

Abstract
Background
Supratentorial primitive neuroectodermal tumor (sPNET) is a malignant brain tumor with poor prognosis. New model systems that replicate sPNET's molecular subtype(s) and maintain cancer stem cell (CSC) pool are needed.
Methods
A fresh surgical specimen of a pediatric sPNET was directly injected into the right cerebrum of Rag2/SCID mice. The xenograft tumors were serially sub-transplanted in mouse brains, characterized histopathologically, and subclassified into molecular subtype through qRT-PCR and immunohistochemical analysis. CSCs were identified through flow cytometric profiling of putative CSC markers (CD133, CD15, CD24, CD44, and CD117), functional examination of neurosphere forming efficiency in vitro, and tumor formation capacity in vivo. To establish a neurosphere line, neurospheres were propagated in serum-free medium.
Results
Formation of intracerebral xenograft tumors was confirmed in 4 of the 5 mice injected with the patient tumor. These xenograft tumors were sub-transplanted in vivo 5 times. They replicated the histopathological features of the original patient tumor and expressed the molecular markers (TWIST1 and FOXJ1) of group 3 sPNET. CD133+ and CD15+ cells were found to have strong neurosphere-forming efficiency in vitro and potent tumor-forming capacity (with as few as 100 cells) in vivo. A neurosphere line BXD-2664PNET-NS was established that preserved stem cell features and expressed group 3 markers.
Conclusion
We have established a group 3 sPNET xenograft mouse model (IC-2664PNET) with matching neurosphere line (BXD-2664PNET-NS) and identified CD133+ and CD15+ cells as the major CSC subpopulations. This novel model system should facilitate biological studies and preclinical drug screenings for childhood sPNET.
Keywords: cancer stem cell, orthotopic xenograft model, supratentorial primitive neuroectodermal tumor
Supratentorial primitive neuroectodermal tumor (sPNET) is a group of highly malignant brain tumors primarily affecting young children. They account for 3%–7% of all childhood brain tumors.1 Many sPNETs exhibit similar histology and immunohistochemical characteristics as medulloblastoma (MB) and are often treated with protocols designed for high-risk MBs.2,3 However, sPNETs are more aggressive tumors and tend to be more refractory to multimodality treatment than MBs. Furthermore, current therapy, particularly craniospinal irradiation, can cause damage in the developing brain that results in long-term neurocognitive toxicity and compromised quality of life for surviving children.4–7
Recent studies have revealed that sPNETs and MBs are molecularly distinct tumors.8,9 Unlike MB, which is characterized by isochromosome 17q (i17q), sPNETs were found to carry a high-level amplicon involving the chromosome 19q13.41 microRNA (miRNA) cluster (C19MC) in 11 of 45 (∼25%) primary CNS-PNETs.10 Compared with the 4 molecular subtypes recently identified in MBs (SHH, WNT, group C, and group D),11 CNS PNETs were recently subcategorized into 3 molecular subtypes in a multicenter, international collaborative study: primitive neural (group 1), oligoneural (group 2), and mesenchymal lineage (group 3).12 Among the 3 subgroups, children with group 3 sPNETs often display metastasis at diagnosis, suggesting that sPNETs expressing the mesenchymal lineage markers are particularly aggressive. Despite the need of a model system for biological studies and preclinical drug screening, no cellular or animal models that replicate this highly aggressive subtype of childhood sPNETs have been developed.
The recent isolation of cancer stem cells (CSCs), also termed “stem-like cancer cell” or tumor-initiating cells, has created a new conceptual Paradigm of how tumors form and why current therapies fail.13,14 Following the isolation of CSCs in MB and glioblastoma, progress has been made in understanding their biology and developing new therapies.15–21 For childhood sPNETs, however, no CSCs have been identified, and it remains unknown if CSCs in sPNET would share similar phenotypic markers of MB stem cells, such as CD13322–26 and CD15,27–29 or stem cell markers identified in other types of normal and/or cancers cells such as CD24/CD44 in breast30–32 and prostate33–35 cancers, and CD117 in hematpoietic36 and osteosarcoma cells.37 Since there is ongoing debate about the relative abundance of CSCs and the specificity of single phenotypic markers in human cancers,38–40 it is also crucial to determine if sPNET stem cells exist that are CD133 and CD15 dual-negative.
Herein, we describe the establishment of a novel patient tumor-derived orthotopic xenograft mouse model of childhood sPNET and a long-term neurosphere line, the molecular grouping of the xenograft tumors during serial sub-transplantation in vivo in mouse brains, and the identification and cell surface marker profiling of CSCs in this model system.
Materials and Methods
Brain Tumor Specimen
A fresh tumor specimen, from a 14-year-old female who underwent craniotomy at Texas Children's Hospital, was obtained in the cryostat laboratory following an Institutional Review Board-approved protocol. Final pathological diagnosis was consistent with primitive neuroectodermal tumor. The tumor tissue (∼1 × 2 × 2 mm3) was quickly transferred in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) on ice to the tissue culture room, where it was washed and mechanically dispersed, as previously described.41,42 After the cell suspension was passed through a 40 μ cell strainer, live tumor cells as single cells and small clumps (∼5–10 cells) were counted with trypan blue staining and resuspended in growth medium (1 × 108/mL), followed by immediate transfer on ice to the animal facility.
