

Abstract
Background
In the model organism Saccharomyces cerevisiae, the transposable elements (TEs) consist of LTR (Long Terminal Repeat) retrotransposons called Ty elements belonging to five families, Ty1 to Ty5. They take the form of either full-length coding elements or non-coding solo-LTRs corresponding to remnants of former transposition events. Although the biological features of Ty elements have been studied in detail in S. cerevisiae and the Ty content of the reference strain (S288c) was accurately annotated, the Ty-related intra-specific diversity has not been closely investigated so far.
Results
In this study, we investigated the Ty contents of 41 available genomes of isolated S. cerevisiae strains of diverse geographical and ecological origins. The strains were compared in terms of the number of Ty copies, the content of the potential transpositionally active elements and the genomic insertion maps. The strain repertoires were also investigated in the closely related Ty1 and Ty2 families and subfamilies.
Conclusions
This is the first genome-wide analysis of the diversity associated to the Ty elements, carried out for a large set of S. cerevisiae strains. The results of the present analyses suggest that the current Ty-related polymorphism has resulted from multiple causes such as differences between strains, between Ty families and over time, in the recent transpositional activity of Ty elements. Some new Ty1 variants were also identified, and we have established that Ty1 variants have different patterns of distribution among strains, which further contributes to the strain diversity.
Keywords: Transposons, Retrotransposons, Ty elements, Ty1, Intra-specific diversity, Genome evolution, Saccharomyces cerevisiae
Background
Transposable elements (TEs) are interspersed repetitive and mobile DNA sequences. They exist in almost all the eukaryotic genomes characterized so far, where they often constitute the largest component. TEs belong to two classes, depending on whether the RNA-mediated ‘copy and paste’ (class I) or DNA-mediated ‘cut and paste’ (class II) mode of transposition is involved. As the result of their ability to proliferate and move to different positions, they give rise to inter- and intra-species genomic differences. The mutational activities of TEs, which result in gene disruption and chromosome rearrangements, also contribute to their hosts’ genetic and phenotypic diversity [1-3]. TE insertions can actually result in novel specific patterns of gene expression by either acting as regulatory elements or disrupting these elements. In addition, TEs can serve as targets for epigenetic modifications [4]. TE induced diversity may therefore have much more complex phenotypic effects than those resulting from point mutations. Since selection processes can operate on TE induced variations, TEs are thought to be particularly powerful agents responsible for adaptive changes and strong drivers of genome evolution [5]. TE activation or reactivation is likely to greatly affect genome evolution, which raises questions as to the difference in “evolvability” existing between organisms showing variable TE contents and even between isolates belonging to the same species.
There exist some extremely marked differences between the TE contents of various species, in terms of the number of copies, the TE repertoires and the respective proportions of full-length, mutated and fossil copies present [6,7]. These differences result partly from host traits (such as their regulation and defense strategies), and partly from features of TEs themselves, which are responsible for their expansion, persistence and extinction in genomes. It has been established that the TE contents of genomes vary not only between species, but also in some cases between populations belonging to the same species [8]. The polymorphism of TE is therefore widely used as an indicator to assess the genetic diversity of organisms with moderate to high TE contents: diagnostic insertion profiles are generated using PCR-based methods such as the Transposon Display, IRAP (Inter-Retrotransposon Amplified Polymorphism) and RBIP (Retrotransposon Based Insertion Polymorphism) methods [9] to obtain phylogenetic markers that are commonly used for mapping, genotyping and taxonomic purposes [10-14]. Since the advent of new sequencing technologies, new approaches have been developed for detecting insertion polymorphism, such as those based on targeted sequencing of TE junctions via sequence capture enrichment procedures [15] and in silico methods designed for investigating sequencing data and genome assemblies in repetitive elements [16-18].
Apart from multicellular organisms, some eucaryotes have relatively poor TE contents. It has been suggested, for example, that hemiascomycetous yeast may have undergone massive TE losses [19], since the TE fraction does not occur in more than 5% of their genomes, and the TE classes and families show a patchy distribution among ‘reservoir species’ and apparently ‘empty species’. Candida albicans harbors many potentially active copies of TE belonging to various families of class I and class II elements, for example, whereas closely related species such as Candida glabrata and Pichia sorbitophila carry only a few degenerate copies and show no traces at all of TEs, respectively [20]. Whether these TE landscapes are characteristic of the species as a whole, rather than being restricted to the strains that were sequenced, still remains to be established by investigating the TE content of a large number of isolates. The inter- and intra-species TE polymorphism has also been previously used to typify industrial strains [21] and to detect some noteworthy aspects of both the evolution of yeast TE and the strain diversity [22-24].
LTR (Long Terminal Repeat) retrotransposons are the main TEs occurring in hemiascomycetes. The putative origins of the present LTR retrotransposon repertoire have been deduced from both the structural features and the inter-species distribution of these elements [22,25,26]. Their evolutionary scenario has been mostly drawn up assuming the occurrence of a process of vertical transmission. Gypsy-like elements are present in all the species in the Hemiascomycete phylum and therefore seem to be the most ancient acquisition [19]; whereas the phylogeny of the elements belonging to the four lineages of Copia-elements (Ty1, Tca2, Ty4 and Ty5) is almost identical to that of their hemiascomycetous hosts [25]. The various Copia-element families may therefore have evolved as the result of successive radiation events from a single ancestral family resembling the present Ty5 elements. Starting with Ty5-like elements encoding a single ORF, some novel features such as an in-frame stop codon (in the Tca2 lineage), a programmed frameshift between the TYA and TYB genes (in the Ty1 lineage) and a change of primer binding site (in the Ty4 lineage) were acquired during the speciation steps. The structural characteristics and the pattern of host distribution of the elements belonging to the Ty4 lineage suggest that these are the youngest elements [19,25]. The possibility that horizontal transmission events may also have occurred was suggested in the case of two elements belonging to the Ty1 lineage: Tsk1 in Lachancea kluyveri[25] and Ty2 in Saccharomyces cerevisiae[22,26].
In comparison with its nearest hemiascomycetous relatives, the model species S. cerevisiae is thought to constitute an exceptional TE reservoir. Only LTR-retrotransposons are present in this species, but it contains more Ty families with full-length copies than other yeast species [20]. These elements called Ty elements account for 1.5% of the genome. Most of the known aspects of LTR-retrotransposon biology have been discovered by studying the Ty elements present in S. cerevisiae[27]. These Ty elements belong to five families, Ty1 to Ty5. Ty3 is a Gypsy-like retrotransposon and Ty1, Ty2, Ty4 and Ty5 are Copia-like retrotransposons. In the reference genome of the S288c strain, the organization of the Ty elements among potentially full-length active copies and solo-LTR resulting from inter-LTR recombination has been thoroughly annotated and described [26,28]. In this strain, the Ty1 family is the largest and most active one: it contains 32 full-length copies and more than 250 solo-LTRs. Previous studies [29-32] and analyses on the genome sequences of several additional S. cerevisiae strains [33-38] have clearly shown the existence of differences in the Ty localization, the number of copies and the relative size of Ty families, depending on the genetic background involved. However, except for the strain K7, these analyses have been restricted to full-length Ty elements and no systematic comparative surveys of the complete Ty landscape associated with solo-LTR elements have yet been carried out with a view to further understanding how Ty elements have contributed to the genotypic and phenotypic diversity of S. cerevisiae.
