

Abstract
A set of aporphine analogs related to nantenine was evaluated for antagonist activity at 5-HT2A and α1A adrenergic receptors.
With regards to 5-HT2A receptor antagonism, a C2 allyl group is detrimental to activity. The chiral center of nantenine is not important for 5-HT2A antagonist activity, however the N6 nitrogen atom is a critical feature for 5-HT2A antagonism.
Compound 12b was the most potent 5-HT2A aporphine antagonist identified in this study and has similar potency to previously identified aporphine antagonists 2 and 3. The ring A and N6 modifications examined were detrimental to α1A antagonism. A slight eutomeric preference for the R enantiomer of nantenine was observed in relation to α1A antagonism.
Keywords: Aporphine, Nantenine, 5-HT2A, α1A, Antagonist, Structure-activity relationship (SAR)
The tetracyclic aporphine template is a privileged scaffold that is endowed with several biological activities.1–8 As central nervous system (CNS) receptor ligands, aporphines have been found to possess high affinity for a number of dopamine receptors (predominantly D1 and D2),9–12 serotonin (5-HT) receptors13–15 and α-adrenergic receptors.6,16 Furthermore, aporphines are known with both agonist and antagonist activity at neuroreceptor sites. The continued exploration of the aporphine template over the last few decades has been driven to some extent by the opportunity/promise for discovery of new ligands with high potency and selectivity for subtypes of the above-mentioned receptors. Such molecules will continue to supplement our toolbox of CNS receptor ligands that will be useful as novel biological probes, as new imaging agents and as leads for drug discovery efforts relevant to psychiatric disorders and drug abuse.
We are primarily interested in evaluating the potential of aporphines as ligands for 5-HT2A receptors. 5-HT2A receptors are implicated in several neuropsychiatric maladies including schizophrenia, depression, anxiety and insomnia.17,18 5-HT2A receptors are also involved in the actions of some stimulant drugs as recent reports have revealed.19–21
Several potent 5-HT2A receptor antagonists are known; in particular compounds with a mixed D2/5-HT2A antagonist profile (eg risperidone, clozapine) are quite prominent and are used clinically to manage schizophrenic symptoms.22–24 However, there are no highly selective 5-HT2A antagonists (>100-fold selectivity vs all other common neuroreceptor targets) clinically available. Nevertheless, such promising compounds (eg eplivanserin) have recently been or are currently being investigated in clinical trials as anti-insomnia medications. Thus, the identification of new highly selective and therapeutically useful 5-HT2A receptor antagonists is still of topical interest.
Our research team has engaged structure-activity relationship (SAR) studies on the aporphine alkaloid nantenine (1, Figure 1) and have identified a number of new nantenine analogs with antagonist activity at the 5-HT2A receptor.25–28 We were guided by previous SAR studies on nantenine for the present study. Nantenine itself is a high affinity α1A adrenergic receptor antagonist with moderate 5-HT2A receptor antagonist potency (see Table 1). Compounds 2 and 3 (Figure 1) are two of the most potent 5-HT2A antagonists we have obtained to date. These compounds lack affinity for the α1A adrenergic receptor. Our prior investigations have mainly focused on the ring A portion of nantenine and in general have indicated a reasonable degree of tolerance for other types and patterns of substitution in the A ring in obtaining high 5-HT2A potency and selectivity vs the α1A receptor. However, the identity and optimal placement of substituents requires further research for maximal potency and selectivity.
Figure 1.
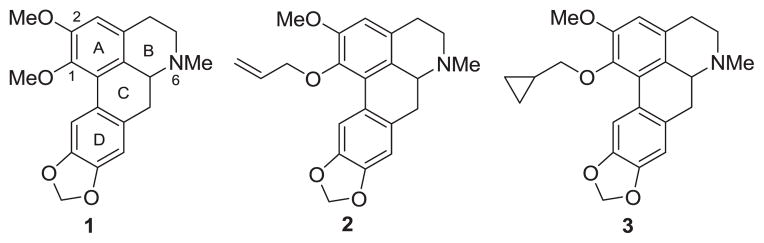
Structures of nantenine (1), compound 2 and compound 3
Table 1.
