, phenylephrine-induced precontraction was not altered by apocinin (0.53 ± 0.039 g, n = 9), tiron (0.49 ± 0.035 g, n = 9), PEG-catalase (0.50 ± 0.049 g, n = 9), angiotensin-(1–7) (0.51 ± 0.042 g, n = 9), or DCPIB (0.50 ± 0.033 g, n = 9), when compared to nonpretreated diabetic rat carotid (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
3.2. Angiotensin-(1–7)-Induced Relaxation
In carotid artery from nontreated control rat, angiotensin-(1–7) evoked a relaxant response (E max = −0.20 ± 0.016 g, n = 9) over the precontraction evoked by phenylephrine (0.30 ± 0.025 g, n = 9). This relaxant response was not altered by endothelium removal. In endothelium-denuded carotid rings from nontreated control rats, this relaxant response was completely inhibited by A779, hydroxocobalamin (Figures 2(a), 2(c), and 2(g)) or L-NNA (E max = −0.03 ± 0.005 g, n = 9) (P < 0.001, one-way ANOVA, Bonferroni post-hoc), but not altered by L-NPA (E max = −0.18 ± 0.011 g, n = 9) or 1400 W (E max = −0.21 ± 0.019 g, n = 9) (P > 0.05, one-way ANOVA, Bonferroni post-hoc). The magnitude of phenylephrine-induced precontraction in nontreated control rat carotid was not altered by endothelium removal (0.33 ± 0.031 g, n = 9) or the pretreatment with A779 (0.34 ± 0.028 g, n = 9), hydroxocobalamin (0.36 ± 0.034 g, n = 9), L-NNA (0.35 ± 0.031 g, n = 9), L-NPA (0.29 ± 0.027 g, n = 9), or 1400 W (0.27 ± 0.026 g, n = 9) (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
Figure 2.
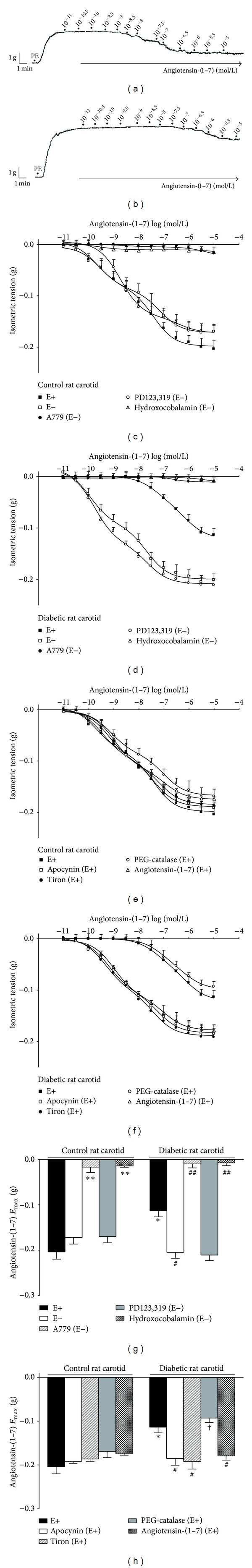
Concentration-response curves for angiotensin-(1–7) in endothelium-intact (E+) or -denuded (E−) carotid rings from control or diabetic rats, over the precontraction induced by phenylephrine (PE). Representative traces from angiotensin-(1–7)-evoked relaxation in E+ control (a) or diabetic (b) rat carotid. Effect of the in vitro pretreatment with A779, PD123,319 or hydroxocobalamin in carotid rings from control (c) or diabetic (d) rats. Effect of the in vitro pretreatment with apocynin, tiron, PEG-catalase, or the chronic (in vivo) treatment with angiotensin-(1–7) in carotid rings from control (e) or diabetic (f) rats. Angiotensin-(1–7) Emax in carotid from control or diabetic rats before or after the in vitro pretreatment with A779, PD123,319, hydroxocobalamin (g), apocynin, tiron, or PEG-catalase or the chronic (in vivo) treatment with angiotensin-(1–7) (h). The values are significantly different (P < 0.01; n = 9) from nonpretreated E+ (∗) or E− (∗∗) carotid rings from nontreated control rats, from nonpretreated E+ (#) or E− (##) carotid rings from nontreated diabetic rats, or from PEG-catalase pretreated E+ (†) carotid rings from nontreated control rats.
Diabetes impaired, but not abolished, the relaxant response induced by angiotensin-(1–7) in rat carotid (E max = −0.11 ± 0.013 g) over the precontraction induced by phenylephrine (0.51 ± 0.029 g, n = 9). This relaxant response was completely restored by endothelium removal (over again, this result required that the characterization of the mediators of angiotensin-(1–7)-induced relaxation was performed in endothelium-denuded carotid rings from diabetic rats, as well as from control rats, to compare the responses). In endothelium-denuded carotid rings from nontreated diabetic rats, the relaxant response evoked by angiotensin-(1–7) was completely inhibited by A779, hydroxocobalamin (Figures 2(b), 2(d), and 2(g)), or L-NNA (E max = −0.01 ± 0.002 g, n = 9) (P < 0.001, one-way ANOVA, Bonferroni post-hoc) but not altered by L-NPA (E max = −0.12 ± 0.009 g, n = 9) or 1400 W (E max = −0.10 ± 0.007 g, n = 9) (P > 0.05, one-way ANOVA, Bonferroni post-hoc). The magnitude of phenylephrine-induced precontraction in nontreated diabetic rat carotid was not altered by endothelium removal (0.48 ± 0.035 g, n = 9) or the pretreatment with A779 (0.49 ± 0.025 g, n = 9), hydroxocobalamin (0.50 ± 0.041 g, n = 9), L-NNA (0.47 ± 0.023 g, n = 9), L-NPA (0.49 ± 0.032 g, n = 9), or 1400 W (0.51 ± 0.039 g, n = 9) (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
In endothelium-denuded carotid rings from nontreated control or diabetic rats, the maximum relaxation induced by angiotensin-(1–7) was lesser than that one induced by angiotensin II (Figures 2(a), 2(b), and 2(e)). However, the maximum relaxation induced by angiotensin II in losartan pretreated endothelium-intact carotid rings from nontreated control or diabetic rats had the same extent as the relaxation evoked by angiotensin-(1–7) in nonpretreated endothelium-intact carotid rings from nontreated control rat (Figure 3).
