

Abstract
Pyrrolopyrimidine containing compounds, also known as 7-deazapurines, are a collection of purine-based metabolites that have been isolated from a variety of biological sources and have diverse functions which range from secondary metabolism to RNA modification. To date, nearly 35 compounds with the common 7-deazapurine core structure have been described. This article will illustrate the structural diversity of these compounds and review the current state of knowledge on the biosynthetic pathways that give rise to them.
Keywords: Biosynthesis of 7-Deazapurines, 7-deazapurines in tRNA, 7-deazapurine in secondary metabolism
1. Introduction
Pyrrolopyrimidine-containing compounds, also known as 7-deazapurines, have been a source of research interest since the discovery of toyocamycin in 1956 [1]. Their broad distribution in biological samples, their potential uses as antibiotic, antifungal, antiviral, and antineoplastic agents, and the cryptic biosynthetic pathways that give rise to them, have fueled significant interest in 7-deazapurines in the intervening five decades since they were first described. This review will catalog known, naturally occurring pyrrolopyrimidines and highlight enzymatic transformations leading to the 7-deazapurine core and the unique tailoring steps required for production of several naturally occurring pyrrolopyrimidines,
2. Biological distribution of 7-deazapurine metabolites
7-Deazapurines are widely distributed in nature and occur in sources as disparate as bacteria and man and function as secondary metabolites or modified bases in RNA. This section will introduce the sources of naturally occurring 7-deazapurine-containing secondary metabolites.
2.1. 7-Deazapurines as secondary metabolites from terrestrial sources
Soil bacteria produce an array of structurally diverse compounds; though not essential for the primary metabolic processes, these so-called ‘secondary metabolites’ confer selective advantage to the producing host. The pioneering studies by Selman Waksman [2] that eventually led to discovery of streptomycin - the first treatment for tuberculosis - underscored a need to examine the biosynthetic potential of soil bacteria and inspired the discovery of a slew of other antibiotics, including compounds that have 7-deazapurine core structures (see Figure 1 for structures and Table 1 for summary).
Figure 1.

Deazapurine-containing secondary metabolites isolated from terrestrial (1–12) and marine (13–29) sources.
Table 1.
Deazapurine secondary metabolites from terrestrial (1–12) and marine (13–29) sources.
| Compound | Source | References |
|---|---|---|
| Toyocamycin (1) | Streptomyces rimosus | [1] |
| Tubercidin | Streptomyces tubercidicus | [4] |
| Sangivamycin | Streptomyces rimosus | [5] |
| Cadeguomycin (4) | Streptomyces hygroscopicus | [122–124] |
| Dapiramycin A (5) | Micromonospora sp. SF-1917 | [125] [126] |
| Epidapiramycin A (6) | Epimerization of Dapiramycin A | [127] |
| Dapiramycin B (7) | Micromonospora sp. SF-1917 | [127] |
| Echiguanine A (8) | Streptomyces sp. MI698-50F1 | [128] |
| Echiguanine B (9) | Streptomyces sp. MI698-50F1 | [128] |
| Kanagawamycin (10) | Actinoplanes kanagawaensis | [129] |
| 5’-Deoxytoyocamycin (11) | Streptomyces sp. A14345 | [130] |
| 5’-O-sulfamoyltubercidin (12) | Streptomyces mirabilis | [131] |
| Tubercidin-5′-α-d-glucopyranose (13) | Tolypotrix tenius | [132] |
| Tubercidin-5′-α-d-glucopyranose (14) | Tolypotrix tenius | [132] |
| 7-Bromo-7-deazaadenine (15) | Echinodictyum | [133] |
| 7-Iodo-5′-deoxytubercidin(16) | Hypnea valendiae | [133] |
| 5’-Deoxtubercidin (17) | Didemnum voeltzkowi | [134] |
| 7-Bromo-5′-deoxytubercidin (18) | Didemnum voeltzkowi | [134] |
| 7-Deazainosine (19) | Aplidium pantherinum | [135] |
| 7-(Methoxycarbonyl)tubercidin (20) | Jaspis johnstoni | [136] |
| 7-Cyano-7-deazaadenine (21) | Jaspis johnstoni | [136] |
| 7-Methoxycarbonyl-7-deazaadenine (22) | Jaspis johnstoni | [136] |
| Mycalisine A (23) | Mycale (marine sponge) | [137] |
| Mycalisine B (24) | Mycale (marine sponge) | [137] |
| Rigidins A-E (25–29) | Eudistoma rigida | [138–140] |
Studies on the identification, therapeutic efficacy, and biosynthesis of 7-deazapurines began with the isolation of an anti Candida agent, toyocamycin (1), from culture filtrates of Streptomyces No. 278. The organism was renamed Streptomyces toyocaensis [1] for the compound that it produces and the structure of toyocamycin was ultimately confirmed by total synthesis in 1968 [3], revealing for the first time a 7-deazapurine core structure in a natural product. Soon thereafter, tubercidin (2) was isolated from culture filtrates of Streptomyces tubercidicus obtained from a soil sample from the Chiba Prefecture in Japan [4]. The third 7-deazapurine, sangivamycin (3), was isolated from culture filtrates of Streptomyces rimosus [5]. Interestingly, S. rimosus (ATCC 14673) also produces toyocamycin [6], suggesting that both compounds are made by a common pathway. Indeed, this was confirmed when the gene cluster involved in the biosynthesis of deazapurines by S. rimosus was uncovered [7]. Each of these three compounds contains a 7-deazaadenosine core structure with the only structural diversification occurring at C-7 of the base. Toyocamycin and sangivamycin bear a cyano or amido substituent at C-7, respectively, while in tubercidin the C-7 substituent is hydrogen.
Toyocamycin is a powerful anti-tumor compound both in vitro and in vivo; however, it also shows high levels of host toxicity [8]. Tubercidin exhibits potent antibiotic activity against Candida albicans and Mycobacterium tuberculosis but does not inhibit the growth of Gram-positive bacteria, and fungi. Tubercidin shows cytotoxic activity towards NF-mouse sarcoma cells in culture [8], cultured mouse fibroblasts [9], and human tumor specimens [10]. Antiviral activity toward Vaccinia, Reovirus III, and Mengiovirus [9], which contain genomes composed of DNA, double stranded RNA, and single stranded RNA, respectively, have also been noted. Sangivamycin is highly cytotoxic to HeLa cells in culture and leukemia L1210 in mice [5].
Toyocamycin, tubercidin, and sangivamycin have served as prototypes for 7-deazapurines; the few instances in which their molecular modes of action have been interrogated are described here. As adenosine analogs, toyocamycin, tubercidin, and sangivamycin would be expected to participate in and possibly interfere with cellular processes involving adenine nucleosides and nucleotides. Indeed, studies with partially purified mammalian adenosine kinases show that toyocamycin and tubercidin are substrates for this enzyme [11]. Furthermore, toyocamycin can be phosphorylated to the mono-, di- and tri-phosphorylated forms in cultured Ehrlich ascites tumor cells and become incorporated into RNA and DNA, indicating that toyocamycin triphosphate is a substrate for mammalian RNA and DNA polymerases and implying that ribonucleotide reductase acts upon toyocamycin nucleotide to produce the corresponding deoxyribonucleotide [12]. Tubercidin, toyocamycin, and sangivamycin triphosphates are not substrates for amino acid aminoacylation, but toyocamycin and sangivamycin are competitive inhibitors of this process [13]. While all three of these deazapurines can be added to the 3′-termini of tRNA by the action of tRNA nucleotidyltransferase, the presence of toyocamycin and sangivamycin in the acceptor stem inhibits the aminoacylation of tRNA [13]. Toyocamycin is incorporated into 45S rRNA [14], which in mammalian systems is cleaved to give ribosomal subunits 5.8S, 18S, and 28S [15]. However, 45S rRNA containing toyocamycin is not further processed to 18S and 45S rRNA. It has been speculated that the presence of toyocamycin in 45S rRNA alters its tertiary structure rendering it unrecognizable to the nucleases that are required for downstream processing. More recently, sangivamycin was demonstrated to be a powerful ATP-competitive inhibitor of protein kinase C (Ki = 11 µM) [16,17].
In summary, toyocamycin, tubercidin, and sangivamycin seem to exert their cytotoxic effects by interfering with cellular metabolism on a variety of levels rather than acting on a specific cellular target. Self-resistance mechanisms have yet to be identified in 7-deazapurine producing organisms; however, since these compounds are isolated from culture filtrates, it is likely that export of the molecules from the cells protects the producing strains.
In the years since the discovery of toyocamycin, a range of structurally diverse deazapurine-based compounds have been isolated from terrestrial or marine sources, often by taking advantage of toxicity of these compounds to various strains of bacteria, fungi, plants, or mammalian cell lines. Table 1 is a comprehensive list of deazapurine secondary metabolites that have been isolated to date. We note that some of these compounds were isolated from marine specimens, such as tunicates; however, it is not clear whether the compounds are produced by the eukaryotic host, or other organisms engaged in symbiotic relationships with them.
2.2. 7-Deazapurines as hypermodified bases in tRNA
In addition to secondary metabolites, 7-deazapurines are also found as modified bases in tRNA (see Figure 2 for structure of queuosine and related modified RNA bases). RNA is one of the most heavily post-transcriptionally modified biological molecules. As of 2011, 109 modifications have been identified of which 93, 31, and 13 are modified bases located in tRNA, rRNA, mRNA, respectively [18]. One of the most highly elaborated RNA bases, queuosine (30), is located in tRNA and contains a 7-deazaguanine core. Pioneering work by Nishimura and coworkers revealed the presence of a non-canonical nucleotide in tyrosyl tRNA, which was designated Q [19]. The corresponding nucleoside of Q is referred to as queuosine, while the free base is called queuine. Queuosine was subsequently found to reside in the wobble position of asparaginyl, tyrosyl, histidyl and aspartyl tRNA, which contain the genetically encoded 5'-GUN-3' in the anticodon loop (where ‘N’ is any canonical nucleoside) [20]. Queuosine-containing tRNAs bind the codons , coding for asparagine, tyrosine, histidine, and aspartate, respectively. Soon after its initial discovery, queuosine was isolated on a preparative scale and its unique cyclopentenediol-appended 7-deazapurine structure was determined [21]. Parallels between the 7-deazaguanine core structure of queuosine and 7-deazapurine containing nucleoside antibiotics were immediately noted. Queuosine, appended with galactose or mannose bound by an O-glycosidic linkage to the C2 position of the cyclopentenediol substituent, are also found in animal Tyr-tRNA and Asp-tRNA, respectively [22,23].