Ke values for analogs at 5-HT2A and α1A receptors
| Compd. | R1 | R2 | X | Ke ± SEMa (nM)
|
Selectivity 5-HT2A/α1A | ||
|---|---|---|---|---|---|---|---|
| 5-HT2A | α1A | ||||||
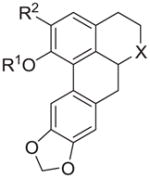
|
12a | H | H | NMe | >3000 | 2950 ± 457 | <1.01 |
| 12b | allyl | H | NMe | 47 ± 5 | 744 ± 74 | 0.06 | |
| 14 | H | allyl | NMe | >3000 | >3000 | - | |
| 15a | Me | allyl | NMe | 485 ± 123 | 566 ± 112 | 0.85 | |
| 15b | allyl | allyl | NMe | 1374 ± 405 | >3000 | <0.45 | |
| 15c | cyclopropylmethyl | allyl | NMe | 963 ± 103 | >3000 | <0.32 | |
| 16a | Me | OMe | O | >3000 | >3000 | - | |
| 16b | allyl | OMe | O | >3000 | >3000 | - | |
| (R)-1 | Me | OMe | NMe | 946 ± 61 | 70 ± 10 | 13.5 | |
| (S)-1 | Me | OMe | NMe | 657 ± 89 | 196 ± 3 | 3.4 | |
| ±-(1)b | Me | OMe | NMe | 850 ± 6 | 36 ± 7 | 23.6 | |
| 2c | allyl | OMe | NMe | 70 ± 15 | >10000 | <0.007 | |
| 3d | cyclopropylmethyl | OMe | NMe | 68 ± 8 | >10000 | <0.007 | |
| prazosin | - | - | - | - | 1.1 ± 0.4 | - | |
| ketanserinb,e | - | - | - | 32 | - | - | |
To continue to expand our understanding of the structural tolerance of aporphines as 5-HT2A receptor antagonists as well as selectivity vs the α1A receptor, we have synthesized and evaluated a new set of aporphine analogs. These compounds were prepared in order to probe three regions in the northern portion of the aporphine template, namely: ring A, the chirality center and the nitrogen atom. As alluded to earlier, our previous studies have indicated that substituents on the ring A moiety are important in controlling the 5-HT2A receptor antagonist activity and selectivity. However, we have not investigated the effect of the chiral center on 5-HT2A antagonism before. This is an important task especially since the existing literature suggests that aporphines exhibit a stereochemical preference for activation of dopamine D1 and D229,30 receptors as well as the related 5-HT1A receptor31 R enantiomers being predominantly agonists and S enantiomers being predominantly antagonists in both cases. With regards to the effect of the N6 nitrogen atom, our prior studies suggest that a basic nitrogen atom is required since the N-acetamide and N-methylsulfonamide derivatives were devoid of activity.25 We sought herein to obtain experimental proof of the absolute requirement for a nitrogen atom for antagonist activity.
Along those lines we have: 1) synthesized and evaluated new ring A analogs containing features of compounds 1, 2 or 3; 2) evaluated nantenine enantiomers - in order to begin to probe the effect of the chirality center of aporphines on receptor antagonism ; and 3) synthesized and evaluated compounds that possess a nitrogen - oxygen isosteric replacement - to investigate the importance of the nitrogen atom on receptor antagonism. Details of these studies are described henceforth.
We were interested in examining the extent to which an allyl group would be accommodated at the C2 position since an allyloxy group seems to be beneficial for antagonism (see data for compound 2, Table 1). The synthetic practicability of obtaining this structural feature via a Claisen rearrangement (a reaction rarely employed with aporphines) supported this impetus.32
To obtain the required ring A analogs Scheme 1 was engaged. Commercially available amine 4 was coupled to bromoacid 5 to give amide 6. Bischler-Napieralski cyclization of 6 was followed immediately by reduction of the dihydroisoquinoline thus formed to the secondary amine 7. Protection of the amine as the N-ethyl carbamate gave compound 8. Microwave-assisted direct arylation33 on 8 afforded compound 9. The benzyl ether 9 was deprotected revealing the phenol functionality in the key intermediate 10. Reduction of 10 with lithium aluminium hydride (LAH) gave compound 12a. Compound 12b was prepared from 10 via allylation to afford compound 11 and subsequent LAH reduction. Claisen rearrangement of the allyl ether 11 provided compound 13. Reduction of 13 gave the phenol analog 14. Analogs 15a–15c were prepared from 13 in two steps via etherification and reduction as shown. Nantenine enantiomers (R)-1 and (S)-1 were prepared by resolution of racemic nantenine as described previously.28 The isochroman analogs 16a and 16b were prepared via a sequence involving oxa Pictet-Spengler cyclization and direct arylation as presented in a recent report.34
Scheme 1.