Although the relaxation induced by angiotensin-(1–7) has not been altered by losartan, apocynin, or tiron in endothelium-intact carotid rings from nontreated control rats, this response was completely restored by losartan, apocynin, or tiron in endothelium-intact carotid rings from nontreated diabetic rat. However, PEG-catalase, DCPIB, or the chronic treatment with angiotensin-(1–7) did not alter the relaxation induced by angiotensin-(1–7) in endothelium-intact carotid rings from nontreated control or diabetic rats. Similarly to the effects of losartan, apocynin, or tiron, the chronic treatment with angiotensin-(1–7) completely restored the relaxation induced by angiotensin-(1–7) in endothelium-intact carotid rings from diabetic rats (Figures 2(e), 2(f), 2(h), 3(b), and 3(d)). In control rat carotid, the magnitude of phenylephrine-induced precontraction was not altered by losartan (0.27 ± 0.022 g, n = 9), apocinin (0.26 ± 0.023 g, n = 9), tiron (0.29 ± 0.027 g, n = 9), PEG-catalase (0.31 ± 0.028 g, n = 9), angiotensin-(1–7) (0.30 ± 0.027 g, n = 9), or DCPIB (0.29 ± 0.021 g, n = 9). Also, in diabetic rat carotid, phenylephrine-induced precontraction was not altered by losartan (0.48 ± 0.033 g, n = 9), apocinin (0.51 ± 0.042 g, n = 9), tiron (0.50 ± 0.044 g, n = 9), PEG-catalase (0.49 ± 0.036 g, n = 9), angiotensin-(1–7) (0.47 ± 0.035 g, n = 9), or DCPIB (0.52 ± 0.039 g, n = 9), when compared to nonpretreated diabetic rat carotid (P > 0.05, one-way ANOVA, Bonferroni post-hoc).
3.3. Levels of O2 −
The FI emitted by DHE-loaded endothelial cells from carotid arteries of nontreated diabetic rats was higher than the FI emitted by control samples. Tiron, apocynin, losartan, or the chronic treatment with angiotensin-(1–7) reduced the FI emitted by DHE-loaded samples from diabetic rats to the control levels obtained in the presence of these chemicals. DCPIB did not alter the FI emitted by DHE-loaded endothelial cells from carotid arteries of nontreated control (FI = 9,758.32 ± 561.29 U, n = 5) or diabetic (FI = 35,094.07 ± 915.43 U, n = 5) rats. The antioxidant effect produced by the chronic treatment with angiotensin-(1–7) in DHE-loaded samples from both control and diabetic rats was completely inhibited by the chronic treatment with A779 (Figure 4).
Figure 4.
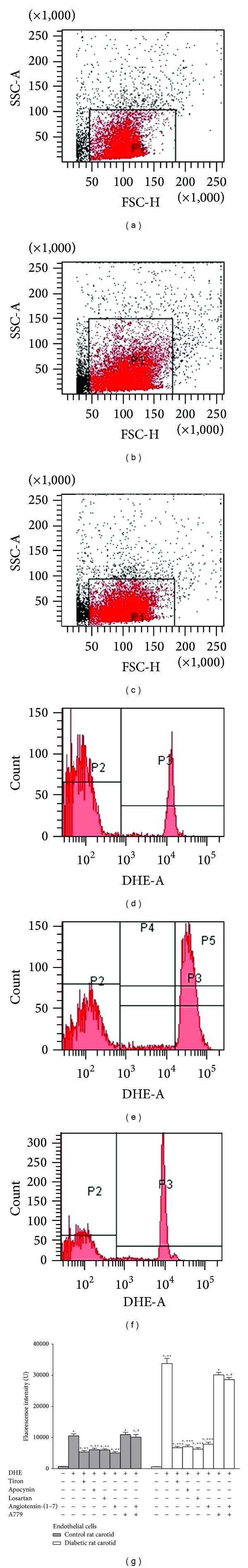
O2 − levels in carotid endothelial cells from control or diabetic rats. Dot plots show the gates of carotid endothelial cells from nontreated control rats (a) or from nontreated (b) or angiotensin-(1–7)-treated (c) diabetic rats. Histograms show the fluorescence emitted by DHE-loaded carotid endothelial cells from nontreated control rats (d) or from nontreated (e) or angiotensin-(1–7)-treated diabetic rats (f). Effects of the in vitro pretreatment with tiron, apocynin, or losartan or the chronic (in vivo) treatment with angiotensin-(1–7) combined or not with A779 on the fluorescence emitted by DHE-loaded endothelial cells samples (g). The values are significantly different (P < 0.01; n = 5) from the respective blank samples from nontreated rats (∗), from nonpretreated DHE-loaded endothelial cells samples from nontreated control (∗∗) or diabetic (∗∗∗) rats, or from the respective DHE-loaded endothelial cells samples from angiotensin-(1–7)-treated rats (#).
3.4. Levels of H2O2
The FI emitted by CDCF-DA-loaded endothelial cells from carotid arteries of nontreated diabetic rats was higher than the FI emitted by control samples. In CDCF-DA-loaded endothelial cells from carotid arteries of nontreated diabetic rats, PEG-catalase, tiron, apocynin, losartan, or the chronic treatment with angiotensin-(1–7) reduced the FI to the control levels obtained in the presence of PEG-catalase, apocynin, or losartan. DCPIB did not alter the FI emitted by CDCF-DA-loaded endothelial cells from carotid arteries of nontreated control (FI = 10,322.04 ±517.81 U, n = 5) or diabetic (FI = 21,085.01 ±639.43 U, n = 5) rats. The antioxidant effect produced by the chronic treatment with angiotensin-(1–7) in CDCF-DA-loaded samples from diabetic rats was completely inhibited by the chronic treatment with A779 (Figure 5).
Figure 5.