The complete genome sequences of 41 S. cerevisiae isolates available were therefore used in this study to examine and compare the Ty-related elements occurring in a whole species. The number of copies and the genomic locations of all the Ty elements were determined. The resulting overall picture yielded some interesting clues about the evolutionary history of Ty-related polymorphism. We also detected new Ty1 variants, and observed that the repertoires of subfamilies corresponding to the closely related Ty1 and Ty2 elements differ from one strain to another.
Results and discussion
Genome-wide detection of Ty elements in various S. cerevisiae genetic backgrounds
The genomic assemblies available for 41 S. cerevisiae strains were sampled: these consisted of the reference strain S288c and a set of 40 additional strains covering a broad range of ecological and geographical origins (Table 1). The large range of geographical origins and habitats was previously found to be associated with considerable genomic variability in the single nucleotide polymorphism (SNP) of the strains [39], which raised questions about the variability of the Ty elements present in these strains.
Table 1.
Strains investigated in this study
| Strain | Location | Source | Reference |
|---|---|---|---|
| AWRI1631 |
South Africa |
Wine |
[40] |
| AWRI796 |
|
Wine |
[40] |
| CBS7960 |
Brazil |
Bioethanol |
* |
| CLIB215 |
New Zeland |
Baker |
* |
| CLIB324 |
Vietnam |
Baker |
* |
| CLIB382 |
Ireland |
Beer |
* |
| EC1118 |
France |
Wine |
[35] |
| FL100 |
|
Laboratory |
* |
| FOSTERSB |
|
Beer (ale) |
[40] |
| FOSTERSO |
|
Beer (ale) |
[40] |
| I14 |
Italy |
Vineyard (soil) |
* |
| IL01 |
US |
Nature (soil) |
* |
| JAY291 |
Brazil |
Bioethanol |
[36] |
| LALVINQA23 |
|
Wine |
[40] |
| M22 |
Italy |
Vineyard |
* |
| NC02 |
US |
Nature (tree exudate) |
* |
| PW5 |
Nigeria |
Fermention (palm wine) |
* |
| RM11 |
US |
Wine |
[41] |
| S288C |
US |
Laboratory |
** |
| SIGMA1278 |
|
Laboratory |
[42] |
| SK1 |
|
Laboratory |
[43] |
| T73 |
Spain |
Wine |
* |
| T7 |
US |
Nature (tree exudate) |
* |
| UC5 |
Japan |
Sake |
* |
| VIN13 |
|
Vineyard |
[40] |
| VL3 |
|
Wine |
[40] |
| WE372 |
South Africa |
Wine |
* |
| Y10 |
Philipines |
Fermentation (coconut) |
* |
| Y12 |
Ivory Coast |
Fermentation (palm wine) |
* |
| Y9 |
Indonesia |
Fermentation (ragi) |
* |
| YJM269 |
|
Fermentation (apple juice) |
* |
| YJM280 |
US |
Clinical |
* |
| YJM320 |
US |
Clinical |
* |
| YJM326 |
US |
Clinical |
* |
| YJM421 |
US |
Clinical |
* |
| YJM428 |
US |
Clinical |
* |
| YJM451 |
US |
Clinical |
* |
| YJM653 |
US |
Clinical |
* |
| YJM789 |
US |
Clinical |
[33] |
| YPS1009 |
US |
Nature (oak exudate) |
* |
| YPS163 | US | Nature (oak exudate) | * |
Variability of the LTR contents
Since LTRs are the most abundant Ty sequences in the reference S288c genome, LTR sequences from Ty1 to Ty5 elements were used as query sequences to screen the 41 genomic sequences. The query sequences were chosen from representative transposition competent elements belonging to each Ty family (Additional file 1). The number of LTR sequences detected in each strain and their distribution among Ty families were determined (Figure 1A and Additional file 2: Table S1). The total number of LTRs detected was found to range from 147 (in the strain T73) to 463 (in the strain SK1), giving a mean number of 315 elements. No clear-cut correlations were detected between the LTR contents and the ecological and geographical origins of the strains, but only a slight bias in the case of the laboratory and clinical strains, which showed the highest LTR contents. The Ty1 LTRs are the most abundant, accounting for 59% of the elements detected both on average and in each individual strain. These results show that LTR sequences belonging to all five Ty families are present and have accumulated in all the strains investigated here. If we take the present number of LTR copies to be an indicator of former transpositional activity, the Ty1 elements can be said to be the most transpositionally active elements in the S. cerevisiae species as a whole. The three-fold difference observed between the maximum and minimum number of LTR copies suggests that the transpositional activity responsible for the process of LTR accumulation observed differs from one strain to another. However, the differences between the strains investigated here were far from being comparable to those observed between the tirant LTR retrotransposon and the helena non-LTR retrotransposon in Drosophila simulans populations [44,45] and between the mPing MITE transposons in various rice strains [46]. Alternatively, some strains may have undergone intense transpositional activities, but if their LTR elements are highly fragmented as the result of successive nested insertions, they may have escaped both resolution during the steps generating the genomic assemblies and detection by our searches. However the latter point cannot be addressed without finishing the genomic sequences manually.
Figure 1.

Differences between strains in the LTR contents. The size of the bars indicates the total number of LTR copies in each strain, and in each Ty1 to Ty5 family. A) All the LTRs detected B) LTRs belonging to Ty coding-elements. The arrowheads indicate the Ty1 relic copies.