Figure 2.

Deazpaurines in tRNA. Queuosine and archaeosine analogs are found in the anticodon loop or the D-loop of tRNA, respectively. Archaeosine is only found in archaea. The site of glutamate esterificaion of glutamylqueusoine remains to be established.
Queuosine is distributed broadly throughout biology in both prokaryotes and eukaryotes [23,24], but it is absent in archaea and, notably, Sacchromyces cerevisiae [25]. Despite its conservation throughout biology, the precise physiological role of this modified base has eluded investigators for decades, but a few key findings are worthy of note. In an experiment where wildtype E. coli were cultured together with a strain that lacks a key gene for the incorporation of the deazapurine into RNA, the deletion strain comprises <1% of the total cell population at deep stationary phase [26], suggesting that the RNA modification may confer growth advantage. In another intriguing report, Tyr-tRNA containing queuosine was demonstrated to suppress read-through of the viral UAG stop codon in the tobacco mosaic virus [27], suggesting a role for the modification in viral infectivity. In the context of viral infections, the genome of phage Dp-1, which is a Streptococcus pneumoniae virulent phage, encodes nearly all of the genes for the biosynthesis of the modified base; this is surprising considering the propensity for compact viral genomes, suggesting a possible role for queuosine in virulence [28]. From its prominent location in the wobble position of tRNA, one may suppose that queuosine could modify the base-pairing characteristics of the tRNA in which it is found. Indeed, His-tRNA containing guanine in the wobble position displays a preference for the codon CAC over CAU while His-tRNA containing queuosine does not [29], suggesting that queuosine could enhance translational efficiency.
Queuosine has been implicated as a requirement in the biosynthesis of tyrosine in mammals [30]. A recent study demonstrated that liver extracts from mice that lack the enzymes for incorporating Q into RNA contained decreased levels of tetrahydrobiopterin, the cofactor required for the conversion of phenylalanine to tyrosine by phenylalanine hydroxylase [31].
Cultured cells derived from various tumors have consistently been observed to contain decreased queuosine levels. Hypomodification by queuosine is often correlated with a higher incidence of tumor metastasis and a lower patient survival rate. The precise meaning of this phenomenon is not well understood [32,33].
Archaeosine (34) is a non-canonical 7-deazapurine nucleoside identified during sequencing of the Met-tRNA from Thermoplasma acidophilum [34] and is widely distributed in archaeal species [35]. It is located at position 15 of the dihydrouridine loop (the D-loop) in select tRNA molecules. As with queuosine, genes for tRNA species that contain archaeosine encode guanosine for the position in which archaeosine is found. The precise physiological role of archaeosine remains unknown but it is thought to enhance stability of the tertiary structure of tRNA [36].
3. Biosynthesis of deazapurines: Insights from radiotracer experiments
The fact that purines are precursors to deazapurines was established by following the fate(s) of various trace radioisotope labeled potential precursors in feeding experiments; efficient uptake of the precursor, however, was not confirmed in every case. Nevertheless, pyrimidines were eliminated as plausible precursors to deazapurines by showing that no radioactivity is incorporated into tubercidin that is isolated from fermentation media of S. tubercidicus cells grown in the presence of [6-14C]-orotic acid. A pre-formed 7-deazapurine was eliminated from consideration by showing that [U-3H]-7-deazaadenine is also not a precursor to tubercidin. Finally, no radioactivity was incorporated into tubercidin when the cells were grown in the presence of [1,4-14C]-succinate, or [1-14C]-propionate, demonstrating that deazapurines are not derived from C3 or C4 carboxylic acid pools [37]. By contrast, 7-deazaadenine base and not the ribosyl moiety of tubercidin isolated from cells grown in the presence of [U-14C]-adenine contained radioactivity, implying that deazapurines are derived from purines. Intriguingly, cells grown in the presence of [2-14C]-adenine and not [8-14C]-adenine incorporated the proffered radiolabel into the 7-deazaadenine base of tubercidin. Collectively, these results suggested that the 7-deazapurine core is derived from a purine nucleoside precursor with the loss of purine carbon 8 [38]. Similar radiotracer results were obtained for the biosynthesis of toyocamycin by S. rimosus [39].
Additional feeding studies established the origin of pyrrole carbons (7 and 8), as well as the cyano carbon in toyocamycin [38]. S. rimosus cells that were grown in the presence of either [1-14C]-ribose or [U-14C]-ribose incorporated radioactivity into the deazapurine base and the ribose moiety of toyocamycin. Moreover, toyocamycin that was isolated from cultures grown in the presence of either [1-3H]-ribose or [3-3H]-ribose revealed tritium incorporation into the deazapurine base only when the proffered ribose was tritiated at carbon 1. Radioactivity in toyocamycin isolated from culture filtrates of cells that were grown in the presence of either [U-14C]-adenosine or adenosine in which a majority of the total 14C radiolabel was located in the ribose was localized carbons 1′, 2′, and 3′. These observations led to the remarkable insight that ribose from a purine nucleoside precursor is rearranged to form carbons 7 and 8, as well as the cyano carbon in toyocamycin, respectively (see Figure 3).
Figure 3.

Summary of radiotracer experiments for the conversion of guanosine to toyocamycin. In general, biosynthesis of all deazapurine-containing compounds from purines entails loss of carbon 8, retention of carbon 2, incorporation of carbons 1′ and 3′. In some cases, the 6-membered ring of the proferred purine undergoes further modifications. Refer to Figs 4, 5 and 7 for the specific reactions in the biosynthesis of toyocamycin.
While the structures of tubercidin, toyocamycin, and sangivamycin resemble adenosine more closely than guanosine, feeding experiments with either [2-14C]-guanine or adenine give rise to radiolabeled deazapurine nucleosides, so it remained unclear which of the two was the true deazapurine precursor because interconversion of purines by endogenous salvage pathways could not be ruled out. Similar biosynthetic insights were obtained in studies aimed at understanding the biosynthesis of queuosine. [40].
The radiotracer experiments showing retention of carbon-2 and loss of carbon-8 in the biosynthesis of the 7-deazapurine backbone suggested a rearrangement analogous to the first step in the biosynthesis of folic acid, pterins, and toxoflavin, which are all synthesized from GTP in a process that requires GTP cyclohydrolase I (GCH I) [41–44]. GCH I catalyzes the conversion of GTP to 7,8-dihydroneopterin triphosphate (H2NTP), in a reaction that leads to loss of carbon-8, retention of carbon-2, and incorporation of the ribose carbons to form a new ring system. Consistent with the radiotracer experiments, dialyzed cell lysates from S. rimosus, which produces both sangivamycin and toyocamycin, contained GCH I activity [45,46]. While the lysate affected the loss of radioactivity from [8-14C]-GTP as formic acid, similar activity was not observed toward [8-14C]-ATP or ITP. Moreover, the activity was not constitutively expressed; it increased dramatically from 30 to 48 h post inoculation, and tracked closely with the production of sangivamycin. These results suggest a common first step in the biosynthetic pathways for folic acid, tetrahydropterin and deazapurines.
In summary, the radiotracer isotope experiments established the outline of the biosynthetic pathway. These results demonstrated that a purine, likely GTP, is the precursor to all deazapurines and that in the course of biosynthesis, C-2 of the purine base is retained but C-8 is eliminated. Moreover, ribose carbons 1'-3' are utilized for the newly formed pyrrole ring. These are summarized in Figure 3.
4. Enzymes involved in the biosynthesis of deazapurines
The enzymatic transformations that lead to the core 7-deazapurine structure are common to all of these compounds. In this section, the biosynthetic steps to the 7-deaza core are discussed, followed by subsections on the unique steps that tailor the 7-deazapurine to queuosine, archaeosine, and toyocamycin/sangivamycin.
4.1. Biosynthesis of the 7-deazapurine core
Nature’s strategy for assembly of the 7-deazapurine core was elucidated in two independent studies that were focused on identifying the biosynthetic pathways for queuosine and the pyrrolopyrimidine nucleosides sangivamycin and toyocamycin, respectively. In the first, comparative genomic approaches led to identification a subset of the genes required for the production of queuosine in Acenitobacter calcoaceticus [47]. Since GTP cyclohydrolase activity had been implicated in queuosine biosynthesis as discussed above, and because genes involved in a single biosynthetic pathway are sometimes co-localized in bacterial genomes, the search for genes in the proximity of GCH I homologs led to discovery of four open reading frames (ykvJ, ykvK, ykvL, and ykvM) that encoded proteins of unknown function. This search was limited to genomes containing the tgt and queA genes, which encode two enzymes involved in late steps of the biosynthesis of queuosine (see next section). The genes, ykvJ, ykvK, ykvL, and ykvM were subsequently renamed queC, queD, queE, and queF for their role in queuosine biosynthesis.