Animal Experiment
Surgical transplantation of tumor cells into mouse cerebrum was performed as we described previously,41,42 following the Institutional Animal Care and Use Committee-approved protocol. The Rag2/SCID mice were bred and maintained in a specific pathogen-free animal facility at Texas Children's Hospital. Mice of both sexes, aged 6–8 weeks, were anesthetized with sodium pentobarbital (50 mg/kg). Tumor cells (1 × 105) were suspended in 2 µL of culture medium and injected into the cerebral hemisphere (1 mm to the right of the midline, 1.5 mm anterior to the lambdoid suture, and 3 mm deep) via a 10-µL 26-gauge Hamilton Gastight 1701 syringe needle. The animals were monitored daily until they developed signs of neurological deficit or became moribund, at which time they were euthanized and their brains removed for histopathological analysis.
Quantitative Real-time PCR
For gene-specific quantitative RT-PCR analysis of the molecular markers that subclassified sPNET into group 1, group 2, and group 3, we amplified 10 ng cDNA synthesized from 1 μg of RNA (TaqMan Reverse Transcription Kit, Applied Biosystems) by use of specific TaqMan probes-primer sets (see Supplementary Table 1) and determined mRNA expression levels relative to actin with the ΔCt method. All RT-PCR assays were done in triplicate.
Whole Genome Gene Expression Profiling
The genome-wide expression analysis was performed using Affymetrix U133 Plus 2.0 array, following the manufacturer's instructions. Total RNA was extracted from intracerebral xenograft tumors at passages I, III, and V with TRIzol reagent (Invitrogen), as described previously.43,44 Normal human cerebral cortex RNAs collected from 2 autopsied brain tissues were included as references. RNA concentrations were measured using Nanodrop 1 000 spectrophotometer. RNA integrity was checked with Agilent 2100 Bioanalyzer. After hybridization, the raw intensity data were log-transformed, normalized, and summarized by Robust Multichip Average (RMA) algorithm. The data were analyzed by BRB ArrayTools, developed by Dr. Richard Simon and the BRB-Array Tools Development Team.45 A heat map was created using the MultiExperiment Viewer, a part of the TM4 Microarray Software Suite.46,47
Immunohistochemical Staining
Immunohistochemical staining was performed using a Vectastain Elite kit (Vector Laboratories) on paraffin sections of whole mouse brains, as described previously.41,42,48 Primary antibodies included the following: glial fibrillary acidic protein (GFAP) (1:200; Dako), vimentin (VMT) (1:200; Dako), Ki67 (1:20; Abcam Inc,), rabbit anti-Nestin (1:500; Chemicon) for animal model characterization; LIN28 (1:400; Cell Signaling Technology) and OLIG2 (1:1 000; Immuno Biological Laboratories) for molecular subtype verification. After slides were incubated with primary antibodies, the appropriate biotinylated secondary antibodies were applied, and the final signal was developed using the 3, 3′-diaminobenzidine substrate kit for peroxidase. The negative control was performed by replacing primary antibodies with phosphate-buffered saline. The immunohistochemical staining was assessed by combining the intensity, scored as negative (–), low (+), medium (++), or strongly positive (+++), and the extent of immunopositivity expressed as the percentage of positive cells.
Neurosphere Assay and the Establishment of a Long-term Neurosphere Line
Single-cell suspensions from xenograft tumors were plated in clonal density and incubated in serum-free stem cell growth medium consisting of Neurobasal media N2 and B27 supplements (0.5x each; Invitrogen), human recombinant basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) (50 ng/mL each; R&D Systems), and penicillin G-streptomycin sulfate (1:100; GIBCO-Invitrogen)41,42
To establish a long-term culture of neurosphere line, the growth culture medium was changed every 3–5 days, and the cells split 1:2 when the media color changed and/or high density (>5/low power field) of big neurospheres (∼50–100 cells) were observed. Mycoplasma contamination was monitored with ATCC Universal Mycoplasma Detection kit).
Immunofluorescent Staining
To examine the multilineage differentiation capacities, preformed neurospheres were resuspended in DMED media supplemented with 10% FBS and plated into chamber slides coated with poly-D-lysine (R&D Systems) to allow for the attachment and outgrowth as monolayer cells. To examine the expression of stem- and differentiation-associated markers of the established neurosphere cell line, cultured neurospheres at passages 9–54 were seeded into chamber slides (BD Falcon) that were precoated with poly-D-lysine. For immunofluorescent staining, cells were fixed and stained with CD133 (1:500; Miltenyi Biote), nestin (1:500; Chemicon), GFAP(1:500; Dako), TuJ-1(1:1 000; Fitzgerald Industries International, Inc.), O4(1:100; R&D Systems), VMT (1:200; Dako), NeuN (1:500; Chemicon), and OTX2 (1:1 000; Chemicon) overnight at 4°C, followed by host-appropriate fluorescein isothiocyanate (FITC) or Texas Red secondary (Jackson Immunoresearch) and costained with DAPI (Sigma). Intensity images were captured using a Nikon fluorescence microscope attached with a color CCD camera.41,42,48 The immunofluorescent staining was assessed by combining the intensity, scored as negative (–), low (+), medium (++) or strongly positive (+++), and the extent of immunopositivity expressed as the percentage of positive cells.