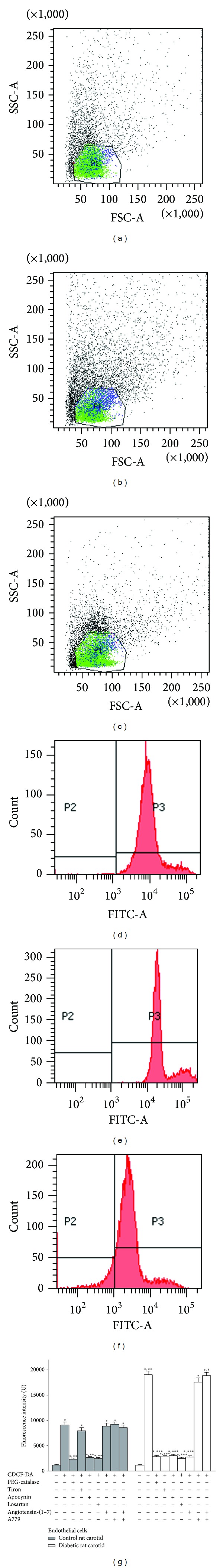
H2O2 levels in carotid endothelial cells from control or diabetic rats. Dot plots show the gates of carotid endothelial cells from nontreated control rats (a) or from nontreated (b) or angiotensin-(1–7)-treated diabetic rats (c). Histograms show the fluorescence emitted by CDCF-DA-loaded carotid endothelial cells from nontreated control rats (d) or from nontreated (e) or angiotensin-(1–7)-treated diabetic rats (f). Effects of the in vitro pretreatment with PEG-catalase, tiron, apocynin, or losartan or the chronic (in vivo) treatment with angiotensin-(1–7) combined or not with A779 on the fluorescence emitted by CDCF-DA-loaded endothelial cells samples (g). The values are significantly different (P < 0.01; n = 5) from the respective blank samples from nontreated rats (∗), from nonpretreated CDCF-DA-loaded endothelial cells samples from nontreated control (∗∗) or diabetic (∗∗∗) rats, or from CDCF-DA-loaded endothelial cells samples from angiotensin-(1–7)-treated diabetic rats (#).
3.5. ACE2, Angiotensin-(1–7), and Mas-Receptors Expression
In the muscular layer from nontreated diabetic rat carotid, ACE2 staining (39.05 ± 3.42%) was not altered, while angiotensin-(1–7) (2.09 ± 1.22%) or Mas receptors (17.63 ± 1.62%) staining was reduced when compared to nontreated control rat carotid (ACE2 = 47.28 ± 4.51%; angiotensin-(1–7) = 81.04 ± 6.29%; Mas = 92.37 ± 7.24%). Mas receptors staining was also detected in the endothelium from nontreated control rat carotid (64.57 ± 6.92%), but it was reduced in the endothelium from nontreated diabetic rat carotid (38.01 ± 2.79%) (P < 0.001, n = 5, one-way ANOVA, Bonferroni post-hoc) (Figure 6).
Figure 6.
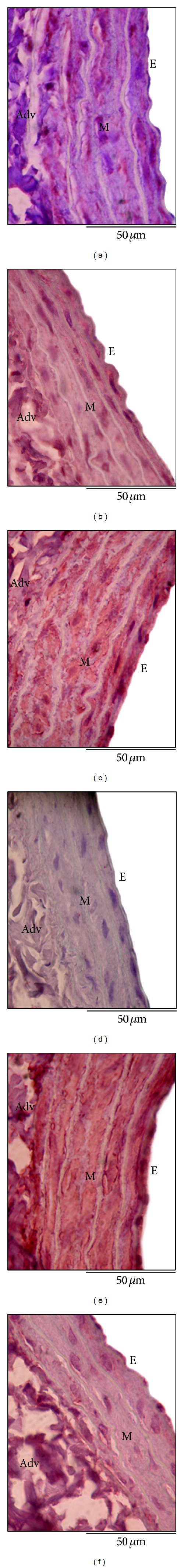
ACE2, angiotensin-(1–7) and Mas receptors staining in carotid arteries from nontreated control or diabetic rats. ACE2 expression in nontreated control (a) or diabetic (b) rat carotid, angiotensin-(1–7) levels in nontreated control (c) or diabetic (d) rat carotid, and Mas receptors expression in nontreated control (e) or diabetic (f) rat carotid. E: endothelium; M: media; Adv: adventitia. The immunostaining is denoted in red (magnification: 100x).
The chronic treatment with angiotensin-(1–7) did not alter ACE2 expression in the muscular layer from control (42.37 ± 3.91%) or diabetic rat carotid (41.25 ± 4.14%). The chronic treatment with DX600 reduced angiotensin-(1–7) staining in the muscular layer from carotid arteries of control rats (4.01 ± 3.17%) to the levels obtained in carotid arteries from nontreated diabetic rats. On the other hand, the chronic treatment with DX600 did not alter angiotensin-(1–7) staining in the muscular layer from carotid arteries of diabetic rats (2.85 ± 1.36%) when compared to nontreated diabetic rats (P < 0.001, n = 5, one-way ANOVA, Bonferroni post-hoc).
The chronic treatment with angiotensin-(1–7) did not alter the expression of Mas receptors neither in the endothelium from control (59.87 ± 5.76%) or diabetic (34.21 ± 3.07%) rat carotid nor in the muscular layer from control (87.23 ± 8.19%) or diabetic (14.95 ± 1.24%) rat carotid when compared to the carotid arteries from the respective nontreated groups (P < 0.001, n = 5, one-way ANOVA, Bonferroni post-hoc).
3.6. Blood Pressure, Carotid Blood Flow, and Carotid Resistance
Blood pressure was not different between nontreated or treated control or diabetic rats. In nontreated diabetic rats, carotid blood flow was reduced and carotid resistance was increased when compared to nontreated control rats. The chronic treatment of control rats with DX600 significantly reduced carotid blood flow and increased carotid resistance to the levels obtained in nontreated diabetic rats. The same effect was observed in carotid blood flow and carotid resistance from control rats that were treated with A779. In diabetic rats, carotid blood flow or carotid resistance was not altered by the chronic treatment with DX600 or A779. The chronic treatment with angiotensin-(1–7) did not alter carotid blood flow or carotid resistance from control rats, but it restored carotid blood flow and carotid resistance from diabetic rats. A779 inhibited the effect of the chronic treatment with angiotensin-(1–7) on carotid blood flow and carotid resistance from diabetic rats (Table 3).
Table 3.