Identification of the genes required for the biosynthesis of toyocamycin and sangivamycin in S. rimosus [7] provided substantial clues to the biosynthesis of the 7-deazapuine core found in nucleoside antibiotics, in queuosine, and in archaeosine. Suhadolnik and coworkers had shown the presence of a toyocamycin nitrile hydratase (TNHase) activity in S. rimosus, which produces both toyocamycin and sangivamycin [6,48]. TNHase catalyzes the conversion of toyocamycin to sangivamycin. Purification of the nitrile hydratase and N-terminal sequencing of the three subunits of the protein led to the identification of three genes encoding TNHase. The remainder of the genes required for the biosynthesis of toyocamycin and sangivamycin were located adjacent to those encoding TNHase, within a single gene cluster of ~ 13 kbp. The entire set of genes involved in toyocamycin and sangivamycin biosynthesis was designated toyA-L (toy designation was employed to denote toyocamycin biosynthesis). As was suspected from the similarities in radiotracer data for biosynthesis of deazapurines (see Section 3), toyM, toyB, and toyC are homologous to the essential genes for the biosynthesis of queuosine, queC, queD, and queE, respectively. In vitro reconstitution of the biosynthesis of 7-cyano-7-deazaguanine (preQ0), a previously identified intermediate in the biosynthesis of queuosine [49], was achieved subsequently in four steps from GTP using recombinantly expressed and purified QueC, QueD and QueE homologs.
The steps from GTP to preQ0 are likely common to almost all 7-deazapurines and will be discussed first. In the sections that follow, the reactions that are unique to the biosynthesis of queuosine, archaeosine and sangivamycin/toyocamycin, will be highlighted. The core reactions involved in the biosynthesis of preQ0 are summarized in Figure 4 and discussed below (Sections 4.1.1–4.1.4).
Figure 4.

Biosynthesis of preQ0. Biosynthesis of 7-cyano-7-deazaguanine core of deazapurines from GTP is accomplished by the successive actions of GTP cyclohydrolase I (GCH I/ToyD), CPH4 synthase (QueD/ToyB), CDG synthase (QueE/ToyC) and preQ0 synthetase (QueC/ToyM). The loss of 3 carbon atoms by the successive actions of GCH I and QueD, elimination of a nitrogen by QueE and incorporation of ammonia by QueC have been demonstrated by high resolution mass spectrometry [56,62]. Loss of the nitrogen as ammonia from CPH4 catalyzed by QueE has not been demonstrated explicitly.
4.1.1. GTP cyclohydrolase I (GCH I)
The role of GCH I in biosynthesis of 7-deazapurines was first demonstrated in vitro with its homolog, ToyD, from the toyocamycin biosynthesis pathway of Streptomyces rimosus [50]. The reaction catalyzed by S. rimosus ToyD is identical to that of the E. coli GCH I. The X-ray crystal structure of the E. coli homolog has revealed the presence of a Zn2+ metal ion, which activates a water molecule in each of the two half reactions that are involved in hydrolytic removal of carbon-8 [50]. Opening of the ribose ring is followed by a series of Amadori rearrangements and subsequent re-cyclization into the pteridine structure of H2NTP [51]. All relevant sequence features that have been shown to be essential for the E. coli GCH I are retained in ToyD.
The discovery that GCH I in S. rimosus is co-localized with the toyocamycin biosynthetic genes is entirely consistent with the radiotracer experiments, where available, which have shown that in the course of conversion of a purine to a deazapurine C-8 of the base is lost whereas C-2 is retained [52,53]; moreover, it accounts for the observation that carbon atoms that are originally part of the sugar are incorporated into the 7-deazapurine core. GCH I is required for biosynthesis of queuosine in E. coli and archaeosine in Haloferax volcanii as well [54].
4.1.2. 6-Carboxy-5,6,7,8-tetrahydropterin synthase (QueD)
The second step in the pathway is catalyzed by 6-carboxy-5,6,7,8-tetrahydropterin (CPH4) synthase (QueD or To ToyB), which converts H2NTP produced by GCH I to 6-carboxy-5,6,7,8-tetrahydropterin (CPH4). The reaction entails the loss of 2′ and 3′ carbons of the substrate as acetaldehyde. The enzyme is homologous to the mammalian 6-pyruvoyltetrahydropterin (PPH4) synthase (mPTPS), which catalyzes the second step in the biosynthesis of tetrahydrobiopterin in eukaryotes [55], namely, the conversion of H2NTP to 6-pyruvoyl-5,6,7,8-tetrahydropterin (PPH4). However, the new activity appears to have evolved in the same scaffold. Intriguingly, CPH4 synthase also catalyzes the conversion of PPH4 and 6-lactoyl-7,8-dihydropterin to CPH4 suggesting the possible intermediacy of these compounds in the catalytic cycle, though chemical and kinetic competence has not been shown yet [56].
4.1.3. CDG synthase (QueE)
The next enzyme in the pathway, 7-carboxy-7-deazaguanine (CDG) synthase (QueE/ToyC), catalyzes the conversion of CPH4 to CDG [56]. CDG synthase is a member of the radical S-adenosyl-L-methionine (SAM) enzyme superfamily [57,58]. Radical SAM enzymes harbor a [4Fe-4S] cluster in which three of the four iron atoms are ligated to cysteine thiolates that are typically located in a conserved CX3CX2C motif. The reduced form of the cluster (+1 oxidation state) donates an electron to affect the reductive cleavage of the C5′-sulfur bond in SAM to generate methionine and 5′-deoxyadenosyl radical (5′-dAdo•). The highly reactive 5′-dAdo• abstracts a hydrogen atom from substrate and the resulting substrate radical undergoes rearrangement resulting in a chemical transformation. Members of this enzyme superfamily catalyze an array of diverse reactions, which are involved in a variety of biological processes including cofactor and antibiotic biosynthesis, tRNA modification, anaerobic oxidation, DNA repair, and protein radical formation [59–61]. The mechanism of CDG synthase remains to be established. However, it is hypothesized that the conversion of CPH4 to CDG is accomplished by H-atom abstraction from either the C-6 or C-7 position of the substrate, and that the resulting activated molecule undergoes a complex radical mediated ring contraction to form CDG [62].
4.1.4. PreQ0 synthetase (QueC)
CDG is converted to 7-cyano-7-deazaguanine (preQ0) by the action of preQ0 synthetase (QueC or ToyM), which catalyzes conversion of the carboxylic acid moiety of CDG to a nitrile with ammonium as a nitrogen source in an ATP-dependent reaction. The putative mechanism for preQ0 synthethase is that the two molecules of ATP are utilized to activate the carboxylate oxygen atoms, allowing for successive half reactions involving addition of ammonia and collapse to the alkyl cyanide. The X-ray crystal structure of QueC from B. subtilis was reported [63] before its enzymatic activity had been established [56]. In QueC, the conserved residues, SGGXDS, comprise a pyrophosphate-binding loop that is found in many enzymes that carry out hydrolysis of the α-β phosphoanhydride bond in ATP [64]. The preQ0 synthetase reaction is a rare example of nitrile production in living systems.
The biosynthetic pathways for 7-deazapurine containing tRNA modifications diverge from those for toyocamycin and sangivamycin from this point forward. The remaining tailoring steps during which queuosine, archaeosine, and toyocamycin/sangivamycin are derived from preQ0 will be discussed next.
4.2. Biosynthesis of queuosine
The pathway to incorporation and maturation of queuosine begins with preQ0, which is formed by the successive actions of GTP cyclohydrolase I, CPH4 synthase, CDG synthase and preQ0 synthetase, as described above. The pathway to queuosine is detailed in Figure 6 and discussed below (Sections 4.2.1–4.2.5).
Figure 6.

Biosynthesis of archaeosine. PreQ0, synthesized as shown in Figure 4, is incorporated into RNA by the archaeal TGT (arcTGT) and elaborated by archaeosine synthase (ArcS) to archaeosine.
4.2.1. PreQ0 reductase (QueF)
PreQ0 reductase catalyzes the NADPH-dependent reduction of the nitrile group in preQ0 to the aminomethyl group found in preQ1 (7-aminomethyl-7-deazaguanine) [65]. QueF is homologous to GCH I and had been annotated initially as an ‘enzyme related to GCH I’ prior to identification of its true biochemical function. QueF, however, lacks His and Cys residues [65] that are required for binding and activation of an essential zinc metal ion in E. coli GCH I [50]. Interestingly, an additional Cys residue, which is also involved in coordination of the metal ion in GCH I, is retained but utilized in covalent catalysis. QueF catalyzes covalent attachment of this Cys thiolate sidechain to the carbon atom of the cyano group of preQ0 to from a thioimide, which is converted to preQ0 in two successive NADPH-dependent reactions [66]. Homology modeling [67] and crystallographic structure determinations have confirmed the structural similarities between QueF and GCH I, showing that it adopts a tunnel fold and the position of the catalytically essential cysteine residue [68].
4.2.2. tRNA:guanine transglycosylase (TGT)
Bacterial TGT catalyzes the posttranscriptional exchange of guanine in the wobble position of tRNA anticodons with the sequence 3′-GUN-5′ (where ‘N’ is any canonical nucleoside) with preQ1 base [49,69]. Modified 7-deazapurine bases in tRNA are unique in that they are incorporated by a base-exchange mechanism without cleavage of the phosphodiester linkage [49]. While preQ1 is the physiological substrate, exogenously supplied guanine and preQ0 are also exchanged for guanine by TGT; by contrast, queuine, the free base of queuosine, is not a substrate for the bacterial TGT. Truncated RNA hairpin stem-loop structures corresponding to the anticodon loop of tRNA are also alternate substrates, so long as a U flanks the 3′-end of the GUN anticodon [70–72]
The mechanism of TGT has been probed by biochemical and structural methods. The most illuminating mechanistic insight was from the structure of the Zymomonas mobilis protein, which when crystalized with tRNA and the catalytically inactive substrate analog 9-deazazguanine revealed the structure of a ternary complex [73]. In the structure the C-1′ of the tRNA base in position 34 is attached to the protein through covalent linkage to the carboxylate moiety of an active Asp. Moreover, the captured covalent intermediate is converted to the product by soaking preQ1 into the crystals, demonstrating its intermediacy in the catalytic cycle [73]. Site-directed and kinetic studies have confirmed the role of the Asp and have demonstrated kinetic competence for the covalent intermediate [74,75].