Variability of the Ty coding-element contents
Previous authors have described the differences between strains in terms of the Ty coding-elements they contain [33-36]. In the reference genome S288c, only 15% of the Ty sequences were found to be full-length Ty elements encoding the proteins TyA (Gag) and TyB (Pol). The remaining sequences correspond to solo-LTRs, which result from inter-LTR recombination events. We therefore investigated the contents of the various genomes in terms of their potentially full-length active Ty element contents. For this purpose, the adjacent LTR sequences were extracted and screened to detect the presence of either TyA or TyB coding sequences in the appropriate orientation. The resulting data show that only 5% of the LTRs detected belong to Ty coding-elements (Figure 1B, Additional file 2: Table S1). Contrary to the overall LTR contents, the abundance of the LTRs belonging to Ty coding-elements is highly variable among the strains (Additional file 3: Figure S1) and only weakly correlated with the total number of LTR copies (Additional file 4: Figure S2, R = 0.415). This variability was observed at several levels. First, the two strains S288c and SIGMA1278 contain a remarkably large number of LTRs corresponding to Ty coding-elements amounting to 99 and 86 copies, respectively, which account for 30% of all the LTRs belonging to coding-elements. Secondly, three strains (NC02, PW5, UC5) have no LTRs belonging to Ty coding-elements. Segments of coding-elements were detected, however, in the genome assemblies of these strains when TYA and TYB sequences were used as query sequences, which suggests that these strains may carry very few intact Ty elements (Additional file 2: Table S1). Lastly, only 21 of the remaining strains have more than five LTR copies corresponding to Ty coding-elements. In the strains showing very few Ty coding-elements, either the transpositional activity responsible for the previous process of LTR accumulation has decreased or the present transpositional activity does not suffice to counterbalance the loss of full-length elements resulting from inter-LTR recombination events. Importantly, we have checked that there is no correlation between the contents in Ty coding-elements and the quality criteria of the surveyed genome assemblies (Additional file 5: Table S2). All the genome assemblies studied here result from sequencing methods generating reads, which size exceeds the size of a single Ty LTR (Additional file 5: Table S2). Nevertheless, for three strains (CLIB382, NC02 and YJM428), one should not exclude that very few coding-Ty have been detected because of the particularly low quality of their assemblies (more than 10,000 scaffolds).
On average, the proportion of LTRs observed in the Ty coding-elements belonging to families Ty1 to Ty5 was the same as in the S288c reference strain (43%, 39%, 5%, 6% and 2%, respectively). The rates of occurrence of LTRs in Ty5 coding-elements are slightly higher (7% on average). In many of the individual strains, however, the above proportions between Ty families were no longer observed. This is partly due to the fact that in several strains, some Ty families lack LTRs belonging to coding-elements. As previously described in the strains YJM789 and EC1118 [33,35], for example, 24 additional strains may lack either Ty3 or Ty4 coding-elements, or both. Another example is given by the Ty1/Ty2 ratio, which is commonly used to compare yeast strains [35,47,48]. Among the strains investigated here, this ratio was found to be variable, either due to no Ty2 coding-elements being detected (in strains CLIB382, I14 and Y12) or to the prevalence of Ty2 full-length elements over Ty1 elements (FOSTERSO, RM11, FL100, CLIB215, IL01 and CLIB382 strains). It is worth noting that the sole Ty1 coding-element detected in RM11 and CLIB215 is in fact an inactive relic (see below), which indicates that Ty1 may be extinct in these two strains. However, the LTR contents attest that Ty1 was recently an active Ty family.
Importantly, the highly variable Ty coding-element contents result in differences in the future Ty expansions among the various isolates. The existence of ‘Ty permissive’ strains, in which full-length potentially functional Ty elements have subsisted, and ‘non Ty permissive’ strains, which are poorly endowed or even devoid of functional Ty elements raises several questions. (i) Are the differences in the Ty coding-elements due to a recent decrease in transpositional activity or to an enhanced host response, leading to the loss of Ty coding-elements? Interestingly, it was reported in a previous study that the ‘Ty permissive’ strain FL100 showed greater transpositional activity than the ‘Ty permissive’ strain S288c, which suggests that the mechanism involved in Ty maintenance may depend on the genetic background [49]. (ii) May the differences in Ty content result from differences between the genetic backgrounds rather than depending on the strains’ preponderant state of propagation (haploid or diploid) or their ecological niches? If so, what are the genetic determinants responsible for the maintenance/deletion rates of functional Ty elements? (iii) Ty elements are known to be stress sensitive [27], and it has been hypothesized that populations showing enhanced TE activity are more likely to survive during environmental fluctuations because they produce a larger number of genomic variants for natural selection processes to work on [50]. It would therefore be interesting to compare the adaptive potential of these strains: how do the various Ty contents affect the adaptation processes and how are they themselves influenced during these processes?
Genome-wide distribution of the Ty elements
Genomes differ not only in their TE content but also in the location of their TE insertions, resulting in different maps of occupied and empty loci. In order to assess the intra-specific polymorphism of Ty insertions, we extracted the neighboring sequences with respect to the Ty elements detected (either LTRs, corresponding to all the Ty insertions, or Ty coding-elements) and mapped them against the S288c reference genome (Additional file 6: Figure S3). It is worth noting that the distributions of the Ty-related insertions detected are consistent with their respective target preferences. Ty1 to Ty4 show a preference for insertion points located near genes transcribed by RNA polymerase III [28,51]. Genes of this kind were found to be present in the flanking sequence of 62% of the elements detected (Additional file 2: Table S1).
Different ways of presenting the resulting data illustrate the various aspects of the polymorphism of Ty insertions. At each Ty insertion locus, we sought to determine whether its occupancy was specific to a given strain or whether it also occurred in other strains. The insertion maps give the number of strains showing a Ty insertion belonging to the same family at the same locus (Additional file 6: Figure S3). This number ranged between one and 41 strains, depending on the locus. The maps show nearly homogeneous patterns of inter-chromosomic distribution between singly and shared occupied loci, regardless of the Ty family involved. Two noticeable exceptions were observed, however: chromosome XI does not carry any highly shared loci in the case of the families Ty2, Ty3 and Ty4, and chromosome XV does not carry any highly shared loci in the case of the Ty4 family. Highly shared loci may correspond to fixed TE insertions occurring either early during the host’s evolution and/or as the result of positive selection processes.
Focusing on Ty coding-elements, we observed that contrary to what occurs with the LTRs, the patterns of locus occupancy are mostly either specific to a given strain or shared by just a few strains (only 18 out of the 130 occupied loci are common to more than five strains). The insertion maps are therefore potentially more polymorphic in the case of Ty coding-elements than in that of LTRs (see below). Few of the loci occupied by a Ty coding-element insertion in a given strain also carry an LTR insertion in the other strains. This finding indicates that recent strain-specific transposition events were the main cause of the insertional polymorphism observed. It also suggests that the loss of Ty coding-elements mediated by inter-LTR recombination events occurred early after the transpositional insertion process. Interestingly, the two Ty coding-element insertions that are common to the largest number of strains are non-functional relics of Ty1 coding-elements located on chromosome IV (one of which was mentioned above, and will be referred to again below). Both copies may encode the TyA protein but lack the TYB gene and the terminal LTR, which may have enabled the excision of their coding region to occur.