Blood pressure (BP), carotid blood flow (F), and carotid resistance (R) from control or diabetic rats.
| Rats | Cardiovascular parameters | ||
|---|---|---|---|
| BP (mmHg) | F (mL/min) | R (U) | |
| Nontreated control | 81.7 ± 5.4 | 6.11 ± 0.48 | 14.1 ± 2.3 |
| Nontreated diabetic | 99.2 ± 8.1 | 2.37 ± 0.21* | 44.6 ± 3.2* |
| DX600-treated control | 76.4 ± 6.2 | 2.09 ± 0.17* | 41.9 ± 3.5* |
| DX600-treated diabetic | 89.2 ± 7.5 | 2.13 ± 0.19 | 45.2 ± 4.2 |
| Angiotensin-(1–7)-treated control | 79.5 ± 6.1 | 5.76 ± 0.32 | 13.2 ± 2.9 |
| Angiotensin-(1–7)-treated diabetic | 97.0 ± 9.1 | 6.29 ± 0.51# | 15.0 ± 2.1# |
| A779-treated control | 81.1 ± 7.4 | 1.99 ± 0.14* | 40.2 ± 4.3* |
| A779-treated diabetic | 98.7 ± 8.3 | 2.44 ± 0.23 | 42.4 ± 4.1 |
| Angiotensin-(1–7) + A779-treated control | 75.6 ± 7.0 | 2.05 ± 0.19∗,† | 38.2 ± 3.8∗,† |
| Angiotensin-(1–7) + A779-treated diabetic | 99.5 ± 9.2 | 1.97 ± 0.11† | 46.3 ± 4.7† |
The values are significantly different (P < 0.01; n = 5) from nontreated control rats (∗), nontreated diabetic rats (#), or the respective rats treated with angiotensin-(1–7) (†).
4. Discussion
The present study has three major new findings. The first one shows that type I-diabetes impairs the functionality of ACE2-angiotensin-(1–7)-Mas axis in rat carotid by a mechanism that involves the endothelial AT1-activated NAD(P)H oxidase-driven generation of O2 − and H2O2: while H2O2 derived from O2 − dismutation inhibits ACE2 activity in generating angiotensin-(1–7) seemingly by activating I Cl,SWELL, O2 − inhibits the nitrergic vasorelaxant effect evoked by angiotensin-(1–7) upon Mas receptors activation. The second new finding shows that the impaired functionality of ACE2-angiotensin-(1–7)-Mas axis increases carotid resistance, which reduces carotid blood flow during type I-diabetes, highlighting the importance of the vasoprotective role assigned to the activation of this axis in both healthy and diseased conditions. Finally, the third new finding shows that the chronic treatment with angiotensin-(1–7) restores the functionality of carotid ACE2-angiotensin-(1–7)-Mas axis, what contributes to restore the carotid resistance and blood flow in diabetic conditions, by a mechanism that involves Mas-mediated antioxidant effects against the endothelial generation of O2 − and H2O2 catalyzed by AT1-activated NAD(P)H oxidase.
The functionality of vascular ACE2 (i.e., the activity of vascular ACE2 in generating angiotensin-(1–7)) was studied by relaxant curves induced by angiotensin II in rat carotid. This approach is supported by the evidences that angiotensin II-evoked relaxation in rat carotid is partially mediated by Mas receptors [19], seemingly upon angiotensin II hydrolysis into angiotensin-(1–7) and that the only enzyme able to convert angiotensin II into angiotensin-(1–7) in vascular tissues is ACE2 [17, 25]. Our findings show that angiotensin II-evoked relaxation curve in rat carotid is characterized by a biphasic response consisting in a residual AT1-mediated contraction induced by nanomolar concentrations of angiotensin II (about 0.15 g), followed by an endothelium-independent relaxation induced by micromolar concentrations of this peptide (about −0.35 g). Our findings also show that DX600 completely abolishes the relaxation induced by angiotensin II without changing the residual contraction mediated by AT1-receptors, which allows us to conclude that the relaxant response induced by angiotensin II in rat carotid occurs upon the ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7). In turn, we found that losartan or A779 partially inhibits the relaxation triggered by angiotensin II: while losartan abrogates the complete relaxation of the residual contraction evoked by angiotensin II by inhibiting this contractile response, A779 does not alter neither the residual contraction evoked by angiotensin II nor the relaxation of this contractile response but abrogates the relaxation beyond the relaxant response that counteracts the residual contraction induced by angiotensin II. Thus, these findings allow us to divide the ACE2-dependent relaxation induced by angiotensin II in rat carotid into two components: (1) the losartan-sensitive relaxant response (about −0.15 g), which occurs upon the dissociation of angiotensin II from AT1-receptors and the subsequent ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7) and (2) the A779-sensitive relaxant response (about −0.2 g), which depends on the activation of Mas receptors by angiotensin-(1–7) generated from angiotensin II. Indeed, the vascular generation of angiotensin-(1–7) from angiotensin II is catalyzed by ACE2, which hydrolyzes only angiotensin II with high catalytic efficiency among the angiotensin peptides [17, 25]. Moreover, Tirapelli et al. [19] had already shown that Mas receptors partially mediate angiotensin II-induced relaxation in rat carotid.
Our results show that, similar to angiotensin II, angiotensin-(1–7) also evokes an endothelium-independent relaxation (about −0.2 g), which is completely mediated by Mas-receptors, NO, and endothelial NOS (eNOS) in rat carotid. In agreement with these findings, we also show that Mas receptors are mostly expressed in the muscular layer from rat carotid, supporting our functional evidence concerning the Mas-mediated endothelium-independent relaxation evoked by angiotensin-(1–7) in this vessel. Indeed, the vasorelaxation mediated by Mas receptors has mostly been described as a NOS-dependent nitrergic mechanism [19, 41–44], since the activation of these receptors by angiotensin-(1–7) leads to the phosphorylation of eNOS [45], which is also expressed in smooth muscle layer from rat carotid [9]. Another important aspect from our results is that the relaxant response induced by angiotensin-(1–7) had the same extent of the Mas-mediated (i.e., the A779-sensitive) relaxation evoked by angiotensin II in rat carotid, confirming that part of the relaxation induced by angiotensin II in this vessel is mediated by angiotensin-(1–7) upon the ACE2-catalyzed hydrolysis of the octapeptide.