Unlike the bacterial TGT, the eukaryotic TGT homolog (QTRT1) catalyzes the incorporation of queuine (the base of queuosine) into tRNA instead of preQ1 [76,77]. Eukaryotes do not synthesize queuosine precursors de novo and obtain queuosine from the diet or from intestinal flora as the free base, queuine. Queuine is a common nutrient factor and is found in many sources, including milk, serum, wheat germ and tomatoes [78]. Purified QTRT1 from several laboratories has been found to contain, varying subunit compositions [79–83]. QTRT1 associates with another protein, QTRTD1, which is a splice variant of QTRT1 and shares significant sequence identity with it; the two associate in vivo and appear to localize to the outer membrane of the mitochondria [84]. Complex formation, either by co-expression [84] or in vitro mixing [85] leads to active protein. The bacterial and mammalian proteins have been proposed to have evolved via divergent evolution, and there is evidence that small changes in active site architecture are responsible for their differential substrate recognition properties [86,87].
4.2.3. S-Adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA)
QueA was first identified as a protein encoded by the queA gene, which in E. coli co-localized in the chromosome with tgt and encoded a protein that could complement a methyl-deficient strain of E. coli K12, which accumulates preQ1 in tRNA [88,89]. The so-called QueA enzyme utilizes SAM as a substrate and the C-4 of the ribosyl moiety of SAM is transferred to the aminomethyl group of preQ1 and isomerized to the cyclopentendiol epoxide [90–92]. The enzyme employs an ordered sequential bi-ter kinetic mechanism, in which tRNA substrate is bound first, followed by SAM, and products are released in the order of adenine, methionine, and modified RNA [93]. As with TGT, truncated tRNA molecules that contain only the anti-codon stem loop are substrates [94]. X-Ray crystal structures of the ligand-free proteins from Thermatoga maritima and Bacillus subtilis have been solved recently [95,96]. The structures should facilitate studies of the fascinating mechanism by which SAM serves as a ribosyl donor instead of its more common role as a methyl group donor in biology.
4.2.4. oQ reductase (QueG)
The final step of queuosine biosynthesis, the conversion of oQ to queuosine, had remained elusive until very recently, but vitamin B12 had been implicated previously as a cofactor required for the reaction [97]. A screen of tRNA from over 1700 E. coli strains from the Keio collection [98], that harbored a deletion in a single gene of unknown function (so-called y-genes), led to the identification of the gene yjeS (renamed queG after the queuosine biosynthesis genes) that encodes oQ reductase [99]. QueG is homologous to a family of vitamin B12 dependent, iron-sulfur cluster containing reductive dehalogenases that catalyze the dechlorination of compounds such as tetrachloroethene to form ethane [100]. The purified, recombinant QueG catalyzes the conversion of oQ to Q in a synthetic, oQ containing RNA stem loop corresponding to the anticodon loop of Tyr-tRNA, as well as isolated cellular tRNA from the ΔqueG strain [99]. Mechanistic studies on this protein are on-going to define the role(s) of the complex cofactors required for conversion of oQ to Q.
4.2.5. Glutamylqueuosine synthetase
A recent investigation into a truncated aminoacyl-tRNA synthetase homolog in E. coli (yadB) that contains a catalytic core but lacks a conserved anticodon recognition domain resulted in the unexpected finding that it does not catalyze the aminoacylation of any tRNA species [101,102]. Rather, YadB catalyzes the ATP-dependent addition of glutamate to the cyclopentendiol moiety of Asp-tRNA (see Fig. 2 for the structure). YadB is not widely distributed in nature, suggesting that it is not essential; the biological function of this modification remains unknown.
4.2.6. Mannosyl/galactosyl queuosine
Queuosine in tRNA isolated from mammalian sources is modified at the 4-position of the cyclopentendiol moiety with a mannosyl or galactosyl group. The identity of the sugar appears to be related to the amino acid encoded by the tRNA, with tRNATyr being galactosylated and tRNAAsp being mannosylated [22,23,103]. The enzyme that catalyzes mannosylation of Asp tRNA was partially purified from rat liver and shown to utilize GDP-mannose as substrate [104]. Microinjection studies with Xenopus laevis show that the modification can occur in vivo given exogenous tRNA [105–107]
4.3. Biosynthesis of archaeosine
The biosynthesis of archaeosine is essentially identical to that of queuosine up to the formation of preQ0. In Haloferax volcanii, knock-out and complementation studies have shown that GCH I is required for appearance of archaeosine in tRNA [54]; moreover, preQ0 has been detected in acid-soluble cell extracts from H. volcanii and archaeosine is present in tRNA isolated from cells grown in Q-deficient media [108]. This section will discuss the two steps that are required for incorporation of preQ0 into tRNA and its amidination to archaeosine (see Figure 6 for summary).
4.3.1. Archaeal tRNA:guanine transglycosylase (arcTGT)
As with bacterial and eukaryotic TGT, the archaeal TGT (arcTGT) catalyzes exchange of guanosine in RNA for archaeosine [108,109]. However, unlike the other proteins, the substrate appears to be preQ0, the 7-cyanoguanine base, and the reaction takes place not at the wobble position of RNA but in the D-loop. ArcTGT is composed of a single type of subunit and appears to be homologous to the bacterial protein. Interestingly, the proteins from archaea appear to have three additional C-terminal domains (Domain C1–3), one of which, domain C3, is homologous to PUA domains (for pseudouridine synthases and archaeosine tRNA transglycosylase) [110]. In some archaea, the C-terminal extension is encoded by a separate gene [111]. While comparison of the apo [111] and tRNA-bound [112] X-ray crystal structure of arcTGT suggested that the PUA domain may be responsible for proper orientation of the substrate, namely, exposing the modification site by disrupting the D-loop, biochemical studies have indicated that deletion of the entire C-terminal region does not impair the reaction [113]. The catalytic efficiency of the protein is reduced, primarily due to an effect on Km.
4.3.2. Archaeosine synthase
The final step in the biosynthesis of archaeosine is catalyzed by archaeosine synthase (ArcS) [114]. The enzyme catalyzes the hydrolysis of glutamine (glutaminase), which is utilized to carry out amidination of preQ0 to generate archaeosine. ATP is not required for the process. ArcS homologs are widely distributed in archaea. Sequence alignments suggest that it is homologous to ArcTGT but lacks the catalytic residues that are essential for the base exchange reaction, as would be expected from the unique reaction that it catalyzes. As with ArcTGT, ArcS has a PUA domain whose function is not understood.
4.4. Biosynthesis of toyocamycin/sangivamycin
PreQ0 is an intermediate in the biosynthesis of toyocamycin/sangivamycin (see Figure 7 for pathway). The identification of the gene cluster for the biosynthesis of these compounds revealed that in addition to enzymes required for the biosynthesis of the 7-deazapurine core, the cluster also houses a series of purine biosynthesis/salvage genes, which presumably encode enzymes involved in appending preQ0 to a new ribose and those for converting the guanine-like preQ0 to the adenine-like base moiety found in toyocamycin. The purine salvage genes are predicted, based on bioinformatics, to encode homologs of guanine/hypoxanthine phosphoribosylpyrophosphate transferase (PRPTase) (ToyH), GMP reductase (ToyE), adenylosuccinate synthetase (ToyG), adenylosuccinate lyase (ToyF), and haloacid dehalogenase (ToyI). These proteins are collectively hypothesized to convert preQ0 to toyocamycin [7]. It will be interesting to determine the extent to which these proteins have diverged from their counterparts in primary metabolism in order to accept substrates that have a substituent appended to the newly installed carbon, which in purines is a nitrogen.
Figure 7.
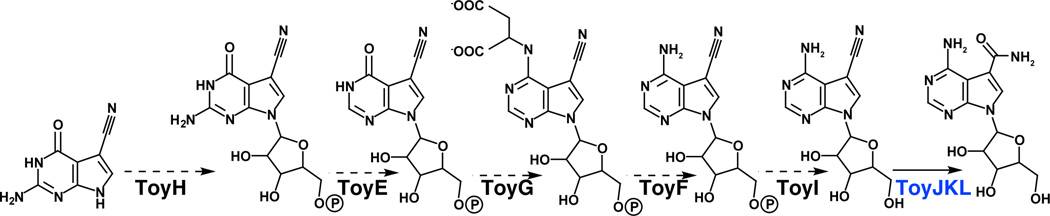
Biosynthesis of toyocamycin and sangivamycin from preQ0. Dashed arrows denote hypothetical reactions that are based on sequence similarity. PreQ0 is presumably converted to toyocamycin by the successive actions of ToyH, ToyE, ToyG, ToyF and ToyI. Toyocamycin nitrile hydratase (ToyJKL) catalyzes the conversion of toyocamycin to sangivamycin.
4.4.1. Toyocamycin nitrile hydratase (TNHase)
Conversion of toyocamycin to sangivamycin is catalyzed by toyocamycin nitrile hydratase (TNHase) [7]. TNHase belongs to a family of metalloenzymes that ligate either a non-heme Fe3+ ion or non-corrin Co3+ ion to three cysteine sulfurs and two backbone amide nitrogens in a conserved active site motif, V-C-(S/T)-L-C-S-C [115]. The presence of Ser or Thr correlates with coordination to iron or cobalt, respectively. Two of the cysteines in the conserved motif (C2 and C3) are post translationally oxidized to cysteine sulfinic (−SO2H) and sulfenic acid (−SOH), respectively (therefore TNHase is expected to ligate cobalt). Most NHases are composed of two subunits and the active site post-translationally modified cysteine residues reside in the α-subunit. TNHase is composed of three subunits - the β and γ subunits are homologous to the N- and C-terminal halves of a typical NHase beta subunit.
5. Regulation of biosynthesis of deazapurines
As with other secondary metabolites produced by microorganisms, it appears that the biosynthesis of deazapurine containing compounds is regulated. The compounds are often isolated from culture filtrates 24–48 h after inoculation, suggesting that the biosynthetic pathways for their production are turned-on in the stationary phase of growth. The biosynthetic cluster for toyocamycin/sangivamycin also encodes a LuxR-like transcriptional activator [7]. However, the mode of regulation, and whether the activation of the pathway is tied to general transcriptional regulators in Streptomyces, remains to be established.