In addition, the loci containing Ty1 insertions are occupied by a larger number of strains (20 strains on average) than the loci where the insertions belong to other Ty families (Additional file 6: Figure S3). The occupancy of the loci containing Ty2 is that which occurs the least commonly among the strains (involving only 7 strains on average). Half of the loci containing Ty1 were observed, for example, in more than 20 other strains, whereas only 11 to 22% of the loci occupied by the families Ty2 to Ty5 are common to more than 20 strains. These differences between Ty families were presented by plotting the number of insertions against the number of strains showing these same insertions at the same locus, normalized by the total number of insertions observed in the whole Ty family (Figure 2). This yielded characteristic patterns reflecting the level of polymorphism of each Ty family. The Ty1 pattern is characterized by a low level of polymorphism between individuals because almost all the same loci are occupied in many strains. By contrast, the Ty2 pattern observed shows that the insertions are equally distributed between single loci and loci with medium and high rates of common occupancy, and the Ty3 insertions preferentially show medium rates of common occupancy. In the case of Ty4, most of the insertions are highly shared, but there are also considerable numbers of single and medium rates of occurrence of common insertion. Ty5 insertions occur at either single or common loci, but the small number of insertions observed (26) makes it difficult to detect a significant pattern of distribution.
Figure 2.

Patterns of insertional polymorphism of the Ty1 to Ty5 families. The number of loci occupied by an LTR insertion was plotted against the number of strains having insertions at the same locus, normalized by the total number of insertions corresponding to each Ty family.
Lastly, in order to show up the likenesses and differences between strains, we compared their profiles of locus occupancy (Figure 3). These profiles were drawn up on the basis of presence/absence matrices (Additional file 7) in the case of both the LTRs (Figure 3) and the coding elements present in each Ty family (Additional file 8: Figure S4). TE distributions are assumed to recapitulate both the history of the TE family and the evolution of the host [50]. TE insertions (and particularly retrotransposon insertions) are therefore widely used as genetic markers for studying evolutionary and population relationships. This emerged particularly clearly here upon looking at the common insertions detected between the closely related lab strains S288c, FL100 and SIGMA1278 (Figure 3 and Additional file 8: Figure S4) [52]. In this context, the fact that the insertion profiles of a given TE family are very similar in all the strains may result from the presence of fixed insertions and reflects the fact that few new insertions have occurred since the divergence of the strains. However, the Ty1 LTR profiles cannot be interpreted on these lines. It is known from the S288c reference strain that at a given insertion locus, several Ty1 insertions are often adjacent and even nested, whereas the insertions from the other Ty families are considerably less numerous and more widely dispersed. The low level of polymorphism observed among Ty1 occupied loci (Figure 2) and the large number of loci common to many strains may therefore result here from the saturation of the Ty1 insertion sites rather than from the presence of fixed insertions. It can be seen from the Ty2 LTR insertion profiles that these sites are the most variable among strains, reflecting a later and probably still ongoing period of activity. These findings are consistent with (i) the hypothesis that S. cerevisiae Ty2 elements have been recently acquired [22,26] and with (ii) the fact that, at least in some strains, Ty2-related sequences are transposed in the form of Ty1/2 hybrids (see below). In the case of these hybrid elements, the transcription rates recorded in [53] and the transposition rates in the S288c background recorded in [54] are particularly high. The profiles of the Ty4-related loci are those showing the largest numbers of insertions common to many strains (Figure 3). However, some strains carry additional insertions (Y9, Y12, Y10, YJM269 and SK1), which suggests that Ty4 activity occurred later on. The Ty3 and Ty5 profiles are the most highly structured ones: the insertion profiles of loci with mean rates of shared occupancy reflect the presence of clearly visible strain clusters. These clusters may have resulted from a period of activity of Ty3 and Ty5 elements that took place after the Ty4 expansion and before the Ty2 expansion. Both the Ty3 and Ty4 profiles suggest that they resulted from several waves of amplification/activation subsequent to periods of inactivity. It was previously suggested that this behavior might explain the distribution of the LTR-retrotransposon families observed in the rice genome [55].
Figure 3.

Differences between strains in the locations of LTR insertions. LTR insertion profiles of Ty1 to Ty5 families: in each strain, each grey rectangle indicates the presence of a Ty insertion at the corresponding locus. Dark grey rectangles indicate insertions in common with the S288c reference strain. Hierarchical clustering analysis was applied to both the strains and the loci. The resulting trees are presented in the case of the strains.
It has been stated that the majority of the Ty insertions are fixed and that the catalog of Ty insertion sites discovered using the S288c reference genome describes the core state of most Ty element locations across strains in S. cerevisiae[26]. The comparisons made here between insertion profiles clearly show which insertions are specific to each of the strains investigated. For example, the strain SK1 was found to have a remarkably large number of Ty1, Ty2 and Ty3 insertions, which is consistent with previous data on its SNP polymorphism [39]. IL01 carries a specific set of Ty3 insertions and Y9 and Y12 have particular Ty4 insertion profiles. It would be interesting to know whether these Ty amplifications may have an impact on the phenotypic diversity of these isolates.
Variability of the Ty1 and Ty2 coding-elements
TE copies of the same element are not identical and the diversity of the TEs themselves may contribute to the strain diversity. Ty1 and Ty2 related sequences are the most abundant sequences detected in the strains investigated here. Based on phylogenetic data, these two families have been found to be closely related [25]. However, their coexistence in S. cerevisiae does not result from a Ty speciation process occurring in the same host, but Ty2 may have been acquired via a process of horizontal transfer from the S. mikatae species [22,26]. The most suitable regions for discriminating elements in these two families are located in the coding regions, especially the Gag coding region [28,56]. Here we sampled more than 400 segments from coding-elements belonging to these two families, thus increasing the set of sequences available for investigating the variability of the Ty1 and Ty2 families. These analyses focused on the extremities of the coding sequences because they are assumed to be more accurately assembled than the internal sequences. Approximately 300 Ty segments were extracted and aligned, corresponding to the first 300 nucleotides downstream and upstream of the LTRs, which have been referred to as TYA300 and TYB300. We performed independent phylogenetic reconstructions using these two sets of sequences (Additional file 9 and Additional file 10). The resulting trees (Figure 4) provide a useful means of displaying not only the sequence diversity but also the distribution of the Ty subfamilies in the various strains. For example, they clearly show the lack of full-length Ty1 observed in RM11 and CLIB215 (Additional file 11: Figure S5).
Figure 4.
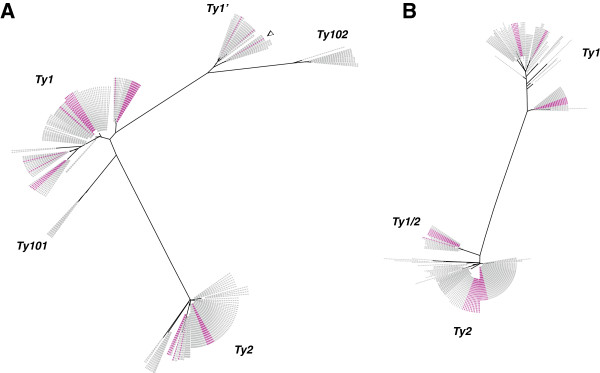
Ty1 and Ty2 coding-element subfamilies. Phylogenic trees were drawn up, based on 300 aligned nucleotide positions. Individual sequence names have been omitted. Branches are drawn to scale. Pink-colored leaves correspond to sequences detected in the strain S288c. Grey leaves correspond to sequences detected in all the remaining strains. The corresponding Ty family or subfamily is indicated for the clusters. The arrowhead indicates the Ty1’ relic copy. A) TYA300 neighbor joining tree based on the 300 nucleotides, in line with the 5′ LTR and the distribution of elements in the strain S288c. B) TYB300 neighbor joining tree based on the 300 nucleotides preceding the 3′ LTR and the distribution of elements in the strain S288c.