Interestingly, our findings show that type I-diabetes inhibits the relaxant response induced by angiotensin II but only attenuates the Mas-mediated endothelium-independent relaxation evoked by angiotensin-(1–7) in rat carotid. Moreover, we observed that the residual relaxation evoked by angiotensin-(1–7) in diabetic rat carotid is mediated by NO and eNOS. Although the muscular expression of eNOS in this vessel is significantly reduced [9], recent evidences have shown that the acute stimulation with nano- to micromolar concentrations of angiotensin-(1–7) increases eNOS expression in rat cerebral ischemic tissues [46], spontaneously hypertensive rat ventricles [47], and human circulating fibrocytes [48]. These findings allow us to suggest that the generation of NO from muscular eNOS in diabetic rat carotid during angiotensin-(1–7)-evoked relaxation may be related to one of the following reasons: (1) the residual expression of eNOS at the muscular layer form diabetic rat carotid is enough to generate the NO levels that mediate this relaxant response or (2) the acute stimulation with nano- to micromolar concentrations of angiotensin-(1–7) may increase the expression of eNOS in this vessel, what would support the NO generation during the relaxation.
According to the present results, the inhibition of angiotensin II-induced relaxation in diabetic rat carotid, taken together with the findings concerning the unchanged muscular expression of ACE2 and the reduced muscular levels of angiotensin-(1–7) in this vessel, allows us to strongly suggest that diabetes impairs the ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7), but not the expression of the carboxypeptidase. This finding is confirmed when we show that the endogenous generation of angiotensin-(1–7) in rat carotid, which is catalyzed by ACE2, is lost in diabetic rats, since the chronic treatment with DX600 reduced angiotensin-(1–7) levels only in carotid arteries from normoglycaemic animals. Moreover, our functional results show that the diabetes-elicited inhibition of ACE2-catalyzed hydrolysis of angiotensin II in rat carotid (and thus, the inhibition of angiotensin II-induced relaxation) involves endothelial pathways completely mediated by AT1-receptors, NAD(P)H oxidase, and O2 − but partially mediated by H2O2. In turn, diabetes-elicited impairment of angiotensin-(1–7)-evoked relaxation involves endothelial mechanisms that are only mediated by AT1-receptors, NAD(P)H oxidase, and O2 −, but not by H2O2. In addition to these findings, we also observed that the Mas-mediated (A779-sensitive) relaxation induced by angiotensin II in PEG-catalase pretreated carotid rings from diabetic rat had the same extent as the relaxation induced by angiotensin-(1–7) in diabetic rat carotid (i.e., about −0.1 g). Taken together, these findings allow us to conclude that H2O2 generated from endothelial AT1-activated NAD(P)H oxidase-derived O2 − inhibits the ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7), while AT1-activated NAD(P)H oxidase-derived O2 − impairs the nitrergic transduction pathways underlying Mas receptors activation in diabetic rat carotid. Confirming these data, we found an increase in the levels of H2O2 produced from AT1-activated NAD(P)H oxidase-derived O2 −, whose generation was also increased in endothelial cells from diabetic rat carotid. Indeed, type I-diabetes increases the endothelial generation of O2 − derived from NAD(P)H oxidase in rat carotid [8, 9]. Moreover, the generation of H2O2 from O2 − can occur by the dismutation of O2 − [49]. In turn, upon AT1-receptors activation by angiotensin II in endothelial cells, Nox1 subunit from vascular NAD(P)H oxidase can generate O2 −, which is latter converted to H2O2 [50, 51]. Finally, type I-diabetes enhances the endothelial expression of AT1-receptors and the reactive oxygen species generation mediated by these receptors in rat carotid, what explains the selective activation of endothelial NAD(P)H oxidase by AT1-receptors in this vessel [8].
The cleavage of angiotensin II by somatic ACE2 can be reduced by increasing the physiological extracellular levels of Cl− [30], due to the presence of a regulatory Cl− binding site at the extracellular region of the carboxypeptidase [52]. Added to these findings, H2O2 generated from NAD(P)H oxidase-derived O2 − can activate I Cl,SWELL in vascular smooth muscle cells, leading to the efflux of Cl− and thus increasing Cl− extracellular levels [31, 32]. Taken together, these findings prompted us to investigate the role of I Cl,SWELL in the modulation of ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7) during the relaxation evoked by angiotensin II in diabetic rat carotid. Interestingly, we observed that the inhibition of I Cl,SWELL partially restored the relaxation evoked by angiotensin II, but not by angiotensin-(1–7), in diabetic rat carotid, similarly to the effect produced by the removal of H2O2. In addition, by showing that DCPIB did not alter O2 − or H2O2 levels in endothelial cells from diabetic rat carotid, we excluded the hypothesis that the inhibition of I Cl,SWELL could lead to the scavenging of reactive oxygen species that modulate angiotensin II-evoked relaxation in this vessel. Thus, our data, supported by the findings from Rushworth et al. [30], Ren et al. [31], Matsuda et al. [32], and Towler et al. [52], strongly suggest that H2O2 generated from endothelial AT1-activated NAD(P)H oxidase-derived O2 − inhibits the ACE2-catalyzed hydrolysis of angiotensin II into angiotensin-(1–7) seemingly by activating I Cl,SWELL in smooth muscle cells from diabetic rat carotid. In turn, O2 − derived from endothelial AT1-activated NAD(P)H oxidase can impair nitrergic signaling pathways during type I-diabetic conditions, like the Mas-mediated relaxation evoked by angiotensin-(1–7) in rat carotid, by converting NO into peroxynitrite (ONOO−) or by inducing to NOS uncoupling [53].
One of the most important aspects from our results is that the chronic treatment with angiotensin-(1–7) restores the functionality of ACE2-Angiotensin-(1–7)-Mas axis (i.e., both angiotensin II- and angiotensin-(1–7)-evoked relaxation) in diabetic rat carotid. According to our findings, this protective effect results from a Mas-mediated mechanism that inhibits the generation of O2 − and H2O2 catalyzed by AT1-activated NAD(P)H oxidase in endothelial cells from diabetic rat carotid. Moreover, we excluded the hypothesis that the restoration of ACE2-Angiotensin-(1–7)-Mas axis functionality in carotid arteries from angiotensin-(1–7)-treated diabetic rats could be resultant from a translational effect of enhancing ACE2 or Mas receptors expression, since they remained unaltered after the treatment. Thus, our results can be explained by the findings from Sampaio et al. [21], who described that the activation of Mas receptors by angiotensin-(1–7) inhibits the AT1-mediated NAD(P)H oxidase assembly and the subsequent reactive oxygen species generation upon angiotensin II stimulation in endothelial cells.