Substantially more is known about the regulation of appearance of queuosine in RNA. The queuosine precursor, preQ1, down-regulates the transcription of queuosine biosynthetic genes through the binding of a preQ1-specific riboswitch [116,117]. To date, two distinct, non-homologous classes of preQ1-binding riboswitches have been confirmed in prokaryotes and several structural studies have provided the molecular basis of the interactions [118–120] that may discriminate for the modified base.
6. Perspectives and future directions
The burst of research in the deazapurine biosynthesis pathway has been spurred in the last 10 years by the availability of genome sequences and emergence of comparative genomic tools. The complete outline of the pathway to various deazapurines has revealed the key biosynthetic steps, and chemical intuition suggests that all known deazapurines (Figure 1) can be biosynthesized from pathways that have in common a GTP cyclohydrolase I, CPH4 synthase and CDG synthase. In the near future, coupling of recent advances in sequencing technologies with bioinformatics tools will hasten discovery of biosynthetic steps that lead to some of the more highly decorated deazapurines. If the present state of the field is any indication, there are numerous unprecedented chemical transformations that will emerge from these studies in the future.
Highlights.
GTP is a precursor to all known 7-deazapurines
7-Deazapurines are widely distributed in all kingdoms of life
Biosynthetic pathways for 7-deazapurines include several novel transformations
Figure 5.

Biosynthesis of queuosine. PreQ0, synthesized as shown in Figure 4 is reduced by preQ0 reductase (QueF), incorporated into tRNA by tRNA:guanine transglycosylase (TGT), and elaborated by epoxyqueuosine synthase (QueA) and oQ reductase (QueG) to the hypermodified base queuosine.
Acknowledgments
V.B. is grateful for National Institutes of Health (NIH) (GM72623) and Burroughs Wellcome Fund for funding various aspects of the work. R.M.M. acknowledges support from Biological Chemistry Training grant from NIH (T32 GM008804) and Science Foundation Arizona for a Graduate Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Nishimura H, Katagiri K, Sato K, Mayama M, Shimaoka N. Toyocamycin, a new anti-candida antibiotics. J. Antibiot. 1956;9:60–62. [PubMed] [Google Scholar]
- 2.Waksman S. Streptomycin: background, isolation, properties, and utilization. Science. 1953;118:259–266. doi: 10.1126/science.118.3062.259. [DOI] [PubMed] [Google Scholar]
- 3.Tolman RL, Robins RK, Townsend LB. Pyrrolo[2,3-d]pyrimidine nucleoside antibiotics. Total synthesis and structure of toyocamycin, unamycin B, vengicide, antibiotic E-212, and Sangivamycin (BA-90912) J Am Chem Soc. 1968;90:524–526. doi: 10.1021/ja01004a076. [DOI] [PubMed] [Google Scholar]
- 4.Anzai K, Nakamura G, Suzuki S. A new antibiotic, tubercidin. J. Antibiot. 1957;10:201–204. [PubMed] [Google Scholar]
- 5.Rao K, Renn D. BA-90912: An antitumor substance. Antimicrob Agents Chemother (Bethesda) 1963;161:77–79. [PubMed] [Google Scholar]
- 6.Uematsu H, Suhadolnik R. In vivo and enzymatic conversion of toyocamycin to sangivamycin by Streptomyces rimosus. Arch Biochem Biophys. 1974;162:614–619. doi: 10.1016/0003-9861(74)90223-9. [DOI] [PubMed] [Google Scholar]
- 7.McCarty RM, Bandarian V. Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem Biol. 2008;15:790–798. doi: 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saneyoshi M, Tokuzen R, Fukora F. Antitumor Activities and Structural Relationship of Tubercidine Toyocamycin and Their Derivatives. Gann. 1965;56:219. [Google Scholar]
- 9.Acs G, Reich E, Mori M. Biological and biochemical properties of the analogue antibiotic tubercidin. Proc Natl Acad Sci USA. 1964;52:493–501. doi: 10.1073/pnas.52.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolberg WH. Effect of tubercidin on nucleoside incorporation in human tumors. Biochem. Pharmacol. 1965;14:1921–1925. doi: 10.1016/0006-2952(65)90290-x. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg B, Klenow H, Hansen K. Some properties of partially purified mammalian adenosine kinase. J Biol Chem. 1967;242:350–356. [PubMed] [Google Scholar]
- 12.Suhadolnik R, Uematsu T, Uematsu H. Toyocamycin: Phosphorylation and incorporation into RNA and DNA and the biochemical properties of the triphosphate. Biochim Biophys Acta. 1967;149:41–49. doi: 10.1016/0005-2787(67)90689-2. [DOI] [PubMed] [Google Scholar]
- 13.Uretsky SC, Acs G, Reich E, Mori M, Altwerger L. Pyrrolopyrimidine nucleotides and protein synthesis. J Biol Chem. 1968;243:306–312. [PubMed] [Google Scholar]
- 14.Tavitian A, Uretsky SC, Acs G. The effect of toyocamycin on cellular RNA synthesis. Biochim Biophys Acta. 1969;179:50–57. doi: 10.1016/0005-2787(69)90121-x. [DOI] [PubMed] [Google Scholar]
- 15.Gurney T. Characterization of mouse 45 S ribosomal RNA subspecies suggests that the first processing cleavage occurs 600 ± 100 nucleotides from the 5“ end and the second 500 ± 100 nucleotides from the 3” end of a 13.9 kb precursor. Nucleic Acids Res. 1985;13:4905–4919. doi: 10.1093/nar/13.13.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osada H, Sonoda T, Tsunoda K, Isono K. A new biological role of sangivamycin: Inhibition of protein kinases. J. Antibiotics. 1989;XLII:102–106. doi: 10.7164/antibiotics.42.102. [DOI] [PubMed] [Google Scholar]
- 17.Loomis C, Bell R. Sangivamycin, a nucleoside analogue, is a potent inhibitor of protein kinase C. J Biol Chem. 1988;263:1682–1692. [PubMed] [Google Scholar]
- 18.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, et al. The RNA Modification Database, RNAMDB:2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.RajBhandary U, Gross H. E. coli tyrosine transfer RNA-primary sequence and direct evidence for base pairing between terminal sequences. (n.d.). [Google Scholar]
- 20.Harada F, Nishimura S. Possible anticodon sequences of tRNAHis, tRNAAsn, and tRNAAsp from Escherichia coli B . Universal presence of nucleoside Q in the first postion of the anticondons of these transfer ribonucleic acids. Biochemistry. 1972;11:301–308. doi: 10.1021/bi00752a024. [DOI] [PubMed] [Google Scholar]
- 21.Kasai H, Oashi Z, Harada F, Nishimura S, Oppenheimer N, Crain P, et al. Structure of the modified nucleoside Q isolated from Escherichia coli transfer ribonucleic acid. 7-(4,5- cis -dihydroxy-1-cyclopenten-3-ylaminomethyl)-7- deazaguanosine. Biochemistry. 1975;14:4198–4208. doi: 10.1021/bi00690a008. [DOI] [PubMed] [Google Scholar]
- 22.Okada N, Shindo-Okada N, Nishimura S. Isolation of mammalian tRNAAsp and tRNATyr by lectin-Sepharose affinity column chromatography. Nucleic Acids Res. 1977;4:415–423. doi: 10.1093/nar/4.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasai H, Nakanishi K, Macfarlane RD, Torgerson DF, Ohashi Z, McCloskey JA, et al. The structure of Q* nucleoside isolated from rabbit liver transfer ribonucleic acid. J Am Chem Soc. 1976;98:5044–5046. doi: 10.1021/ja00432a071. [DOI] [PubMed] [Google Scholar]
- 24.White BN, Tener GM. Activity of a transfer RNA modifying enzyme during the development of Drosophila and its relationship to the su(s) locus. J Mol Biol. 1973;74:635–651. doi: 10.1016/0022-2836(73)90054-5. [DOI] [PubMed] [Google Scholar]
- 25.Kasai H, Kuchino Y, Nihei K, Nishimura S. Distribution of the modified nucleoside Q and its derivatives in animal and plant transfer RNA's. Nucleic Acids Res. 1975;2:1931–1940. doi: 10.1093/nar/2.10.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noguchi S, Nishimura Y, Hirota Y, Nishimura S. Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. Function and biosynthesis of queuosine in tRNA. J Biol Chem. 1982;257:6544–6550. [PubMed] [Google Scholar]
- 27.Bienz M, Kubli E. Wild-type tRNATyrG reads the TMV RNA stop codon, but Q base-modified tRNATyrQ does not. Nature. 1981;294:188–190. doi: 10.1038/294188a0. [DOI] [PubMed] [Google Scholar]
- 28.Sabri M, Häuser R, Ouellette M, Liu J, Dehbi M, Moeck G, et al. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J Bacteriol. 2011;193:551–562. doi: 10.1128/JB.01117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier F, Suter B, Grosjean H, Keith G, Kubli E. Queuosine modification of the wobble base in tRNAHis influences “ in vivo ” decoding properties. Embo J. 1985;4:823–827. doi: 10.1002/j.1460-2075.1985.tb03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marks T, Farkas WR. Effects of a diet deficient in tyrosine and queuine on germfree mice. Biochem Biophys Res Commun. 1997;230:233–237. doi: 10.1006/bbrc.1996.5768. [DOI] [PubMed] [Google Scholar]