The TYA300 tree (Figure 4A) reveals how large the Ty1’ subfamily is. This divergent subfamily differs from Ty1 mainly in its variant TYA sequence. It was initially described in the strain S288c [28], where it belongs to a minor group consisting of only two potentially active elements. Another 20 copies of potentially active Ty1’ members were detected here in the strains FL100, Y9, Y12, YJM320, YJM421, YJM269 and YJM789. In Y12 and YJM269, there are even as many Ty1’ elements as canonical Ty1 elements; whereas no potentially functional Ty1’ copies were detected in some of the ‘Ty permissive’ strains (SIGMA1278, SK1 and CBS7960). The Ty1’ subfamily also includes one of the two Ty relics mentioned above. This relic, which lacks the TYB gene and the 3′ LTR, was detected on chromosome IV (coord. 800,000) of 15 of the strains investigated, some of them apparently devoid of active Ty1’ copies. This particular Ty1’ copy was therefore produced prior to the separation of these strains. Altogether, these findings support for strain specific extinctions or amplifications of this Ty variant to have occurred.
The TYA300 tree shows the presence of two clusters that do not include any elements belonging to the reference strain S288c and therefore correspond to new Ty1 variants. The variant we have called Ty101 is related to Ty1’ (89% identity). The corresponding element was detected in 17 strains at the same location (around position 999,000 on chromosome IV), whereas a Ty2-Ty1 tandem is present at this position in S288c. This variant is the second of the degenerate non-functional Ty elements mentioned above: it has undergone chromosomal rearrangements resulting in the loss of TYB, and the TYA sequence is preceded by an LTR with the opposite orientation. The pattern of organization and the sequence of this variant are highly conserved among the 17 strains (99.5% identity). It is worth noting that this fossil element harbors a single nucleotide substitution at the primer binding site. It should therefore be possible to initiate reverse transcription by using the acceptor stem of the tRNA encoded by tT(UGU)H rather than the tRNAs encoded by the IMT genes. The other variant, which we have called Ty102, shows 92% identity with the TYA300 from Ty1 elements and a set of specific SNPs (Additional file 9). One copy of this variant is present in seven strains (CBS7960, FL100, T73, Y10, YJM269, YJM280 and YJM653) at various genomic locations, which suggests that unlike Ty101, it is still transpositionally active.
Like the TYA300 tree, the TYB300 tree (Figure 4B) shows the existence of a clear-cut phylogenetic separation between the Ty1 and the Ty2 sequences. However, the two clades do not suffice to be able to differentiate accurately between all the Ty1 and Ty2 elements: several annotated Ty1 elements belonging to the strain S288c are included in two clusters containing Ty2. These elements correspond to the Ty1/2 hybrids previously described [56]. There are two types of hybrids: those with a Ty2 TYB terminal segment which is longer than 300 pb and could not be distinguished here from real Ty2 elements, and ‘short’ hybrids with a 60-pb long Ty2 segment. The latter elements belong to a distinct cluster containing seven S288c elements and 14 elements belonging to other strains: eight SIGMA1278 elements, three YJM789 elements and one FL100, one CLIB324 and one YJM320 element. It is worth noting that a few additional strains, some of which are apparently not closely related to S288c, were found to carry hybrid Ty1/2 elements, which indicates that Ty2 sequences hitchhiking Ty1 elements and thus enabling them to propagate is not just an oddity which is specific to S288c. Further investigations are now required to check whether hybrid elements of this kind are specific to a whole subset of strains.
The present findings confirm the complexity of the structure of the Ty1- and Ty2-related subfamilies. They show the existence of considerable diversity, in the form of hybrid elements and divergent subfamilies, two of which correspond to newly described variants. Ty variants and hybrids may constitute innovations that improve the maintenance of these elements with time and during the evolution of the host. This is consistent with the fact that the Ty1’ Gag gene is known to have evolved in response to functional constraints [28]. It has also been established that during the ‘life cycle’ of a TE family [6], mutations lead to the occurrence of variant TE copies. Inter-element recombination processes occurring during retrotransposition events have also been found to drive the divergence among related LTR-retrotransposons [54,57]. This raises questions about the cohabitation in the same genome, of these variants sharing components of both TE and cellular origin. In this context, it is rather striking that Ty101 is not a currently successful element because reverse-transcription priming of this element by a distinct tRNA might have been expected to provide it with a selective advantage toward Ty1, Ty2 and Ty3. In addition, the patchy distribution of Ty1’ and Ty102 may reflect the fact that they encounter variable levels of success, possibly depending on the background of the strain. As regards the origins of the various Ty1 variants, it is of particular interest that based on the TYA sequence, Ty1 and Ty1’ were found to be as divergent as Ty1 and Ty2 elements; whereas the coding elements belonging to the Ty3, Ty4 and Ty5 families show much less diversity (data not shown). As in the case of the Ty2 family, this finding suggests that a horizontal transfer process may have been responsible for the origin of Ty1’ rather than a process of speciation taking place within the same host.
Conclusions
Based on the whole genome data available for various S. cerevisiae strains, an initial overall picture of the intra-specific genetic diversity of this important model organism was compiled in this study, focusing on its Ty retrotransposon content. The results presented here show the considerable differences, which exist between these strains in terms of the number of full-length Ty elements, which may in turn act on the future variability of the strains. Some of the strains investigated were found to show considerable insertion polymorphism. As Ty insertions are known to alter the rates of expression of adjacent genes [58-61], it would be worth performing further studies in order to assess the potential impact of this polymorphism on the phenotypic characteristics of these strains. Finally, the differences observed here in the composition of the Ty1 subfamilies may be attributable to differences between the strain dependent Ty maintenance strategies involved. This initial approach was necessary to be able to further investigate and understand the effects of Ty elements on the S. cerevisiae genome and the interactions between these elements, which govern the equilibrium between Ty loss and expansion.