Interestingly, even if type I-diabetes subregulates the endothelial expression of Mas receptors in rat carotid, the activation of this residual population of receptors during the chronic treatment with Mas agonists can trigger antioxidant effects that are enough to overcome the impairment of the local ACE2-angiotensin-(1–7)-Mas axis functionality. These findings have important implications to the therapeutic efficacy of Mas agonists on attenuating vascular dysfunctions and complications, since we showed that the activation of Mas receptors triggers a positive feedback over the ACE2-angiotensin-(1–7)-Mas axis when the functionality of this vasoprotective axis is impaired, even during diseases that subregulate the vascular expression of these receptors, such as type I-diabetes. Indeed, the chronic treatment of type I-diabetic rats with angiotensin-(1–7) or the nonpeptide Mas agonist AVE-0991 was efficacious in restoring the contractile and relaxation functions of mesenteric bed, carotid, and renal arteries [20].
Finally, we found that the chronic treatment with DX600 or A779 reduced carotid blood flow by increasing carotid resistance in normoglycaemic rats to the levels obtained with nontreated diabetic rats. On the other hand, carotid resistance and blood flow in diabetic rats were not altered after the chronic treatment with DX600 or A779. Moreover, while the chronic treatment with angiotensin-(1–7) did not alter carotid resistance and blood flow in normoglycaemic rats, it restored these parameters in diabetic rats, and this protective effect of angiotensin-(1–7) was inhibited when angiotensin-(1–7) was coadministrated with A779. Taken together, these findings point the vasoprotective role assigned to the ACE2-angiotensin-(1–7)-Mas axis functionality in maintaining carotid resistance and blood flow and confirm that the impairment of the functionality of this axis damages carotid function in diabetic conditions. Considering that carotid blood flow rate is determined, in a higher extent, by the resistance of smaller proximal arteries and arterioles (such as internal and external carotid arteries) than the carotid resistance, our findings allow us to suggest that the impairment of ACE2-angiotensin-(1–7)-Mas axis functionality reaches not only carotid artery but also the proximal resistance vessels.
5. Conclusions
This is the first study that shows that the endothelial AT1-activated NAD(P)H oxidase-driven generation of O2 − and H2O2 in carotid arteries from type I-diabetic rats impairs the functionality of the local vasoprotective ACE2-angiotensin-(1–7)-Mas axis, which in turn impairs carotid blood flow. In this mechanism, H2O2 derived from O2 − dismutation inhibits ACE2 activity in generating angiotensin-(1–7) seemingly by activating I Cl,SWELL, while O2 − inhibits the nitrergic vasorelaxant effect evoked by angiotensin-(1–7) upon Mas receptors activation. Furthermore, we originally showed that the chronic treatment of diabetic rats with angiotensin-(1–7) restores the functionality of carotid ACE2-angiotensin-(1–7)-Mas axis by triggering a positive feedback on this axis, played by a residual population of endothelial Mas-receptors that blunts the endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species in rat carotid. Finally, we found that the Mas-mediated antioxidant effects evoked by the chronic treatment with angiotensin-(1–7) also restores carotid resistance and blood flow in diabetic rats, pointing the important contribution of the ACE2-angiotensin-(1–7)-Mas axis in maintaining carotid function (Figure 7). These findings have relevant implications to support the therapeutic efficacy of Mas agonists on preventing or attenuating diabetic endothelial dysfunction in carotid arteries and its underlying cerebrovascular complications by enhancing the vasoprotective role of the local ACE2-angiotensin-(1–7)-Mas axis.
Figure 7.
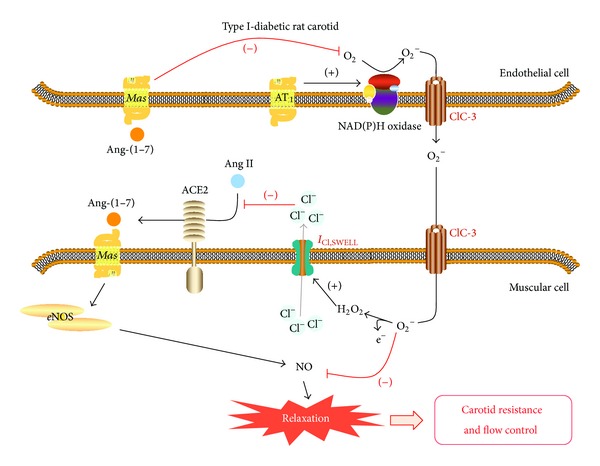
Conclusive graphical abstract. Endothelial AT1-activated NAD(P)H oxidase-driven generation of O2 − and H2O2 in carotid arteries from type I-diabetic rats impairs the functionality of the local vasoprotective ACE2-angiotensin-(1–7)-Mas axis, which in turn impairs carotid blood flow. In this mechanism, H2O2 derived from O2 − dismutation inhibits ACE2 activity in generating angiotensin-(1–7) by activating I Cl,SWELL, while O2 − inhibits the nitrergic vasorelaxant effect evoked by angiotensin-(1–7) upon Mas receptors activation. The chronic treatment of diabetic rats with angiotensin-(1–7) restores the functionality of carotid ACE2-angiotensin-(1–7)-Mas axis by triggering a positive feedback on this axis, played by a residual population of endothelial Mas-receptors that blunts the endothelial AT1-activated NAD(P)H oxidase-driven generation of reactive oxygen species in rat carotid. Mas-mediated antioxidant effects evoked by the chronic treatment with angiotensin-(1–7) also restores carotid resistance and blood flow in diabetic rats, pointing the important contribution of the ACE2-angiotensin-(1–7)-Mas axis in maintaining carotid function.
Acknowledgments
The authors thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Protocol no. 2009/01005-0, no. 2012/00640-7, and no. 2012/09019-3) for the financial support. The authors also thank Professor Dr. Leandra Náira Zambelli Ramalho and Professor Dr. Fernando Morgan de Aguiar Corrêa from the Faculdade de Medicina de Ribeirão Preto/Universidade de São Paulo, for their assistance in the immunohistochemical and blood pressure experiments, respectively.
Conflict of Interests
The authors declare there is no conflict of interests regarding the publication of this paper.