- 31.Rakovich T, Boland C, Bernstein I, Chikwana VM, Iwata-Reuyl D, Kelly VP. Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J Biol Chem. 2011;286:19354–19363. doi: 10.1074/jbc.M111.219576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dirheimer G, Baranowski W, Keith G. Variations in tRNA modifications, particularly of their queuine content in higher eukaryotes. Its relation to malignancy grading. Biochimie. 1995;77:99–103. doi: 10.1016/0300-9084(96)88111-9. [DOI] [PubMed] [Google Scholar]
- 33.Huang B, Wu R. Relationship of the queuine content of transfer ribonucleic acids to histopathological grading and survival in human lung cancer. Cancer Res. 1992 [PubMed] [Google Scholar]
- 34.Kilpatrick M, Walker R. The nucleotide sequence of the tRNAMet from the archaebacterium Thermoplasma acidophilum. Zentralbl. Bakteriol. Mikrobiol. Hyg. 1 Abt. Orig. C. 1982;3:78–89. [Google Scholar]
- 35.Edmonds CG, Crain PF, Gupta R, Hashizume T, Hocart CH, Kowalak JA, et al. Posttranscriptional modification of tRNA in thermophilic archaea ( Archaebacteria) J Bacteriol. 1991;173:3138–3148. doi: 10.1128/jb.173.10.3138-3148.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregson JM, Crain PF, Edmonds CG, Gupta R, Hashizume T, Phillipson DW, et al. Structure of the archaeal transfer RNA nucleoside G*-15 (2-amino-4,7-dihydro- 4-oxo-7-beta-D-ribofuranosyl-1 H -pyrrolo[2,3- d ]pyrimidine-5-carboximi damide (archaeosine)) J Biol Chem. 1993;268:10076–10086. [PubMed] [Google Scholar]
- 37.Smulson M, Suhadolnik R. The biosynthesis of the 7-deazaadenine ribonucleoside, tubercidin, by Streptomyces tubercidicus. J Biol Chem. 1967;242:2872–2876. [PubMed] [Google Scholar]
- 38.Suhadolnik RJ, Uematsu T. Biosynthesis of the pyrrolopyrimidine nucleoside antibiotic, toyocamycin. VII. Origin of the pyrrole carbons and the cyano carbon. J Biol Chem. 1970;245:4365–4371. [PubMed] [Google Scholar]
- 39.Uematsu T, Suhadolnik RJ. Nucleoside antibiotics. VI. Biosynthesis of the pyrrolopyrimidine nucleoside antibiotic toyocamycin by Streptomyces rimosus. Biochemistry. 1970;9:1260–1266. doi: 10.1021/bi00807a030. [DOI] [PubMed] [Google Scholar]
- 40.Kuchino Y, Kasai H, Nihei K, Nishimura S. Biosynthesis of the Modified Nucleoside Q in Transfer RNA. Nuc. Acids Res. 1976;3:393–398. doi: 10.1093/nar/3.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buff K, Dairman W. Biosynthesis of biopterin by two clones of mouse neuroblastoma. Mol. Pharmacol. 1975;11:87–93. [PubMed] [Google Scholar]
- 42.Levenberg B, Kaczmarek D. Enzymic release of carbon atom 8 from guanosine triphosphate, an early reaction in the conversion of purines to pteridines. Biochim Biophys Acta. 1966;117:272–275. doi: 10.1016/0304-4165(66)90179-6. [DOI] [PubMed] [Google Scholar]
- 43.Guroff G, Strenkoski CA. Biosynthesis of pteridines and of phenylalanine hydroxylase cofactor in cell-free extracts of Pseudomonas species (ATCC 11299a) J Biol Chem. 1966;241:2220–2227. [PubMed] [Google Scholar]
- 44.Burg A, Brown G. The biosynthesis of folic acid. 8. Purification and properties of the enzyme that catalyzes the production of formate from carbon atom 8 of guanosine triphosphate. J Biol Chem. 1968;243:2349–2358. [PubMed] [Google Scholar]
- 45.Elstner E, Suhadolnik R. Guanosine triphosphate-8-formylhydrolase. Methods Enzymol. 1975;43:515–520. doi: 10.1016/0076-6879(75)43113-5. [DOI] [PubMed] [Google Scholar]
- 46.Elstner EF, Suhadolnik RJ. The biosynthesis of the nucleoside antibiotics. IX. Purification and properties of guanosine triphosphate 8-formylhydrolase that catalyzes production of formic acid from the ureido carbon of guanosine triphosphate. J Biol Chem. 1971;246:6973–6981. [PubMed] [Google Scholar]
- 47.Reader JS, Metzgar D, Schimmel P, de Crécy-Lagard V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J Biol Chem. 2004;279:6280–6285. doi: 10.1074/jbc.M310858200. [DOI] [PubMed] [Google Scholar]
- 48.Uematsu T, Suhadolnik R. Toyocamycin nitrile hydrolase. Methods Enzymol. 1975;43:759–762. doi: 10.1016/0076-6879(75)43143-3. [DOI] [PubMed] [Google Scholar]
- 49.Okada N, Noguchi S, Kasai H, Shindo-Okada N, Ohgi T, Goto T, et al. Novel Mechanism of post-transcriptional modification of tRNA. J Biol Chem. 1979;254:3067–3073. [PubMed] [Google Scholar]
- 50.Auerbach G, Herrmann A, Bracher A, Bader G, Gutlich M, Fischer M, et al. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. Proc Natl Acad Sci U S A. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bracher A, Eisenreich W, Schramek N, Ritz H, Gotze E, Herrmann A, et al. Biosynthesis of pteridines: NMR studies on the reaction mechanisms of GTP cyclohydrolase I, pyrovoyltetrahydropterin synthase, and sepiapterin reductase. J Biol Chem. 1998;273:28132–28141. doi: 10.1074/jbc.273.43.28132. [DOI] [PubMed] [Google Scholar]
- 52.Reynolds J, Brown G. The biosynthesis of folic acid. IV. Enzymatic synthesis of dihydrofolic acid from guanine and ribose compounds. J Biol Chem. 1964;239:317–325. [PubMed] [Google Scholar]
- 53.Reynolds J, Brown G. Enzymatic formation of the pteridine moiety of folic acid from guanine compounds. J Biol Chem. 1962;237:2713–2715. [PubMed] [Google Scholar]
- 54.Phillips G, El Yacoubi B, Lyons B, Alvarez S, Iwata-Reuyl D, de Crécy-Lagard V. Biosynthesis of 7-deazaguanosine-modified tRNA nucleosides: a new role for GTP cyclohydrolase I. J Bacteriol. 2008;190:7876–7884. doi: 10.1128/JB.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takikawa S, Curtius HC, Redweik U, Ghisla S. Purification of 6-pyruvoyltetrahydropterin synthase from human liver. Biochem Biophys Res Commun. 1986;134:646–651. doi: 10.1016/s0006-291x(86)80468-5. [DOI] [PubMed] [Google Scholar]
- 56.McCarty RM, Somogyi A, Bandarian V. Escherichia coli QueD is a 6-carboxy-5,6,7,8-tetrahydropterin synthase. Biochemistry. 2009;48:2301–2303. doi: 10.1021/bi9001437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frey PA, Hegeman AD, Ruzicka FJ. The Radical SAM Superfamily. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 58.Sofia H, Chen G, Hetzler B, Reyes-Spindola J, Miller N. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frey PA, Hegeman AD. Enzymatic reaction mechanisms. 2007:831. [Google Scholar]
- 60.Booker SJ. Anaerobic functionalization of unactivated C-H bonds. Curr Opin Chem Biol. 2009;13:58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vey JL, Drennan CL. Structural insights into radical generation by the radical SAM superfamily. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCarty RM, Somogyi A, Lin G, Jacobsen NE, Bandarian V. The deazapurine biosynthetic pathway revealed: in vitro enzymatic synthesis of preQ0 from guanosine 5'-triphosphate in four steps. Biochemistry. 2009;48:3847–3852. doi: 10.1021/bi900400e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cicmil N, Huang RH. Crystal structure of QueC from Bacillus subtilis : an enzyme involved in preQ1 biosynthesis. Proteins. 2008;72:1084–1088. doi: 10.1002/prot.22098. [DOI] [PubMed] [Google Scholar]
- 64.Bork P, Koonin EV. A P-loop-like motif in a widespread ATP pyrophosphatase domain: implications for the evolution of sequence motifs and enzyme activity. Proteins. 1994;20:347–355. doi: 10.1002/prot.340200407. [DOI] [PubMed] [Google Scholar]
- 65.Van Lanen SG, Reader JS, Swairjo MA, de Crécy-Lagard V, Lee B, Iwata-Reuyl D. From cyclohydrolase to oxidoreductase: discovery of nitrile reductase activity in a common fold. Proc Natl Acad Sci USA. 2005;102:4264–4269. doi: 10.1073/pnas.0408056102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee B, Van Lanen S, Iwata-Reuyl D. Mechanistic studies of Bacillus subtilis QueF, the nitrile oxidoreductase involved in queuosine biosynthesis. Biochemistry. 2007;46:12844–12854. doi: 10.1021/bi701265r. [DOI] [PubMed] [Google Scholar]
- 67.Swairjo M, Reddy R, Lee B, Van Lanen S, Brown S, de Crecy-Lagard V, et al. Crystallization and preliminary X-ray characterization of the nitrile reductase QueF: a queuosine-biosynthesis enzyme. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:945–948. doi: 10.1107/S1744309105029246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim Y, Zhou M, Moy S, Morales J, Cunningham MA, Joachimiak A. High-resolution structure of the nitrile reductase QueF combined with molecular simulations provide insight into enzyme mechanism. J Mol Biol. 2010;404:127–137. doi: 10.1016/j.jmb.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okada N, Nishimura S. Isolation and characterization of a guanine insertion enzyme, a specific tRNA transglycosylase, from Escherichia coli. J. Biol Chem. 1979;254:3061–3066. [PubMed] [Google Scholar]
- 70.Curnow AW, Garcia GA. tRNA-guanine transglycosylase from Escherichia coli. Minimal tRNA structure and sequence requirements for recognition. J Biol Chem. 1995;270:17264–17267. doi: 10.1074/jbc.270.29.17264. [DOI] [PubMed] [Google Scholar]