One of the main problems which still remain to be solved is that of the assembling of the large repetitive sequences of which TEs consist. It was not possible here to determine the differences between strains in terms of the mutated and potentially non-autonomous full-length Ty elements they contain. However, several studies have shown that processes of competition and complementation between autonomous and non-autonomous TE elements may play an important role in TE dynamics [6,54,57,62]. Recent and still ongoing progress in high throughput sequencing methods may soon make it possible to perform routine sequencing on long reads with a view to assembling these long repetitive sequences without any need for laborious manual finishing. Another important topic that we will then be able to address is the resolution of Ty-related gross chromosomal rearrangements such as translocations in the genomes of each strain and their contribution to the diversity and evolution of S. cerevisiae.
Methods
Strains and genome assemblies
The geographical and ecological origins and references relating to the genome assemblies of the 41 strains investigated here are presented in Table 1. Further information about the surveyed genome assemblies are summarized in Additional file 5: Table S2.
Ty coding-element detection
Sequences containing Ty were detected in the genomic assemblies of 41 strains by performing similarity searches with the BLAST suite of programs [63]. Ty segments corresponding to the five Ty families were identified independently using query sequences from typical full-length elements (Additional file 1).
This first round of searches did not make it possible to discriminate between elements from the Ty1 and Ty2 families. In addition, in the 288c reference sequences, 18 out of the 32 full-length Ty1 elements are in fact Ty1/2 hybrids presumably resulting from recombination events occurring during reverse transcription processes in heterozygous virus-like particles [56]. These hybrid elements have inherited their TyB segment and their 240 bp long LTR-U3 segments from Ty2. The sequences detected with the Ty1 and Ty2 queries were therefore compared with a set of sequences from thoroughly characterized Ty1 and Ty2 elements, excluding the hybrid elements (Additional file 1). The best alignment score was used to assign the sequence affiliation to the Ty1 or Ty2 family. Importantly, in the cases where only the LTR or the 3′ extremity of an element were detected, the fact that Ty2 sequences can be propagated by both Ty1- and the Ty2-mediated processes makes it impossible to distinguish between Ty2 elements and Ty1/2 hybrids.
Mapping Ty coding-elements
The regions (2,500 nt long) flanking the Ty elements detected were retrieved from the assemblies of the strains investigated. The Repeat Masker program (http://www.repeatmasker.org.) was used to mask Ty-related sequences in order to map these flanking regions unambiguously along the S288c reference genome by performing similarity searches. The distributions of the Ty elements detected in each strain were compared in order to detect the existence of loci common to several strains as well as specific/singly occupied loci. These data were used to generate “presence/absence matrices” with which to construct heat maps with the R package. Sequences and coordinates of tRNA and RNA polymerase III transcribed genes were downloaded at http://yeastmine.yeastgenome.org/ (06/2012).
Sequence analyses
The search results were parsed using dedicated python scripts. Multiple sequence alignments were performed with ClustalW2 [64]. Phylogenetic trees were drawn up using the neighbor-joining method (with the Hasegawa-Kishino-Yano 85 substitution model) with Seaview [65]. The trees were then drawn with FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files.
Competing interests
The authors declare that they do not have any competing interests.
Authors’ contributions
AF and CBG designed the experiments and analysed the data. AF performed the experiments. CBG and JS wrote the manuscript. JS planned the study, participated in its design and acted as the coordinator. All the authors have read and approved the manuscript.
Supplementary Material
Query sequences used in similarity searches.
Number of total LTRs and LTRs from Ty coding-elements per strain.
Distributions of LTR contents in the 41 strains. Boxplots representing distributions of LTR contents (total LTRs and LTRs from coding-Tys) in the 41 strains.
Correlations between the number of LTR copies and the number of LTRs belonging to Ty coding-elements in each strain. Each point corresponds to one of the investigated strains.
Characteristics of the genomic assemblies.
Chromosomal locations of the Ty insertions. An individual map was drawn up for each Ty1 to Ty5 family. The horizontal axes correspond to the 16 concatenated chromosomes. Alternate white and yellow boxes mark out the chromosome boundaries. Along the chromosomes, the vertical bars indicate the position of the loci corresponding to Ty insertions. Blue bars correspond to the presence of LTR, and red bars correspond to the presence of Ty coding-elements. The size of the bars is proportional to the number of strains carrying a Ty insertion at the same locus. Grey bars indicate the position of RNA polymerase III transcribed genes. The arrowheads indicate the Ty1 relic copies.
Presence/absence matrices.
Differences between strains in the locations of Ty coding-element insertions. Ty coding-element insertion profiles in families Ty1 to Ty5: in each strain, each grey rectangle indicates the presence of a Ty insertion at the corresponding locus. Dark grey rectangles indicate insertions in common with the S288c reference strain. Hierarchical clustering was applied to both the strains and the loci. The resulting trees are presented in the case of the strains.
TYA300 multiple alignments.
TYB300 multiple alignments.
Distribution of strain sequences in the TYA300 and TYB300 trees. Distribution of RM11 (purple) and CLIB215 (blue) sequences in TYA300 (A) and in TYB300 (B) trees. The arrowheads indicate the Ty1’ relic copies.
Contributor Information
Claudine Bleykasten-Grosshans, Email: bleykasten@unistra.fr.
Anne Friedrich, Email: anne.friedrich@unistra.fr.
Joseph Schacherer, Email: schacherer@unistra.fr.
Acknowledgements
We thank Justin Fay and Leonid Kruglyak for making the genomic sequences of some of the strains investigated available to the scientific community. We thank Sophie Siguenza for her help with the tree drawing. This research was supported by an ANR grant (2011-JSV6-004-01).