Flow Cytometry (FCM) Analysis
Approximately 106 xenograft tumor cells were incubated with fluorochrome-conjugated antibodies for 15–30 minutes at 4°C in 100 µL of Hanks’ balanced saline solution (HBSS) containing 2% FBS and 5–20 µL of each undiluted antibody. After washing 3 times in HBSS, cells were suspended in HBSS supplemented with 5% FBS and subsequently analyzed with a LSR II (Becton Dickinson). The following antibodies were used: CD133-APC (1:10; Milteny), CD15-FITC, CD117-FITC, CD24-FITC (1:10; Abcam), and CD44-APC (1:5; BD Bioscience). Isotype controls: APC-H7 Mouse IgG2b, k Isotype Control (1:20; BD Bioscience), Mouse IgG1 FITC Isotype Control (1:10; Abcam), Mouse IgM FITC Isotype control (1:10 or 1:5; Invitrogen). For double staining, the tumor cells incubated with the first antibodies (usually conjugated with APC) were washed and incubated with the second antibodies, which had been conjugated with FITC. Positive staining with cell-surface-specific antibodies was defined as the emission of a level of fluorescence that exceeded the levels obtained by 99% of cells from the same starting population when these were stained with control fluorochrome-conjugated antibodies.
Florescence-activated Cell Sorting of CD133+ and/or CD15+ cells
Xenograft tumor cells were labeled with CD133-APC (Milteny) at 4°C for 10 minutes per manufacturer's instructions. These cells were then washed with HBSS and stained again with CD15-FITC (Abcam). The double-stained cells were resuspended in serum-free stem cell growth medium (as detailed under the section of Neurosphere Assay)22,25, and flow-sorted with Cytomation MoFlo (Dako). Side scatter and forward scatter profiles were used to eliminate cell doublets. Dead cells were excluded by propidium iodide staining.
In Vitro and in Vivo Limiting Dilution Assay for the Examination of Self-renewal Capacity
To examine self-renewal capacity of dual-positive (CD133+/CD15+), mono-positive (CD133+/CD15− and CD133−/CD15+), and dual-negative (CD133−/CD15−) cells in vitro, the purified subpopulations of tumor cells were plated in clonal density and incubated in the stem cell growth medium in 96-well plates for 14 days before being counted under a Nikon Eclipse phase contrast microscope. To examine their tumor-forming efficiency in vivo, the 4 subpopulations of florescence-activated cell sorting- (FACS) purified xenograft tumors were counted with trypan blue. The dual- and mono-positive CD133 and CD15 cells were resuspended in serum-free stem cell growth medium and injected into the right cerebrum of Rag2/SCID mice (10, 102, and 103 cells/mouse) in 2 µL of medium. The dual-negative (CD133−/CD15−) cells were resuspended in DMEM media supplemented with 10% FBS and injected at 102, 103, and 104 cells/mouse (n = 5 per group) as well.
Statistical Analysis
Comparisons between 2 groups were done using the t test and 2-way analysis of variance. Differences of animal survival times were compared through log-rank analysis followed by post-hoc Holmes pairwise comparisons using SigmaStat 3.5 (Systat Software) and graphed with SigmaPlot 11 (Systat Software). Limiting dilution analyses of FACS-purified CD133+ and/or CD15+ tumor cells in vivo were performed based on Bonnefoix et al.,49 using the limdil function of the “statmod” package (author G.K. Smyth, http://bioinf.wehi.edu.au/software/limdil/), part of the R statistical software project (http://www.r-project.org). Xenograft tumor-forming frequencies were compared using likelihood ratio tests.38,50 P < .05 was considered as statistically significant.
Results
Tumorigenicity and Sub-transplantability of Primary sPNET Cells in SCID Mice
We utilized mechanical dispersion techniques to prepare cell suspension from a fresh sPNET specimen and injected these tumor cells (1 × 105/mouse) into the right cerebrum of Rag2/SCID mice within 60 minutes of tumor resection. All 5 mice developed signs of neurological deficit or became moribund within 48–76 (66 ± 12.6) days post injection. In 4 of 5 mice, the growth of xenograft tumor was confirmed (Fig. 1A). Grossly, the mouse brains were enlarged often revealing a huge intracerebral (IC) xenograft tumor (Fig. 1A). This model was therefore designated as intracerebral xenograft model 2664 (IC-2664PNET).
Fig. 1.