- 71.Nakanishi S, Ueda T, Hori H, Yamazaki N, Okada N, Watanabe K. A UGU sequence in the anticodon loop is a minimum requirement for recognition by Escherichia coli tRNA-guanine transglycosylase. J Biol Chem. 1994;269:32221–32225. [PubMed] [Google Scholar]
- 72.Curnow AW, Kung FL, Koch KA, Garcia GA. tRNA-guanine transglycosylase from Escherichia coli: gross tRNA structural requirements for recognition. Biochemistry. 1993;32:5239–5246. doi: 10.1021/bi00070a036. [DOI] [PubMed] [Google Scholar]
- 73.Xie W, Liu X, Huang RH. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat Struct Biol. 2003;10:781–788. doi: 10.1038/nsb976. [DOI] [PubMed] [Google Scholar]
- 74.Garcia GA, Chervin SM, Kittendorf JD. Identification of the rate-determining step of tRNA-guanine transglycosylase from Escherichia coli. Biochemistry. 2009;48:11243–11251. doi: 10.1021/bi901501a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kittendorf JD, Sgraja T, Reuter K, Klebe G, Garcia GA. An essential role for aspartate 264 in catalysis by tRNA-guanine transglycosylase from Escherichia coli. J. Biol Chem. 2003;278:42369–42376. doi: 10.1074/jbc.M304323200. [DOI] [PubMed] [Google Scholar]
- 76.Okada N, Harada F, Nishimura S. Specific replacement of Q base in the anticodon of tRNA by guanine catalyzed by a cell-free extract of rabbit reticulocytes. Nucleic Acids Res. 1976;3:2593–2603. doi: 10.1093/nar/3.10.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shindo-Okada N, Okada N, Ohgi T, Goto T, Nishimura S. Transfer ribonucleic acid guanine transglycosylase isolated from rat liver. Biochemistry. 1980;19:395–400. doi: 10.1021/bi00543a023. [DOI] [PubMed] [Google Scholar]
- 78.Katze JR, Basile B, McCloskey JA. Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: wide distribution in nature. Science. 1982;216:55–56. doi: 10.1126/science.7063869. [DOI] [PubMed] [Google Scholar]
- 79.Deshpande KL, Seubert PH, Tillman DM, Farkas WR, Katze JR. Cloning and characterization of cDNA encoding the rabbit tRNA-guanine transglycosylase 60-kilodalton subunit. Arch Biochem Biophys. 1996;326:1–7. doi: 10.1006/abbi.1996.0039. [DOI] [PubMed] [Google Scholar]
- 80.Slany RK, Müller SO. tRNA-guanine transglycosylase from bovine liver. Purification of the enzyme to homogeneity and biochemical characterization. Eur J Biochem. 1995;230:221–228. doi: 10.1111/j.1432-1033.1995.0221i.x. [DOI] [PubMed] [Google Scholar]
- 81.Morris RC, Brooks BJ, Eriotou P, Kelly DF, Sagar S, Hart KL, et al. Activation of transfer RNA-guanine ribosyltransferase by protein kinase C. Nucleic Acids Res. 1995;23:2492–2498. doi: 10.1093/nar/23.13.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walden TL, Howes N, Farkas WR. Purification and properties of guanine, queuine-tRNA transglycosylase from wheat germ. J Biol Chem. 1982;257:13218–13222. [PubMed] [Google Scholar]
- 83.Howes NK, Farkas WR. Studies with a homogeneous enzyme from rabbit erythrocytes catalyzing the insertion of guanine into tRNA. J Biol Chem. 1978;253:9082–9087. [PubMed] [Google Scholar]
- 84.Boland C, Hayes P, Santa-Maria I, Nishimura S, Kelly VP. Queuosine formation in eukaryotic tRNA occurs via a mitochondria-localized heteromeric transglycosylase. J Biol Chem. 2009;284:18218–18227. doi: 10.1074/jbc.M109.002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y-C, Kelly VP, Stachura SV, Garcia GA. Characterization of the human tRNA-guanine transglycosylase: confirmation of the heterodimeric subunit structure. Rna. 2010;16:958–968. doi: 10.1261/rna.1997610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stengl B, Reuter K, Klebe G. Mechanism and substrate specificity of tRNA-guanine transglycosylases (TGTs): tRNA-modifying enzymes from the three different kingdoms of life share a common catalytic mechanism. Chembiochem. 2005;6:1926–1939. doi: 10.1002/cbic.200500063. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y-C, Brooks AF, Goodenough-Lashua DM, Kittendorf JD, Showalter HD, Garcia GA. Evolution of eukaryal tRNA-guanine transglycosylase: insight gained from the heterocyclic substrate recognition by the wild-type and mutant human and Escherichia coli tRNA-guanine transglycosylases. Nucleic Acids Res. 2011;39:2834–2844. doi: 10.1093/nar/gkq1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Okada N, Noguchi S, Nishimura S, Ohgi T, Goto T, Crain PF, et al. Structure determination of a nucleoside Q precursor isolated from E. coli tRNA-7-(aminomethyl)-7-deazaguanosine. Nucleic Acids Res. 1978;5:2289–2296. doi: 10.1093/nar/5.7.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reuter K, Slany R, Ullrich F, Kersten H. Structure and organization of Escherichia coli genes involved in biosynthesis of the deazaguanine derivative queuine, a nutrient factor for eukaryotes. J Bacteriol. 1991;173:2256–2264. doi: 10.1128/jb.173.7.2256-2264.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kinzie S, Thern B, Iwata-Reuyl D. Mechanistic studies of the tRNA-modifying enzyme QueA: a chemical imperative for the use of AdoMet as a “ribosyl” donor. Org Lett. 2000;2:1307–1310. doi: 10.1021/ol005756h. [DOI] [PubMed] [Google Scholar]
- 91.Slany RK, Bösl M, Kersten H. Transfer and isomerization of the ribose moiety of AdoMet during the biosynthesis of queuosine tRNAs, a new unique reaction catalyzed by the QueA protein from Escherichia coli. Biochimie. 1994;76:389–393. doi: 10.1016/0300-9084(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 92.Slany RK, Bösl M, Crain PF, Kersten H. A new function of S-adenosylmethionine: the ribosyl moiety of AdoMet is the precursor of the cyclopentenediol moiety of the tRNA wobble base queuine. Biochemistry. 1993;32:7811–7817. doi: 10.1021/bi00081a028. [DOI] [PubMed] [Google Scholar]
- 93.Van Lanen SG, Iwata-Reuyl D. Kinetic mechanism of the tRNA-modifying enzyme S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) Biochemistry. 2003;42:5312–5320. doi: 10.1021/bi034197u. [DOI] [PubMed] [Google Scholar]
- 94.Van Lanen SG, Kinzie SD, Matthieu S, Link T, Culp J, Iwata-Reuyl D. tRNA modification by S-adenosylmethionine:tRNA ribosyltransferase-isomerase. Assay development and characterization of the recombinant enzyme. J Biol Chem. 2003;278:10491–10499. doi: 10.1074/jbc.M207727200. [DOI] [PubMed] [Google Scholar]
- 95.Grimm C, Ficner R, Sgraja T, Haebel P, Klebe G, Reuter K. Crystal structure of Bacillus subtilis S-adenosylmethionine:tRNA ribosyltransferase-isomerase. Biochem Biophys Res Commun. 2006;351:695–701. doi: 10.1016/j.bbrc.2006.10.096. [DOI] [PubMed] [Google Scholar]
- 96.Mathews I, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Axelrod H, et al. Crystal structure of S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) from Thermotoga maritima at 2.0 Å resolution reveals a new fold. Proteins. 2005;59:869–874. doi: 10.1002/prot.20419. [DOI] [PubMed] [Google Scholar]
- 97.Frey B, McCloskey J, Kersten W, Kersten H. New function of vitamin B12: cobamide-dependent reduction of epoxyqueuosine to queuosine in tRNAs of Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1988;170:2078–2082. doi: 10.1128/jb.170.5.2078-2082.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miles ZD, McCarty RM, Molnar G, Bandarian V. Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc Natl Acad Sci USA. 2011;108:7368–7372. doi: 10.1073/pnas.1018636108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wohlfarth G, Dieket G, Banerjee R. Reductive dehalogenases. 1999 [Google Scholar]
- 101.Blaise M, Becker HD, Keith G, Cambillau C, Lapointe J, Giegé R, et al. A minimalist glutamyl-tRNA synthetase dedicated to aminoacylation of the tRNAAsp QUC anticodon. Nucleic Acids Res. 2004;32:2768–2775. doi: 10.1093/nar/gkh608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salazar JC, Ambrogelly A, Crain PF, McCloskey JA, Söll D. A truncated aminoacyl-tRNA synthetase modifies RNA. Proc Natl Acad Sci USA. 2004;101:7536–7541. doi: 10.1073/pnas.0401982101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Costa A, Païs de Barros JP, Keith G, Baranowski W, Desgrès J. Determination of queuosine derivatives by reverse-phase liquid chromatography for the hypomodification study of Q-bearing tRNAs from various mammal liver cells. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004;801:237–247. doi: 10.1016/j.jchromb.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 104.Okada N, Nishimura S. Enzymatic synthesis of Q nucleoside containing mannose in the anticodon of tRNA: isolation of a novel mannosyltransferase from a cell-free extract of rat liver. Nucleic Acids Res. 1977;4:2931–2938. doi: 10.1093/nar/4.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carbon P, Haumont E, de Henau S, Keith G, Grosjean H. Enzymatic replacement in vitro of the first anticodon base of yeast tRNAAsp: application to the study of tRNA maturation in vivo, after microinjection into frog oocytes. Nucleic Acids Res. 1982;10:3715–3732. doi: 10.1093/nar/10.12.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carbon P, Haumont E, Fournier M, de Henau S, Grosjean H. Site-directed in vitro replacement of nucleosides in the anticodon loop of tRNA: application to the study of structural requirements for queuine insertase activity. Embo J. 1983;2:1093–1097. doi: 10.1002/j.1460-2075.1983.tb01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haumont E, Nicoghosian K, Grosjean H, Cedergren RJ. The nucleotide sequence of mannosyl-Q-containing tRNAAsp from Xenopus laevis oocytes. Biochimie. 1984;66:579–582. doi: 10.1016/0300-9084(84)90154-8. [DOI] [PubMed] [Google Scholar]