References
- Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet. 2008;14:587–617. doi: 10.1146/annurev.genet.42.110807.091549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Usaite R, Germann SM, Lisby M, Botstein D, Regenberg B. Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc Natl Acad Sci USA. 2010;14(43):18551–18556. doi: 10.1073/pnas.1014023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2013;14(1):49–61. doi: 10.1038/nrg3374. [DOI] [PubMed] [Google Scholar]
- Wang X, Weigel D, Smith LM. Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet. 2013;14(2):e1003255. doi: 10.1371/journal.pgen.1003255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenais B, Caruso A, Hiard S, Casse N. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene. 2012;14(1):7–15. doi: 10.1016/j.gene.2012.07.042. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Capy P. Population genetics models of competition between transposable element subfamilies. Genetics. 2006;14(2):785–793. doi: 10.1534/genetics.105.052241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ. Transposable elements and factors influencing their success in eukaryotes. J Hered. 2009;14(5):648–655. doi: 10.1093/jhered/esp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira C, Fablet M, Lerat E, Boulesteix M, Rebollo R, Burlet N, Akkouche A, Hubert B, Mortada H, Biemont C. A comparative analysis of the amounts and dynamics of transposable elements in natural populations of Drosophila melanogaster and Drosophila simulans. J Environ Radioact. 2012;14:83–86. doi: 10.1016/j.jenvrad.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Grzebelus D. Transposon insertion polymorphism as a new source of molecular markers. J Fruit Ornam Plant Res. 2006;14(14):21–19. [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T. et al. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;14(7):1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro MP, Fontdevila A. Osvaldo and Isis retrotransposons as markers of the Drosophila buzzatii colonisation in Australia. BMC Evol Biol. 2011;14:111. doi: 10.1186/1471-2148-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampicinini G, Cervella P, Biemont C, Sella G. Insertional variability of four transposable elements and population structure of the midge Chironomus riparius (Diptera) Mol Genet Genomics. 2011;14(3–4):293–305. doi: 10.1007/s00438-011-0646-8. [DOI] [PubMed] [Google Scholar]
- Zerjal T, Rousselet A, Mhiri C, Combes V, Madur D, Grandbastien MA, Charcosset A, Tenaillon MI. Maize genetic diversity and association mapping using transposable element insertion polymorphisms. Theor Appl Genet. 2012;14(8):1521–1537. doi: 10.1007/s00122-012-1807-9. [DOI] [PubMed] [Google Scholar]
- McLain AT, Meyer TJ, Faulk C, Herke SW, Oldenburg JM, Bourgeois MG, Abshire CF, Roos C, Batzer MA. An alu-based phylogeny of lemurs (infraorder: Lemuriformes) PLoS One. 2012;14(8):e44035. doi: 10.1371/journal.pone.0044035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DA, Batzer MA. Reading TE leaves: new approaches to the identification of transposable element insertions. Genome Res. 2011;14(6):813–820. doi: 10.1101/gr.110528.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Lu G, Zhao Q, Liu X, Han B. Genome-wide analysis of transposon insertion polymorphisms reveals intraspecific variation in cultivated rice. Plant Physiol. 2008;14(1):25–40. doi: 10.1104/pp.108.121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Sen SK, Konkel MK, Batzer MA. Computational methods for the analysis of primate mobile elements. Methods Mol Biol. 2010;14:137–151. doi: 10.1007/978-1-60327-367-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabot F, Picault N, El-Baidouri M, Llauro C, Chaparro C, Piegu B, Roulin A, Guiderdoni E, Delabastide M, McCombie R. et al. Transpositional landscape of the rice genome revealed by paired-end mapping of high-throughput re-sequencing data. Plant J. 2011;14(2):241–246. doi: 10.1111/j.1365-313X.2011.04492.x. [DOI] [PubMed] [Google Scholar]
- Dujon B. Yeasts illustrate the molecular mechanisms of eukaryotic genome evolution. Trends Genet. 2006;14(7):375–387. doi: 10.1016/j.tig.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Bleykasten-Grosshans C, Neuveglise C. Transposable elements in yeasts. C R Biol. 2011;14(8–9):679–686. doi: 10.1016/j.crvi.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Legras JL, Karst F. Optimisation of interdelta analysis for Saccharomyces cerevisiae strain characterisation. FEMS Microbiol Lett. 2003;14(2):249–255. doi: 10.1016/S0378-1097(03)00205-2. [DOI] [PubMed] [Google Scholar]
- Liti G, Peruffo A, James SA, Roberts IN, Louis EJ. Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex. Yeast. 2005;14(3):177–192. doi: 10.1002/yea.1200. [DOI] [PubMed] [Google Scholar]
- Dunn B, Sherlock G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008;14(10):1610–1623. doi: 10.1101/gr.076075.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B, Richter C, Kvitek DJ, Pugh T, Sherlock G. Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res. 2012;14(5):908–924. doi: 10.1101/gr.130310.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuveglise C, Feldmann H, Bon E, Gaillardin C, Casaregola S. Genomic evolution of the long terminal repeat retrotransposons in hemiascomycetous yeasts. Genome Res. 2002;14(6):930–943. doi: 10.1101/gr.219202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr M, Bensasson D, Bergman CM. Evolutionary genomics of transposable elements in Saccharomyces cerevisiae. PLoS One. 2012;14(11):e50978. doi: 10.1371/journal.pone.0050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage P, Todeschini AL. Happy together: the life and times of Ty retrotransposons and their hosts. Cytogenet Genome Res. 2005;14(1–4):70–90. doi: 10.1159/000084940. [DOI] [PubMed] [Google Scholar]
- Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;14(5):464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- Wilke CM, Adams J. Fitness effects of Ty transposition in Saccharomyces cerevisiae. Genetics. 1992;14(1):31–42. doi: 10.1093/genetics/131.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Castillo-Davis CI, Oshiro G, Liang D, Richards DR, Zhou Y, Hartl DL. Genetic diversity in yeast assessed with whole-genome oligonucleotide arrays. Genetics. 2003;14(1):79–89. doi: 10.1093/genetics/163.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel A, Dapprich J, Kunkel M, Gresham D, Pratt SC, Dunham MJ. Global mapping of transposon location. PLoS Genet. 2006;14(12):e212. doi: 10.1371/journal.pgen.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan SJ, Scheifele LZ, Martinez-Murillo F, Irizarry RA, Boeke JD. Transposon insertion site profiling chip (TIP-chip) Proc Natl Acad Sci USA. 2006;14(47):17632–17637. doi: 10.1073/pnas.0605450103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, McCusker JH, Hyman RW, Jones T, Ning Y, Cao Z, Gu Z, Bruno D, Miranda M, Nguyen M. et al. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci USA. 2007;14(31):12825–12830. doi: 10.1073/pnas.0701291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Forgan AH, Pretorius IS, Chambers PJ. Comparative genome analysis of a Saccharomyces cerevisiae wine strain. FEMS Yeast Res. 2008;14(7):1185–1195. doi: 10.1111/j.1567-1364.2008.00434.x. [DOI] [PubMed] [Google Scholar]
- Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S. et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA. 2009;14(38):16333–16338. doi: 10.1073/pnas.0904673106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso JL, Carazzolle MF, Mieczkowski PA, Duarte FM, Netto OV, Missawa SK, Galzerani F, Costa GG, Vidal RO, Noronha MF. et al. Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production. Genome Res. 2009;14(12):2258–2270. doi: 10.1101/gr.091777.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V. et al. Population genomics of domestic and wild yeasts. Nature. 2009;14(7236):337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akao T, Yashiro I, Hosoyama A, Kitagaki H, Horikawa H, Watanabe D, Akada R, Ando Y, Harashima S, Inoue T. et al. Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7. DNA Res. 2011;14(6):423–434. doi: 10.1093/dnares/dsr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;14(7236):342–345. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Desany BA, Riches D, Affourtit JP, Forgan AH, Pretorius IS, Egholm M, Chambers PJ. Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae. PLoS Genet. 2011;14(2):e1001287. doi: 10.1371/journal.pgen.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Pratt SC, Seidel HS, Kruglyak L. Population genomic analysis of outcrossing and recombination in yeast. Nat Genet. 2006;14(9):1077–1081. doi: 10.1038/ng1859. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B. et al. Genotype to phenotype: a complex problem. Science. 2010;14(5977):469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant KT, Wei W, Mancera E, Argueso JL, Schlattl A, Delhomme N, Ma X, Bustamante CD, Korbel JO, Gu Z. et al. The baker‘s yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 2010;14(9):e1001109. doi: 10.1371/journal.pgen.1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fablet M, McDonald JF, Biemont C, Vieira C. Ongoing loss of the tirant transposable element in natural populations of Drosophila simulans. Gene. 2006;14:54–62. doi: 10.1016/j.gene.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Granzotto A, Lopes FR, Lerat E, Vieira C, Carareto CM. The evolutionary dynamics of the Helena retrotransposon revealed by sequenced Drosophila genomes. BMC Evol Biol. 2009;14:174. doi: 10.1186/1471-2148-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR. Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci USA. 2006;14(47):17620–17625. doi: 10.1073/pnas.0605421103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibeas JI, Jimenez J. Genomic complexity and chromosomal rearrangements in wine-laboratory yeast hybrids. Curr Genet. 1996;14(5):410–416. doi: 10.1007/s002940050150. [DOI] [PubMed] [Google Scholar]
- Dunn B, Levine RP, Sherlock G. Microarray karyotyping of commercial wine yeast strains reveals shared, as well as unique, genomic signatures. BMC Genomics. 2005;14(1):53. doi: 10.1186/1471-2164-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch ES, Schacherer J, Bleykasten-Grosshans C, Souciet JL, Potier S, de Montigny J. Influence of genetic background on the occurrence of chromosomal rearrangements in Saccharomyces cerevisiae. BMC Genomics. 2009;14:99. doi: 10.1186/1471-2164-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosid E, Brodsky L, Kalendar R, Raskina O, Belyayev A. Diversity of long terminal repeat retrotransposon genome distribution in natural populations of the wild diploid wheat Aegilops speltoides. Genetics. 2012;14(1):263–274. doi: 10.1534/genetics.111.134643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hani J, Feldmann H. tRNA genes and retroelements in the yeast genome. Nucleic Acids Res. 1998;14(3):689–696. doi: 10.1093/nar/26.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Ruderfer DM, Gresham D, Dolinski K, Botstein D, Kruglyak L. Genome-wide analysis of nucleotide-level variation in commonly used Saccharomyces cerevisiae strains. PLoS One. 2007;14(3):e322. doi: 10.1371/journal.pone.0000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Benard L, Springer M, Lesage P. Differential effects of chromatin and Gcn4 on the 50-fold range of expression among individual yeast Ty1 retrotransposons. Mol Cell Biol. 2002;14(7):2078–2088. doi: 10.1128/MCB.22.7.2078-2088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleykasten-Grosshans C, Jung PP, Fritsch ES, Potier S, de Montigny J, Souciet JL. The Ty1 LTR-retrotransposon population in Saccharomyces cerevisiae genome: dynamics and sequence variations during mobility. FEMS Yeast Res. 2011;14(4):334–344. doi: 10.1111/j.1567-1364.2011.00721.x. [DOI] [PubMed] [Google Scholar]
- Baucom RS, Estill JC, Leebens-Mack J, Bennetzen JL. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009;14(2):243–254. doi: 10.1101/gr.083360.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, McDonald JF. Evidence for the role of recombination in the regulatory evolution of Saccharomyces cerevisiae Ty elements. J Mol Evol. 1998;14(1):14–20. doi: 10.1007/PL00006358. [DOI] [PubMed] [Google Scholar]
- Du J, Tian Z, Bowen NJ, Schmutz J, Shoemaker RC, Ma J. Bifurcation and enhancement of autonomous-nonautonomous retrotransposon partnership through LTR Swapping in soybean. Plant Cell. 2010;14(1):48–61. doi: 10.1105/tpc.109.068775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder GS, Rose AB, Pearlman RE. Transposable element sequences involved in the enhancement of yeast gene expression. Proc Natl Acad Sci USA. 1985;14(16):5428–5432. doi: 10.1073/pnas.82.16.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants F, Potier S, Souciet JL, de Montigny J. Delta sequence of Ty1 transposon can initiate transcription of the distal part of the URA2 gene complex in Saccharomyces cerevisiae. FEMS Microbiol Lett. 1997;14(1):69–74. doi: 10.1111/j.1574-6968.1997.tb10269.x. [DOI] [PubMed] [Google Scholar]
- Todeschini AL, Morillon A, Springer M, Lesage P. Severe adenine starvation activates Ty1 transcription and retrotransposition in Saccharomyces cerevisiae. Mol Cell Biol. 2005;14(17):7459–7472. doi: 10.1128/MCB.25.17.7459-7472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant G, Pennetier C, Lesage P. Remodeling yeast gene transcription by activating the Ty1 long terminal repeat retrotransposon under severe adenine deficiency. Mol Cell Biol. 2008;14(17):5543–5554. doi: 10.1128/MCB.00416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabot F, Schulman AH. Parasitism and the retrotransposon life cycle in plants: a hitchhiker‘s guide to the genome. Heredity. 2006;14(6):381–388. doi: 10.1038/sj.hdy.6800903. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;14(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;14(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;14(2):221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Query sequences used in similarity searches.
Number of total LTRs and LTRs from Ty coding-elements per strain.
Distributions of LTR contents in the 41 strains. Boxplots representing distributions of LTR contents (total LTRs and LTRs from coding-Tys) in the 41 strains.
Correlations between the number of LTR copies and the number of LTRs belonging to Ty coding-elements in each strain. Each point corresponds to one of the investigated strains.
Characteristics of the genomic assemblies.
Chromosomal locations of the Ty insertions. An individual map was drawn up for each Ty1 to Ty5 family. The horizontal axes correspond to the 16 concatenated chromosomes. Alternate white and yellow boxes mark out the chromosome boundaries. Along the chromosomes, the vertical bars indicate the position of the loci corresponding to Ty insertions. Blue bars correspond to the presence of LTR, and red bars correspond to the presence of Ty coding-elements. The size of the bars is proportional to the number of strains carrying a Ty insertion at the same locus. Grey bars indicate the position of RNA polymerase III transcribed genes. The arrowheads indicate the Ty1 relic copies.
Presence/absence matrices.
Differences between strains in the locations of Ty coding-element insertions. Ty coding-element insertion profiles in families Ty1 to Ty5: in each strain, each grey rectangle indicates the presence of a Ty insertion at the corresponding locus. Dark grey rectangles indicate insertions in common with the S288c reference strain. Hierarchical clustering was applied to both the strains and the loci. The resulting trees are presented in the case of the strains.
TYA300 multiple alignments.
TYB300 multiple alignments.
Distribution of strain sequences in the TYA300 and TYB300 trees. Distribution of RM11 (purple) and CLIB215 (blue) sequences in TYA300 (A) and in TYB300 (B) trees. The arrowheads indicate the Ty1’ relic copies.