Histopathological characteristics of the orthotopic xenograft mouse model IC-2664PNET. (A) Gross appearance and cross section of a mouse brain showing the growth of intracerebral xenograft tumor that had spread into the lateral (*) and 4th ventricle (arrowhead). (B) Log-rank analysis of animal survival times of IC-2664PNET during serial sub-transplantations of xenograft tumors in vivo in mouse brains (P < .05 between passages I and III, P > .05 in all other pairwise comparisons). (C) H&E staining showing the histology of the original patient tumor as compared with that of the xenograft tumors at passages I and III. (D) Immunohistochemical staining of the original patient tumor and xenograft tumors (passages I and III) for markers of cell proliferation (Ki67), glial differentiation (GFAP), neural progenitor marker (nestin), and vimentin (VMT) (magnification 40x). (E) Images showing the invasive growth of the xenograft tumor cells (arrow heads) in vivo.
To determine the sub-transplantability of the developed xenograft tumors, we injected the xenograft tumor cells (1 × 105) harvested from the donor mice into the brains of 5–10 recipient mice, as we described previously.41,42 Starting from passage II, a tumorigenicity of 100% was reproducibly maintained for more than 5 passages. Compared with the median survival times of 69 days in mice receiving the primary patient tumor (passage I), mice injected with xenograft tumor cells at passages II, III, IV, and V survived for 63, 51, 42, and 53 days, respectively (P < .01 between passages I and III) (Fig. 1B).
Replication of the Histopathological Characters of the Primary Tumor
To evaluate whether the xenograft tumors replicated the histopathological phenotypes of the parent tumor, particularly during repeated sub-transplantations, H&E staining of paraffin sections from the xenograft tumors was compared with those from the original patient tumor. Xenograft tumors from the initial injections and subsequent sub-transplantations showed similar, if not identical, histological characters of the primary tumor including the high cellular density, increased mitotic index, and high nucleus-cytoplasm ratio (Fig. 1C) as well as invasion into neighboring normal mouse brain and spread through cerebrospinal fluid (Fig. 1A and E).
Further examination using immunohistochemical staining also revealed a striking similarity including high proliferation index (50%–70%) as revealed by Ki-67 positivity, strong posivity (+++) of glial marker GFAP in isolated (1%–5%) tumor cells, high-level (+++) expression of neuronal precursor marker nestin (>90%), and strongly positive reaction (+++) for VMT in the majority (>95%) of tumor cells (Fig. 1D).
Determination of Molecular Subtypes of the Established Xenograft Tumors
Recent transcription and DNA copy number profiling of identified 3 molecular subgroups in CNS PNET, ie, primitive neural (group 1), oligoneural (group 2), and mesenchymal lineage (group 3).12 To determine the molecular subtypes of our xenograft model, we applied qRT-PCR to analyze the expression of marker genes in group 1 (LIN28A, LIN28B, and NKX6-2), group 2 (OLIG1, OLIG2, PDGFA, and NCAM2), and group 3 (MSX1, TWIST1, TGFB3, TGFBR3, and FOXJ1). When compared with normal fetal brain, the only genes consistently overexpressed in the xenograft tumors during serial passage (passages I, III and V) were TWIST1 and FOXJ1, which are 2 markers of group 3 (Fig. 2A). Immunohistochemical staining of group 1 marker LIN28 and group 2 marker OLIG2 failed to detect positive reactions as well (Fig. 2B), further validating the mRNA analysis results. These data categorized our IC-2664PNET into group 3, of which patients had the highest incidence of metastasis at diagnosis.12
Fig. 2.

Molecular subgrouping of IC-2664PNET xenografts. (A) Quantitative RT-PCR analyses of 3 CNS-PNET xenografts, 2664-I, 2664-III and 2664-V, using group-specific PNET cell lineage markers. Data are shown as mean expression relative to fetal brain of 3 independent replicas. Error bars represent the SEM. (B) Immunohistochemical analyses of the 3 xenograft tumors (from passages I to V) showing double-negative staining of the group 1 marker LIN28 and group 2 marker OLIG2. (C) Hierarchical cluster map of cell lineage markers during serial subtransplantations in vivo in mouse brains.
To further assess the expression of additional cell lineage genes to confirm that IC-2664PNE was a group 3 tumor during the serial sub-transplantation in vivo in mouse brains, we performed whole genome gene expression profiling of passage I, III and V xenograft tumors and plotted the cell lineage markers, which had been identified in CNS PNETs,12 into a hierarchical cluster map (Fig. 2C). Most of the primitive neural markers (10 of 12 genes) in IC-2664PNT xenograft tumors were below 2 folds when compared with the normal childhood cerebral tissues; only HOX4C and MEIS1 were overexpressed (>2 folds). For the oligoneural markers, none of the 7 genes was overexpressed (<1 fold). Analysis of the 9 mesenchymal makers identified 3 overexpressed genes COL5A1 (>2 folds), ZIC2 (>3.6 folds), and COL1A2 (>5.9 folds). The remaining 6 genes were not overexpressed (<1 fold). It is noteworthy that levels of TWIST1 and FOXJ1 were not as high as those detected with qRT-PCR (Fig. 2A), probably due to the differences of detection sensitivities and dynamic ranges between qRT-PCR and gene expression chips. The overall expression levels of these marker genes during serial sub-transplantations were relatively stable, and the correlation coefficient reached 0.879 between passages I and III, 0.888 between passages III and V, and 0.636 between passages I and V. Altogether, these data support the subclassification of IC-2664PNT into group 3 and suggested that coexpression of different lineage markers was possible.