References
- 1.Glowinska-Olszewska B, Hyniewicz A, Jeznach M, et al. Relationship between circulating endothelial progenitor cells and endothelial dysfunction in children with type 1 diabetes: a novel paradigm of early atherosclerosis in high-risk young patients. European Journal of Endocrinology. 2013;168:153–161. doi: 10.1530/EJE-12-0857. [DOI] [PubMed] [Google Scholar]
- 2.Dalla Pozza R, Beyerlein A, Thilmany C, et al. The effect of cardiovascular risk factors on the longitudinal evolution of the carotid intima medial thickness in children with type 1 diabetes mellitus. Cardiovascular Diabetology. 2011;10, article 53 doi: 10.1186/1475-2840-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polak JF, Backlund J-YC, Cleary PA, et al. Progression of carotid artery intima-media thickness during 12 years in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) study. Diabetes. 2011;60(2):607–613. doi: 10.2337/db10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurks R, Eisinger MJ, Goovaerts I, et al. Early endothelial dysfunction in young type 1 diabetics. European Journal of Vascular and Endovascular Surgery. 2009;37(5):611–615. doi: 10.1016/j.ejvs.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Yousif MHM, Benter IF, Akhtar S. The role of tyrosine kinase-mediated pathways in diabetes-induced alterations in responsiveness of rat carotid artery. Autonomic and Autacoid Pharmacology. 2005;25(2):69–78. doi: 10.1111/j.1474-8673.2004.00333.x. [DOI] [PubMed] [Google Scholar]
- 6.Yousif MHM, Benter IF, Hares N, Canatan H, Akhtar S. Phosphoinositide 3-kinase mediated signalling contributes to development of diabetes-induced abnormal vascular reactivity of rat carotid artery. Cell Biochemistry and Function. 2006;24(1):13–22. doi: 10.1002/cbf.1278. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y, Vanhoutte PM. Oxidative stress and COX cause hyper-responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. British Journal of Pharmacology. 2008;154(3):639–651. doi: 10.1038/bjp.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pernomian L, Santos Gomes M, Baraldi Araujo Restini C, Naira Zambelli Ramalho L, Renato Tirapelli C, Maria De Oliveira A. The role of reactive oxygen species in the modulation of the contraction induced by angiotensin II in carotid artery from diabetic rat. European Journal of Pharmacology. 2012;678(1–3):15–25. doi: 10.1016/j.ejphar.2011.12.036. [DOI] [PubMed] [Google Scholar]
- 9.Pernomian L, Gomes MS, Corrêa FMA, Restini CBA, Ramalho LNZ, de Oliveira AM. Diabetes confers a vasoprotective role to the neurocompensatory response elicited by carotid balloon injury: consequences on contralateral carotid tone and blood flow. European Journal of Pharmacology. 2013;708:124–138. doi: 10.1016/j.ejphar.2013.02.053. [DOI] [PubMed] [Google Scholar]
- 10.Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Medical Science Monitor. 2005;11(6):RA194–RA205. [PubMed] [Google Scholar]
- 11.Migdalis IN, Gerolimou B, Kozanidou G, Hatzigakis SM, Karmaniolas KD. Effect of fosinopril sodium on early carotid atherosclerosis in diabetic patients with hypertension. Journal of Medicine. 1997;28(5-6):371–380. [PubMed] [Google Scholar]
- 12.Igarashi M, Hirata A, Yamaguchi H, et al. Candesartan inhibits carotid intimal thickening and ameliorates insulin resistance in balloon-injured diabetic rats. Hypertension. 2001;38(6):1255–1259. doi: 10.1161/hy1101.095537. [DOI] [PubMed] [Google Scholar]
- 13.Baguet J-P, Asmar R, Valensi P, Nisse-Durgeat S, Mallion JM. Effects of candesartan cilexetil on carotid remodeling in hypertensive diabetic patients: The MITEC Study. Vascular Health and Risk Management. 2009;5:175–183. doi: 10.2147/vhrm.s3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertension Research. 2009;32(7):533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fressato de Godoy MA, Pernomian L, de Oliveira AM, Rattan S. Biosynthetic pathways and the role of the MAS receptor in the effects of angiotensin-(1-7) in smooth muscle. International Journal of Hypertension. 2012;2012:6 pages. doi: 10.1155/2012/121740.121740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. The Journal of Biological Chemistry. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 17.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. The Journal of Biological Chemistry. 2002;277(17):14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 18.Santos RAS, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirapelli CR, Fukada SY, de Godoy MAF, de Oliveira AM. Analysis of the mechanisms underlying the vasorelaxant action of angiotensin II in the isolated rat carotid. Life Sciences. 2006;78(23):2676–2682. doi: 10.1016/j.lfs.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Benter IF, Yousif MHM, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. American Journal of Physiology—Heart and Circulatory Physiology. 2007;292(1):H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- 21.Sampaio WO, de Castro CH, Santos RAS, Schiffrin EL, Touyz RM. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50(6):1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- 22.Yousif MHM, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacological Research. 2012;66:269–275. doi: 10.1016/j.phrs.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Pernomian L, Gomes MS, de Oliveira AM. Balloon catheter injury abolishes phenylephrine-induced relaxation in the rat contralateral carotid. British Journal of Pharmacology. 2011;163(4):770–781. doi: 10.1111/j.1476-5381.2011.01275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pernomian L, Gomes MS, Restini CBA, Pupo AS, de Oliveira AM. Cross-talk with β2-adrenoceptors enhances ligand affinity properties from endothelial alpha1D-adrenoceptors that mediates carotid relaxation. Journal of Pharmacology and Pharmacotherapeutics. 2013;65:1337–1346. doi: 10.1111/jphp.12105. [DOI] [PubMed] [Google Scholar]