Synthesis of ring A analogs
Reagents and conditions: (a) 1,1′-carbonyldiimidazole (CDI), THF, 0 °C - rt, 5 h, 80% ; (b) trifluoromethanesulfonic acid, pyridine, DCM, 0 °C - rt, 4 h; (c) NaBH4, MeOH, 0 °C, 2 h, 88% over two steps; (d) Ethyl chloroformate, K2CO3, DCM, rt, 3 h, 85% ; (e) Pd(OAc)2, di-tert-butyl(methyl)phosphonium tetrafluoroborate, K2CO3, (CH3)3CCOOH, DMSO, 135 °C, microwaves, 6 min, 50% ; (f) H2/Pd, rt, 8 h, 95% ; (g) alkyl bromide, KI, K2CO3, acetone, 70 °C, 6 h, 60–70% ; (h) LAH, THF, 0 °C, 10 h, 50–60% ; (i) N,N-diethylaniline, 215 °C, microwaves, 6 min, 90%
All analogs were screened at 10 μM in multi-well format for intrinsic (agonist) and antagonist activity at the human 5-HT2A receptor using Fluorescence Imaging Plate Reader (FLIPR) -based (Molecular devices, Sunnydale, CA) functional assays that detect receptor-mediated mobilization of internal calcium with a calcium sensitive fluorescent dye as reported previously.25–27 A similar set of assays was performed for the α1A - adrenergic receptor. Data from these evaluations are presented in Table 1.
As shown in Table 1, compound 12a lacked any appreciable activity at either receptor. The placement of an allyloxysubstitutent at C2 (ie compound 12b) resulted in a significant increase in 5-HT2A antagonist activity (> 60-fold as compared to the parent phenol 12a). Antagonism of the α1A receptor also increased although the magnitude of this increase was less (4-fold increase as compared to 12b). However, when 12b is compared to nantenine (1), a significant decrease in α1A antagonist activity was seen. The above data tends to suggest that the C2 methoxyl substituent of nantenine is not required for 5-HT2A antagonist activity.
Compound 14 lacked antagonist activity for both receptors indicating that an allyl substituent at C2 is not well tolerated at either receptor. A comparison of 15a with 1 reveals a slight improvement in 5-HT2A antagonism but a decrease in α1A antagonist activity upon replacement of the C2 methoxyl group with an allyl substituent. The antagonist activity of 15a was higher at both receptors than compound 14 which is indicative of a greater tolerance for an alkoxy substituent than a phenol at C1. Compound 15b had a diminished affinity at both receptors as compared to 15a. When 15b is compared to compound 12b, a significant decrease in antagonist activity of 15b at both receptors manifests. This suggests that an allyl substituent at C2 is not well accommodated at either receptor. Compound 15c had activity and selectivity that was similar to 15b. If a comparison of the 15b/15c pair is made with the 2/3 pair of compounds it may be surmised that C1 allyloxy and C1 cyclopropylmethyloxy groups endow the aporphine template with very similar 5-HT2A antagonist potency irrespective of the identity of the C2 substituent. That is, it appears that the allyl and cyclopropylmethyl functionalities are bioisosteric with respect to 5-HT2A receptor antagonism. From our previous studies, an allyloxy or cyclopropylmethyloxy substituent (ie compounds 2 and 3 respectively) imparted high 5-HT2A antagonist activity and selectivity to the nantenine template. The analysis of compounds 15a, 15b and 15c showed a reversal in this trend and again points to a considerable lack of tolerance for a C2 allyl group at the 5-HT2A receptor.