Preservation of Tumor cells with Self-renewal and Multipotent Capacities
Since no cell surface markers have been identified in sPNETs, we utilized neurosphere assay, a functional assay that can reliably examine the presence of cells with stem cell features,22,25 to determine if IC-2664PNET contains cells with strong self-renewal capacity. We plated single xenograft tumor cells at clonal density (500 cells/mL in 6-well plates) in serum-free medium containing EGF and bFGF that favor the growth of normal neural as well as brain tumor stem cells.41,42,51 Formation of neurospheres, ranging from 10.7% to 12.4%, was seen in 14 days (Fig. 3A). Immunofluorescent staining of the neurospheres identified cells expressing CD133 and nestin (Fig. 3C), 2 markers associated with normal and brain tumor stem cells.22,26,52
Fig. 3.

Formation and multipotent differentiation of neurospheres from IC-2664 sPNET cells. (A) Time-course images showing the in vitro formation of neurospheres from single sPNET cells in the presence of EGF and bFGF. (B) Attachment of preformed neurosphere on poly-D lysine coated chamber slide 24 hours after the serum-free medium was replaced with DMEM supplemented with 10% FBS. (C) Immunofluorescent staining of neurospheres before (upper panel) and after (lower panel) serum-induced differentiation. Markers of CSC (CD133), neural progenitor (nestin), glial (GFAP), neuronal (TuJ), and oligodendrocyte differentiation were included.
To examine if the IC-2664PNET neurospheres possess multilineage differentiation capacity, we transferred neurospheres at the end of day 14 from serum-free medium into regular DMEM media supplemented with 10% FBS. The attachment of neurospheres to the poly-D-lysine coated chamber slides started within 4–6 hours of incubation (Fig. 3B). Immunofluorescent staining in the attached monolayer cells identified cells expressing glial marker GFAP, neuronal marker Tuj1, and oligodendrocyte marker O4, demonstrating their multipotency (Fig. 3C).
Expression of Cell Surface and Phenotypic Markers Associated with (Cancer) Stem Cells
Although CD133+ cells were identified in the neurospheres of IC-2664PNET, it is still controversial whether CD133 is the only cell surface marker of brain tumor stem cells because some CD133− cells have been shown to possess stem cell features in glioma and other human cancers.40,53–55 To gain a better understanding of the expression profile of putative CSC phenotypic markers in IC-2664PNET and to identify the subpopulation(s) that contributed to the sustained growth of orthotopic xenograft tumors during serial sub-transplantations, we applied flow cytometric analysis to profile the tumor cells derived from passage I, III and V xenografts using panel cell surface markers associated with either normal neuronal or cancerous stem cells, including CD133, CD15, CD24, CD44, and CD117.22,26–29,32,34,36,37 When these markers were analyzed alone (mono-positive), high levels of CD133+ (47.9%) and CD15+ (65.3%) cells were detected at passage I xenograft tumors and maintained in passage III (38.1% and 40%) and passage V (41.1% and 32.5%), respectively (Fig. 4A). These results suggested that CD133+ and CD15+ cells maintained their self-renewal capacity during serial in vivo sub-transplantations. The trace amount of CD44+ and CD117+ cells (< 0.2%) that were detected at passage I did not increase in passage III and V tumors (Fig. 4A), suggesting that they played a less significant role in sustaining the long-term sub-transplantation of IC-2664PNET xenografts.22,28
Fig. 4.

Flow cytometric (FCM) profile of putative CSC markers and examination of self-renewal capacity. (A) Flow cytometric (FCM) analysis of putative CSC markers during serial in vivo sub-transplantation (passages I to V) using fluorochrome-conjugated antibodies specific to CD133, CD15, CD44, CD24, and CD117 in freshly isolated IC-2664PNET cells. Appropriate isotype set was used as a negative control. (B) Profiles of CD133/CD15 and CD44/CD24 double-staining in IC-2664PNET. (C) Representative images showing the formation of neurospheres from the 4 subpopulations of FACS-purified cells double-stained with antibodies against CD133 and CD15. (D) Quantitative comparison of neurosphere-forming capacity in FACS-purified subpopulations of CD133+ and/or CD15+ cells as compared with CD133−/CD15− cells through limited dilution (P < .05). (E) Log-rank analysis of animal survival in mice engrafted with 1000 FACS-purified dual- and mono-positive CD133 and CD15 cells as compared with dual-negative (CD133−/CD15−) tumor cells (P < .05).