- 108.Watanabe M, Matsuo M, Tanaka S, Akimoto H, Asahi S, Nishimura S, et al. Biosynthesis of archaeosine, a novel derivative of 7-deazaguanosine specific to archaeal tRNA, proceeds via a pathway involving base replacement on the tRNA polynucleotide chain. J Biol Chem. 1997;272:20146–20151. doi: 10.1074/jbc.272.32.20146. [DOI] [PubMed] [Google Scholar]
- 109.Bai Y, Fox D, Lacy J, Van Lanen S, Iwata-Reuyl D. Hypermodification of tRNA in Thermophilic Archaea. J Biol Chem. 2000;275:28731–28738. doi: 10.1074/jbc.M002174200. [DOI] [PubMed] [Google Scholar]
- 110.Ishitani R, Nureki O, Kijimoto T, Watanabe M, Kondo H, Nameki N, et al. Crystallization and preliminary X-ray analysis of the archaeosine tRNA-guanine transglycosylase from Pyrococcus horikoshii. Acta Crystallogr D Biol Crystallogr. 2001;57:1659–1662. doi: 10.1107/s0907444901011994. [DOI] [PubMed] [Google Scholar]
- 111.Ishitani R, Nureki O, Fukai S, Kijimoto T, Nameki N, Watanabe M, et al. Crystal structure of archaeosine tRNA-guanine transglycosylase. J Mol Biol. 2002;318:665–677. doi: 10.1016/S0022-2836(02)00090-6. [DOI] [PubMed] [Google Scholar]
- 112.Ishitani R, Nureki O, Nameki N, Okada N, Nishimura S, Yokoyama S. Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell. 2003;113:383–394. doi: 10.1016/s0092-8674(03)00280-0. [DOI] [PubMed] [Google Scholar]
- 113.Sabina J, Söll D. The RNA-binding PUA domain of archaeal tRNA-guanine transglycosylase is not required for archaeosine formation. J Biol Chem. 2006;281:6993–7001. doi: 10.1074/jbc.M512841200. [DOI] [PubMed] [Google Scholar]
- 114.Phillips G, Chikwana VM, Maxwell A, El Yacoubi B, Swairjo MA, Iwata-Reuyl D, et al. Discovery and characterization of an amidinotransferase involved in the modification of archaeal tRNA. J Biol Chem. 2010;285:12706–12713. doi: 10.1074/jbc.M110.102236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yamada H, Shimizu S, Kobayashi M. Hydratases involved in nitrile conversion: screening, characterization and application. Chem Rec. 2001;1:152–161. doi: 10.1002/tcr.5. [DOI] [PubMed] [Google Scholar]
- 116.Meyer MM, Roth A, Chervin SM, Garcia GA, Breaker RR. Confirmation of a second natural preQ1 aptamer class in Streptococcaceae bacteria. Rna. 2008;14:685–695. doi: 10.1261/rna.937308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roth A, Winkler WC, Regulski EE, Lee BWK, Lim J, Jona I, et al. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat. Struct. Mol. Biol. 2007;14:308–317. doi: 10.1038/nsmb1224. [DOI] [PubMed] [Google Scholar]
- 118.Klein DJ, Edwards TE, Ferré-D'Amaré AR. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol. 2009;16:343–344. doi: 10.1038/nsmb.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jenkins JL, Krucinska J, McCarty RM, Bandarian V, Wedekind JE. Comparison of a preQ1 riboswitch aptamer in the metabolite-bound and free states with implications for gene regulation. J Biol Chem. 2011 doi: 10.1074/jbc.M111.230375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spitale RC, Torelli AT, Krucinska J, Bandarian V, Wedekind JE. The structural basis for recognition of the preQ0 metabolite by an unusually small riboswitch aptamer domain. J Biol Chem. 2009;284:11012–11016. doi: 10.1074/jbc.C900024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu RT, Okabe T, Kim S-H, Suzuki H, Tanaka N. Enhancement of pyrimidine nucleoside uptake into K562 and YAC-1 cells by cadeguomycin. J. Antibiot. 1985;38:1588–1595. doi: 10.7164/antibiotics.38.1588. [DOI] [PubMed] [Google Scholar]
- 122.Wu R, Okabe T, Namikoshi M, Okuda S, Nishimura T, Tanaka N. Cadeguomycin, A novel nucleoside analog antibiotic. J Antibiot (Tokyo) 1982:279–284. doi: 10.7164/antibiotics.35.279. [DOI] [PubMed] [Google Scholar]
- 123.Tanaka N, Wu R, Okabe T, Yamashita H, Shimazu A, Nishimura T. Cadeguomycin, a noval nucleoside analog antibiotic. I. The producing organism, production and isolation of cadeguomycin. J Antibiot (Tokyo) 1982;35:272–278. doi: 10.7164/antibiotics.35.272. [DOI] [PubMed] [Google Scholar]
- 124.Shomura T, Nishizawa N, Iwata M, Yoshida J, Ito M, Amano S, et al. Studies on a new nucleoside antibiotic, dapiramicin. I. Producing organism, assay method and fermentation. J. Antibiot. 1983;36:1300–1304. doi: 10.7164/antibiotics.36.1300. [DOI] [PubMed] [Google Scholar]
- 125.Seto H, Otake N, Koyama M, Ogino H. ScienceDirect - Tetrahedron Letters : The structure of a novel nucleoside antibiotic, dapiramicin A. Tetrahedron Letters. 1983 [Google Scholar]
- 126.Nishizawa N, Kondo Y, Koyama M, Omoto S, Iwata M, Tsuruoka T, et al. Studies on a new nucleoside antibiotic, dapiramicin. II. Isolation, physicochemical and biological characterization. J. Antibiot. 1984;37:1–5. doi: 10.7164/antibiotics.37.1. [DOI] [PubMed] [Google Scholar]
- 127.Nishioka H, Sawa T, Nakamura H, Iinuma H, Ikeda D, Sawa R, et al. Isolation and structure determination of novel phosphatidylinositol kinase inhibitors, echiguanines A and B, from Streptomyces sp. J. Nat. Prod. 1991;54:1321–1325. doi: 10.1021/np50077a014. [DOI] [PubMed] [Google Scholar]
- 128.Naruto S, Uno H, Tanaka A, Kotani H, Takase Y. Kanagawamicin - a new aminonucleoside analog antibiotic from Actinoplanes kanagawaensis. Heterocycles. 1983;20:27–32. [Google Scholar]
- 129.Isaac B, Ayer S, Letendre L, Stonard R. Herbicidal nucleosides from microbial sources. J. Antibiotics. 1991;44:729–732. doi: 10.7164/antibiotics.44.729. [DOI] [PubMed] [Google Scholar]
- 130.Iwata M, Sasaki T, Iwamatsu H, Miyadoh S, Tachibana K, Matsumoto K, et al. A new herbicidal antibiotic, SF2494 (5'-O-sulfamoyltubercidin) produced by Streptomyces mirabilis. Sci Reports of Meiji Seika Kaisha. 1987;26:17–22. [Google Scholar]
- 131.Stewart JB, Bornemann V, Chen JL, Moore RE, Caplan FR, Karuso H, et al. Cytotoxic, fungicidal nucleosides from blue green algae belonging to the Scytonemataceae. J. Antibiot. 1988;41:1048–1056. doi: 10.7164/antibiotics.41.1048. [DOI] [PubMed] [Google Scholar]
- 132.Kazlauskas R, Murphy P, Wells R, Lambert-Baird J, Jamieson D. Halogenated pyrrolo[2,3-d]pyrimidine nucleosides from marine organisms. Aust J. Chem. 1983;36:165–170. [Google Scholar]
- 133.Mitchell SS, Pomerantz SC, Concepción GP, Ireland CM. Tubercidin analogs from the ascidian Didemnum voeltzkowi. J. Nat. Prod. 1996;59:1000–1001. doi: 10.1021/np960457f. [DOI] [PubMed] [Google Scholar]
- 134.Zabriskie TM, Ireland CM. The isolation and structure of modified bioactive nucleosides from Jaspis johnstoni. J. Nat. Prod. 1989;52:1353–1356. doi: 10.1021/np50066a032. [DOI] [PubMed] [Google Scholar]
- 135.Chenon MT, Pugmire RJ, Grant DM, Panzica RP, Townsend LB. Carbon-13 magnetic resonance. XXV. A basic set of parameters for the investigation of tautomerism im purines. Established from carbon-13 magnetic resonance studies using certain purines and pyrrolo[2,3-d]pyrimidines. J Am Chem Soc. 1975;97:4627–4636. doi: 10.1021/ja00849a027. [DOI] [PubMed] [Google Scholar]
- 136.Kato Y, Fusetani N, Matsunaga S. Bioactive marine metabolites IX. Mycalisines A and B, novel nucleosides which inhibit cell division of fertilized starfish eggs, from the marine sponge sp. Tetrahedron Lett. 1985;26:3483–3486. [Google Scholar]
- 137.Kobayashi J, Cheng J, Kikuchi Y, Ishibashi M. Rigidin, a novel alkaloid with calmodulin antagonistic activity from the okinawan marine tunicate Eudistoma cf. rigida. Tetrahedron Letters. 1990 [Google Scholar]
- 138.Tsuda M, Nozawa K, Shimbo K, Kobayashi J. Rigidins B-D, new pyrrolopyrimidine alkaloids from a tunicate Cystodytes species. J. Nat. Prod. 2003;66:292–294. doi: 10.1021/np020393a. [DOI] [PubMed] [Google Scholar]
- 139.Davis RA, Christensen LV, Richardson AD, da Rocha RM, Ireland CM. Rigidin E, a new pyrrolopyrimidine alkaloid from a Papua New Guinea tunicate Eudistoma species. Marine Drugs. 2003;1:27–33. [Google Scholar]