Both compounds 16a and 16b (that lack the N6 moiety) were devoid of antagonist activity. This supports previous SAR evidence that a basic N6 atom is critical for affinity to both receptors. This is also in line with previous molecular docking studies which suggest that the protonated N6 atom is involved in a H-bonding interaction with an aspartate residue in the 5-HT2A receptor binding pocket.25
Evaluation of (R)-1 and (S)-1 indicates that the chiral center of nantenine is not critical for 5-HT2A antagonism although the (S) enantiomer is slightly more potent. Interestingly, there seems to be a reversal of this trend at the α1A receptor; the (R)-enantiomer seems to be slightly more potent than the (S)-enantiomer at the α1A receptor (approximately 3-fold).
In summary, this study has revealed some useful qualitative information concerning the antagonism of aporphines at the 5-HT2A receptor. The data suggest that the C2 position is not tolerant of an allyl moiety. However, a C1 allyloxy substituent is well tolerated when the C2 substituent is hydrogen implying that the C2 methoxyl group of nantenine is not required for high 5-HT2A antagonist potency. This modification also improves selectivity vs the α1A receptor (though this selectivity is moderate as compared to that seen in 2 and 3). Of note, the most potent 5-HT2A aporphine antagonist identified in this study was compound 12b which rivals 2 and 3 in terms of 5-HT2A antagonist potency.
The chiral center of nantenine does not engender any significant preference for either enantiomer towards 5-HT2A antagonism. Somewhat unsurprisingly, the N6 nitrogen is critical for antagonist activity of nantenine analogs at both receptors.
The evaluation of this set of compounds has further expanded our fundamental knowledge concerning the viability of the aporphine template for development as selective 5-HT2A receptor antagonists. For certain, evaluation of larger series of analogs will enable a better understanding of the extent to which the SAR information extracted up to this point may be generalized. Compound 12b identified herein as well as compounds 2 and 3 identified earlier, are useful starting points for further SAR exploration and optimization studies. We are continuing in this vein and will report our findings in due course.
Supplementary Material
Acknowledgments
This publication was made possible by Grant Numbers 1SC1GM092282 and G12RR003037 from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions.
Footnotes
Experimental procedures on synthesis of all new compounds, procedure for biological assays and NMR spectral data on all analogs.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Mohamed SM, Hassan EM, Ibrahim NA. Nat Prod Res. 2010;24:1395. doi: 10.1080/14786410902906959. [DOI] [PubMed] [Google Scholar]
- 2.Kim KH, Piao CJ, Choi SU, Son MW, Lee KR. Planta Med. 2010;76:1732. doi: 10.1055/s-0030-1249972. [DOI] [PubMed] [Google Scholar]
- 3.Yu B, Cook C, Santanam N. J Med Food. 2009;12:1074. doi: 10.1089/jmf.2008.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang H, Wei YB, Zhang C, Ning FX, Qiao W, Huang SL, Ma L, Huang ZS, Gu LQ. Eur J Med Chem. 2009;44:2523. doi: 10.1016/j.ejmech.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 5.Tang H, Wang XD, Wei YB, Huang SL, Huang ZS, Tan JH, An LK, Wu JY, Chan AS, Gu LQ. Eur J Med Chem. 2008;43:973. doi: 10.1016/j.ejmech.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Cassels BK, Asencio M. Nat Prod Commun. 2008;3:643. [PubMed] [Google Scholar]
- 7.Yang Z-d, Zhang X, Du J, Ma Z-j, Guo F, Li S, Yao X-j. Nat Prod Res. 2012;26:387. doi: 10.1080/14786419.2010.487188. [DOI] [PubMed] [Google Scholar]
- 8.Ayers S, Zink DL, Mohn K, Powell JS, Brown CM, Murphy T, Brand R, Pretorius S, Stevenson D, Thompson D, Singh SB. Planta Med. 2007;73:296. doi: 10.1055/s-2007-967124. [DOI] [PubMed] [Google Scholar]