CD133 and CD15 have been successfully used individually in isolating CSCs in various human cancers, including MB and glioblastoma.24,27,28 It remains unknown, however, if these 2 markers are coexpressed in the same tumor cells or are mutually exclusive. To examine the relationship between CD133+ and CD15+ cells in sPNET CSCs, double-staining with CD133 and CD15 was performed, and our results showed that dual-positive (CD133+/CD15+) cells accounted for 41.1% at passage I, 25.1% at passage III, and 30.5% at passage V. Since CD133+/CD15− mono-positive cells ranged from 4.3% at passage I to 12.9% at passage V and CD133−/CD15+ cells changed from 15.7% at passage I to 13.1% at passage III and 3.7% at passage V, these data suggested that CD133 and CD15 coexpressed in the majority of sPNET stem cells (Fig. 4B). Breast and prostate CSCs were shown to be CD24low/CD44high.31,34 This subpopulation, however, was absent (0%) in our IC-2664PNET xenograft cells during the serial sub-transplantations, although there were 3.9%–6.2% CD44low/CD24low cells and >93% CD44low/CD24high cells (Fig. 4B). Because only a tiny piece of tumor tissue from the original patient was obtained for this project, we were unable to examine if the original patient tumor also encompassed the similar subpopulations of putative CSCs. Altogether, our data showed that dual-positive (CD133+/CD15+) cells were the major subpopulation of CSCs in IC-2664PNET.
In Vitro self-renewal Capacity of CD133+ and/or CD15+ sPNET Cells
To determine if dual-positive sPNET cells have stronger self-renewal capacities than those of the mono-positive cells, we isolated dual-positive (CD133+/CD15+), mono-positive (CD133+/CD15−, CD133−/CD15+), and double negative (CD133−/CD15−) cells through FACS from xenografts (passage III) and plated them in serum-free medium that favors the growth of neurosphere in 96-well plates through limited dilutions starting from 1 000 cells/well. Formation of neurospheres was examined under phase-contrast microscope, and those composed of more than 50 cells were counted (Fig. 4C). As shown in Figure 4D, the neurosphere-forming efficiency from the double positive (CD133+/CD15+) cells was 8.7%, higher than that from the CD133−/CD15+ cells (3%) and CD133+/CD15− cells (2.1%) (P < .05); and the double-negative (CD133−/CD15−) cells formed the least number of neurospheres (1.8%) among the 4 subpopulations (P < .01). These data suggested that double-positive (CD133+/CD15+) cells have stronger self-renewal capacity in vitro than the mono-positive cells. Worthy of note is that the overall neurosphere-forming efficiency of our FACS-purified sPNET cells was slightly lower than that obtained from unfractionated tumor cells in this study. This phenomenon has been previously reported in FACS-purified CD133+ glioma cells.39 It remains to be determined if the process of FACS itself exerted any adverse effects on the survival and proliferation of tumor cells.
In Vivo Tumor-forming Efficiency of Dual- and Mono-positive CD133 and CD15 Cells
Formation of transplantable xenograft tumor is a recognized gold standard assay of CSC self-renewal.56 To examine tumor-forming capacities of purified CSC subpopulations, we injected FACS-purified double-positive (CD133+/CD15+), mono-positive (CD133+/CD15− and CD133−/CD15+) cells at 10, 102 and 103 cells/mouse into the right cerebrum of Rag2/SCID mice and compared their tumor-forming efficiency and animal survival times with those of the dual-negative (CD133−/CD15−) cells (102, 103, and 104 cells/mouse) (Table 1). In mice injected with 10 cells, no tumor formation was observed in any of the groups after 280 days of observation. When transplanted with 100 cells, tumor formation was confirmed in 3 of 5 (60%) mice in CD133+/CD15+ and CD133−/CD15+ groups, 2 of 5 (40%) mice in the CD133+/CD15− group, and 0 of 5 (0%) mice in the dual-negative group. Injection of 1 000 cells led to tumor formation in all 5 (100%) mice receiving CD133+/CD15+ and CD133+/CD15− cells and in 4 of 5 (80%) mice injected with CD133−/CD15+ cells. Surprisingly, 3 of 5 (60%) mice engrafted with 1000 dual-negative (CD133−/CD15−) cells and 5 of 5 (100%) mice receiving 10 000 cells also formed xenograft tumors, suggesting that there may exist another subpopulation(s) of CSCs that were truly CD133 and CD15 negative. It may also be possible that our gate setting during FACS did not completely exclude the CD133+ and/or CD155+ cells from the dual-negative fraction.
Table 1.
Tumor-forming efficiency of dual- and mono-positive CD133/CD15 tumor cells
| Group | Cells | Cell Number |
Confidence Intervals 1/(Stem Cell Frequency) |
|||||
|---|---|---|---|---|---|---|---|---|
| 10 | 100 | 1000 | 10 000 | Lower | Estimate | Upper | ||
| A | CD133+/CD15+ | 0/5 | 3/5 | 4/4 | nd | 387 | 127 | 41.8 |
| B | CD133−/CD15+ | 0/5 | 3/5 | 4/5 | nd | 993 | 370 | 137.9 |
| C | CD133+/CD15− | 0/5 | 2/5 | 5/5 | nd | 625 | 203 | 66.3 |
| D | CD133−/CD15− | nd | 0/5 | 3/5 | 5/5 | 3848 | 1262 | 414.5 |
Abbreviation: nd, not done.