- 25.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochemical Journal. 2004;383(1):45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedersen KB, Sriramula S, Chhabra KH, Xia H, Lazartigues E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2011;301(5):R1293–R1299. doi: 10.1152/ajpregu.00339.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson MJ, Barnes JC, Drew GM, et al. Pharmacological profile of GR117289 in vitro: a novel, potent and specific non-peptide angiotensin AT1 receptor antagonist. British Journal of Pharmacology. 1992;107(4):1173–1180. doi: 10.1111/j.1476-5381.1992.tb13425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Decher N, Lang HJ, Nilius B, Brüggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of I[CI,swell] and prevents swelling-induced shortening of guinea-pig atrial action potential duration. British Journal of Pharmacology. 2001;134(7):1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng W, Baki L, Baumgarten CM. Endothelin signalling regulates volume-sensitive Cl- current via NADPH oxidase and mitochondrial reactive oxygen species. Continuing Education in Anaesthesia, Critical Care and Pain. 2010;88(1):93–100. doi: 10.1093/cvr/cvq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rushworth CA, Guy JL, Turner AJ. Residues affecting the chloride regulation and substrate selectivity of the angiotensin-converting enzymes (ACE and ACE2) identified by site-directed mutagenesis. FEBS Journal. 2008;275(23):6033–6042. doi: 10.1111/j.1742-4658.2008.06733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren Z, Raucci FJ, Jr., Browe DM, Baumgarten CM. Regulation of swelling-activated Cl- current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovascular Research. 2008;77(1):73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS. Activation of swelling-activated chloride current by tumor necrosis factor-α requires ClC-3-dependent endosomal reactive oxygen production. The Journal of Biological Chemistry. 2010;285(30):22864–22873. doi: 10.1074/jbc.M109.099838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonaventura D, de Lima RG, Vercesi JA, da Silva RS, Bendhack LM. Comparison of the mechanisms underlying the relaxation induced by two nitric oxide donors: Sodium nitroprusside and a new ruthenium complex. Vascular Pharmacology. 2007;46(3):215–222. doi: 10.1016/j.vph.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Bonaventura D, Lunardi CN, Rodrigues GJ, Neto MA, Bendhack LM. A novel mechanism of vascular relaxation induced by sodium nitroprusside in the isolated rat aorta. Nitric Oxide—Biology and Chemistry. 2008;18(4):287–295. doi: 10.1016/j.niox.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Du J, Daniels DH, Asbury C, et al. Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. The Journal of Biological Chemistry. 2006;281(49):37416–37426. doi: 10.1074/jbc.M605063200. [DOI] [PubMed] [Google Scholar]
- 36.Robertson SJ, Kania KD, Hladky SB, Barrand MA. P-glycoprotein expression in immortalised rat brain endothelial cells: comparisons following exogenously applied hydrogen peroxide and after hypoxia-reoxygenation. Journal of Neurochemistry. 2009;111(1):132–141. doi: 10.1111/j.1471-4159.2009.06306.x. [DOI] [PubMed] [Google Scholar]
- 37.Gao H, Yallampalli U, Yallampalli C. Protein restriction to pregnant rats increases the plasma levels of angiotensin II and expression of angiotensin II receptors in uterine arteries. Biology of Reproduction. 2012;86(3, article 68) doi: 10.1095/biolreprod.111.095844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marshall AC, Shaltout HA, Nautiyal M, Rose JC, Chappell MC, Diz DI. Fetal betamethasone exposure attenuates angiotensin-(1-7)-Mas receptor expression in the dorsal medulla of adult sheep. Peptides. 2013;44:25–31. doi: 10.1016/j.peptides.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giani JF, Miquet JG, Muñoz MC, et al. Upregulation of the angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas receptor axis in the heart and the kidney of growth hormone receptor knock-out mice. Growth Hormone & IGF Research. 2012;22:224–233. doi: 10.1016/j.ghir.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Accorsi-Mendonça D, Corrêa FMA, Paiva TB, de Souza HP, Laurindo FRM, de Oliveira AM. The balloon catheter induces an increase in contralateral carotid artery reactivity to angiotensin II and phenylephrine. British Journal of Pharmacology. 2004;142(1):79–88. doi: 10.1038/sj.bjp.0705732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1-7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27(3):523–528. doi: 10.1161/01.hyp.27.3.523. [DOI] [PubMed] [Google Scholar]
- 42.Ren Y, Garvin JL, Carretero OA. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension. 2002;39(3):799–802. doi: 10.1161/hy0302.104673. [DOI] [PubMed] [Google Scholar]
- 43.Zhi J-M, Chen R-F, Wang J, Jiao X-Y, Zhao R-R. Comparative studies of vasodilating effects of angiotensin-(1-7) on the different vessels. Acta Physiologica Sinica. 2004;56(6):730–734. [PubMed] [Google Scholar]
- 44.Lemos VS, Silva DMR, Walther T, Alenina N, Bader M, Santos RAS. The endothelium-dependent vasodilator effect of the nonpeptide Ang(1-7) mimic AVE 0991 is abolished in the aorta of Mas-knockout mice. Journal of Cardiovascular Pharmacology. 2005;46(3):274–279. doi: 10.1097/01.fjc.0000175237.41573.63. [DOI] [PubMed] [Google Scholar]
- 45.Sampaio WO, Dos Santos RAS, Faria-Silva R, Da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 2007;49(1):185–192. doi: 10.1161/01.HYP.0000251865.35728.2f. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Lu J, Shi J, et al. Central administration of angiotensin-(1-7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats. Neuropeptides. 2008;42(5-6):593–600. doi: 10.1016/j.npep.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Costa MA, Lopez Verrilli MA, Gomez KA, et al. Angiotensin-(1-7) upregulates cardiac nitric oxide synthase in spontaneously hypertensive rats. American Journal of Physiology—Heart and Circulatory Physiology. 2010;299(4):H1205–H1211. doi: 10.1152/ajpheart.00850.2009. [DOI] [PubMed] [Google Scholar]
- 48.Wang K, Hu X, Du C, Tu S, Zhang F, Xie X. Angiotensin-(1-7) suppresses the number and function of the circulating fibrocytes by upregulating endothelial nitric oxide synthase expression. Molecular and Cellular Biochemistry. 2012;365(1-2):19–27. doi: 10.1007/s11010-012-1223-0. [DOI] [PubMed] [Google Scholar]
- 49.Mori H, Iwahashi H. Superoxide dismutase enhanced the formation of hydroxyl radicals in a reaction mixture containing xanthone under UVA irradiation. Bioscience, Biotechnology and Biochemistry. 2007;71(12):3014–3018. doi: 10.1271/bbb.70412. [DOI] [PubMed] [Google Scholar]
- 50.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HHHW, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radical Biology and Medicine. 2008;45(9):1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honjo T, Otsui K, Shiraki R, et al. Essential role of NOXA1 in generation of reactive oxygen species induced by oxidized low-density lipoprotein in human vascular endothelial cells. Endothelium. 2008;15(3):137–141. doi: 10.1080/10623320802125433. [DOI] [PubMed] [Google Scholar]
- 52.Towler P, Staker B, Prasad SG, et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. The Journal of Biological Chemistry. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szabo C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. British Journal of Pharmacology. 2009;156(5):713–727. doi: 10.1111/j.1476-5381.2008.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]