- 9.Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL. J Med Chem. 2008;51:983. doi: 10.1021/jm701045j. [DOI] [PubMed] [Google Scholar]
- 10.Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL. Bioorg Med Chem Lett. 2008;18:3971. doi: 10.1016/j.bmcl.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Toth M, Berenyi S, Csutoras C, Kula NS, Zhang K, Baldessarini RJ, Neumeyer JL. Bioorg Med Chem. 2006;14:1918. doi: 10.1016/j.bmc.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 12.Linnanen T, Brisander M, Mohell N, Johansson AM. Bioorg Med Chem Lett. 2001;11:367. doi: 10.1016/s0960-894x(00)00655-7. [DOI] [PubMed] [Google Scholar]
- 13.Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL. Bioorg Med Chem Lett. 2007;17:4128. doi: 10.1016/j.bmcl.2007.05.057. [DOI] [PubMed] [Google Scholar]
- 14.Hedberg MH, Jansen JM, Nordvall G, Hjorth S, Unelius L, Johansson AM. J Med Chem. 1996;39:3491. doi: 10.1021/jm960188q. [DOI] [PubMed] [Google Scholar]
- 15.Cannon JG, Flaherty PT, Ozkutlu U, Long JP. J Med Chem. 1995;38:1841. doi: 10.1021/jm00011a002. [DOI] [PubMed] [Google Scholar]
- 16.Ivorra MD, Valiente M, Martinez S, Madrero Y, Noguera MA, Cassels BK, Sobarzo EM, D’Ocon P. Planta Med. 2005;71:897. doi: 10.1055/s-2005-871281. [DOI] [PubMed] [Google Scholar]
- 17.Marek GJ, Carpenter LL, McDougle CJ, Price LH. Neuropsychopharmacology. 2003;28:402. doi: 10.1038/sj.npp.1300057. [DOI] [PubMed] [Google Scholar]
- 18.de Angelis L. Curr Opin Investig Drugs. 2002;3:106. [PubMed] [Google Scholar]
- 19.Orejarena MJ, Lanfumey L, Maldonado R, Robledo P. Int J Neuropsychopharmacol. 2011;14:927. doi: 10.1017/S1461145710001215. [DOI] [PubMed] [Google Scholar]
- 20.Dhonnchadha BAN, Fox RG, Stutz SJ, Rice KC, Cunningham KA. Behav Neurosci. 2009;123:382. doi: 10.1037/a0014592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham KA, Anastasio NC, Fox RG, Stutz SJ, Bubar MJ, Swinford SE, Watson CS, Gilbertson SR, Rice KC, Rosenzweig-Lipson S, Moeller FG. ACS Chem Neurosci. 2013;4:110. doi: 10.1021/cn300072u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meltzer HY, Massey BW. Curr Opin Pharmacol. 2011;11:59. doi: 10.1016/j.coph.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Horacek J, Bubenikova-Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, Hoschl C. CNS Drugs. 2006;20:389. doi: 10.2165/00023210-200620050-00004. [DOI] [PubMed] [Google Scholar]
- 24.Worrel JA, Marken PA, Beckman SE, Ruehter VL. Am J Health Syst Pharm. 2000;57:238. doi: 10.1093/ajhp/57.3.238. [DOI] [PubMed] [Google Scholar]
- 25.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Bioorg Med Chem. 2010;18:5562. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhary S, Pecic S, Le GO, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2009;19:2530. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhary S, Ponnala S, Legendre O, Gonzales JA, Navarro HA, Harding WW. Bioorg Med Chem. 2011;19:5861. doi: 10.1016/j.bmc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legendre O, Pecic S, Chaudhary S, Zimmerman SM, Fantegrossi WE, Harding WW. Bioorg Med Chem Lett. 2010;20:628. doi: 10.1016/j.bmcl.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell A, Baldessarini RJ, Gao Y, Zong R, Neumeyer JL. Neuropharmacology. 1990;29:527. doi: 10.1016/0028-3908(90)90064-x. [DOI] [PubMed] [Google Scholar]
- 30.Gao Y, Zong R, Campbell A, Kula NS, Baldessarini RJ, Neumeyer JL. J Med Chem. 1988;31:1392. doi: 10.1021/jm00402a024. [DOI] [PubMed] [Google Scholar]
- 31.Cannon JG, Moe ST, Long JP. Chirality. 1991;3:19. doi: 10.1002/chir.530030105. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Zhang H, Ye N, Zhang J, Wu Q, Sun P, Li L, Zhen X, Zhang A. J Med Chem. 2010;53:1319. doi: 10.1021/jm9015763. [DOI] [PubMed] [Google Scholar]
- 33.Chaudhary S, Pecic S, Le GO, Harding WW. Tetrahedron Lett. 2009;50:2437. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapadia N, Harding W. Tetrahedron. 2013;69:8914. doi: 10.1016/j.tet.2013.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.