We next performed the limiting-dilution analysis38,50 and found that the frequency of xenograft initiating (tumorigenic) cells was significantly different among the 4 groups (P = .02). Xenograft initiating (tumorigenic) cells were estimated to be 0.79% (1 in 127 cells) in the dual-positive (CD133+/CD15+) cells, 0.49% (1 in 203 cells) in the CD133+/CD15− cells, 0.27% (1 in 370 cells) in the CD133−/CD15+ cells, and 079% (1 in 1 262 cells) in the dual-negative (CD133−/CD15−) cells (Table 1). Pairwise tests for the differences among these 4 groups of cells identified significant differences (P < .05) between dual-positive and dual-negative and between CD133-mono-positive and dual-negative cells. The differences between the remaining groups were not significant (P > .05). Since the frequencies of tumorigenic cells in the dual-positive (CD133+/CD15+) and mono-positive (CD133+/CD15−) cells were much higher (9.3 and 6.2 folds, respectively) than that in the dual-negative (CD133−/CD15−) cells; these data suggested that dual-positive and CD133 mono-positive cells were the major populations of CSCs in IC-2664PNT.
The median survival times of mice injected with 100 cells and with confirmed tumor formation were 86 days in the CD133+/CD15+ group, significantly shorter than the 129 and 186 days found in the CD133−/CD15+ and CD133+/CD15− groups, respectively (P = .04). In mice receiving 1000 cells, the median survival times of animals in dual-positive (CD133+/CD15+) groups were 60 days, significantly shorter (P < .05) than 87 days in the CD133−/CD15+ group, 94 days in the CD133+/CD15− group, and 98 days in the 3 mice of the CD133−/CD15− group (Fig. 4E). Taken together, these data confirmed that dual-positive (CD133+/CD15+) cells possess the strongest self-renewal capacity in vivo and that new phenotypic markers are needed to identify the tumor initiating cells in CD133−/CD15−.
The Establishment of a Neurosphere Line BXD-2664PNET-NS
To establish a neurosphere line, the neurosphere cultures were serially passaged every 3–5 days when the media color changed and/or high density (>5/low power field) of big neurospheres (composed of 50–100 cells) (Fig. 5A) were observed. The culture was passaged more than 54 times in vitro and was designated as Baylor xenograft derived 2664sPNET neurosphere line (BXD-2664PNET-NS). Mycoplasma contamination was not detected in cells collected at passages 39 and 56. Periodic chromosomal examination of the cultured cells using mouse- and human-specific antibodies against major histocompatibility antigens ruled out the contamination of mouse cells in the established culture. Serial analysis during in vitro growth confirmed the presence of CD133+ and/or CD15+ cells in the cultured cells (Fig. 5B). Immunocytofluorescent analyses were performed in the cultured neurosphere cells during the serial passage of BXD-2664PNET-NS (at passages 6, 9, 19, 29, 39, and 54) to further examine expression of stem cell and differentiation markers (Fig. 5C, Table 2). High levels (+++) of stem cell markers (nestin and OTX2) were consistently detected from passages 6–54 in most of the tumor cells (>90%), while the strong (+++) expression of the immature neuronal marker (Tuj-1) varied from 90% before passage 9 to 25% at passage 19. The expression levels of VMT increased gradually from medium (++) level in 25% cells at passage 6 to strong (+++) positivity in more than 75% of the cells at passages 39 and 54. Markers of mature glia cells (GFAP), neuron (NeuN) and oligodendroglial cells (O4) were not detected from passages 6 to 54. Altogether, these data suggested that the major stem cell features were maintained in the BXD-2664PNET-NS line.
Fig. 5.

Establishment of a long-term cultured neurosphere line BXD-2664PNET-NS. (A) Images showing the morphology of neurospheres at different sizes. The cultures usually split when large neurospheres (arrow) were readily seen. (B) Profiles showing the FCM analysis of CD133+ and CD15+ subpopulations in the cultured neurospheres during serial passages through double staining. Neurospheres predigested with trypsin to dissociate into single cells were allowed to recover overnight before being double-stained with fluorochrome-conjugated antibodies specific to human CD133 and CD15. (C) Representative images showing the immunofluorescent staining of the cultured neurosphere cells. Following incubation with the primary antibodies against markers associated with stem cells (nestin), mature glial cells (GFAP), immature neuron (TuJ), oligodendrocyte (O4) and vimentin (VMT), fluorochrome-conjugated secondary antibodies (red for nestin, and green for the rest of the antibodies) were applied. The slides were counterstained with DAPI to highlight cell nuclears in blue. Original magnification: x 40.
Table 2.
Summary of immunofluorescence staining in neurosphere cells during serial in vitro passages.
| Marker | Target proteins | BXD-2664PNET (passage) |
|||||
|---|---|---|---|---|---|---|---|
| p6 | p9 | p19 | p29 | p39 | p54 | ||
| OTX2 | Stem cell | +++4 | +++4 | +++4 | +++4 | +++4 | +++4 |
| Nestin | Stem cell | ++4 | ++4 | ++4 | ++4 | ++4 | ++4 |
| GFAP | Mature astrocyte | – | – | – | – | – | – |
| NeuN | Mature neurons | +1 | ++2 | ++1 | +3 | – | – |
| Tuj-1 | Immature neurons | +++4 | +++4 | +++1 | +++2 | +++3 | +++2 |
| O4 | Oligodendroglia cell | – | – | – | – | – | – |
| Vimentin | Neurofilament | ++1 | ++3 | ++3 | ++3 | +++4 | +++4 |
To determine if the established neurosphere cell line can still be categorized into group 3, we examined the mRNA expression of molecular classifiers through qRT-PCR using known groups 1, 2 and 3 PNET samples as positive controls. To gain an even better understanding of the changes of gene expression during long-term in vitro growth, we performed serial analysis of the cells from passages 6, 9, 19, 29, 39, 49, and 57. As summarized in Fig 6, markers of groups 1 and 2 were not expressed in the cells. The expression of group 3 markers was elevated, but not until passage 39. These data suggested that this neurosphere line BXD-2664PNT maintained the features of group 3 sPNET, but the earlier passages (before 39) may have better preserved the nondifferentiated phenotype of cancer stem cells, hence meeting the goal of representing cancer stem cell populations.
Fig. 6.
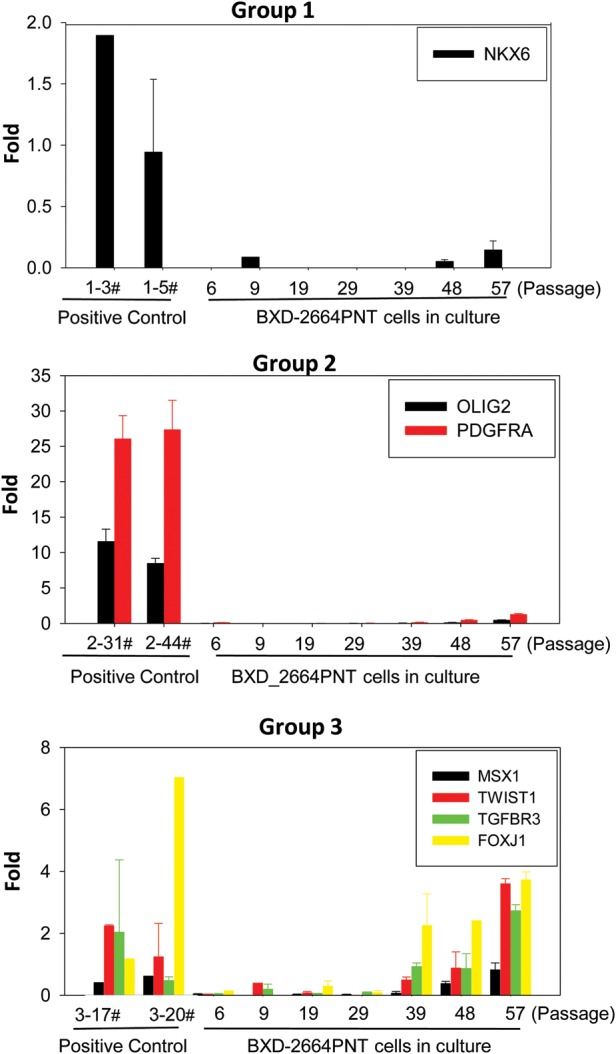
Quantitative examination of molecular classifiers in the established neurosphere line BXD-2664PNT-NS during serial in vitro passages through qRT-PCR. Known groups 1, 2, and 3 sPNET samples were included as positive controls. The levels of mRNA expression in all the samples were normalized to that of a fetal brain and graphed as fold changes (mean ± SD).
To determine if the neurosphere line is tumorigenic, we implanted 1 × 104 and 1 × 105 cells from passage 28 into the right cerebrum of NOD SCID mice and examined tumor formation on H&E stained serial sections of whole mouse brains when the host animals developed signs of sickness. Growth of intracerebral xenograft tumor was observed in 1 of 5 (20%) mice receiving 1 × 104 cells and in 2 in 4 (50%) mice implanted with 1 × 105 tumor cells.
Discussion
We reported here the establishment and characterization of a novel orthotopic xenograft mouse model of sPNET, the isolation of CSCs from the xenograft tumors, and the parallel development of a neurosphere cell line. Since this model was established through direct injection of fresh surgical specimens and has been strictly sub-transplanted in vivo into mouse brains, the xenograft tumors replicated the histopathological features, rapid proliferation, and highly invasive growth patterns of the original patient tumor. More importantly, we demonstrated that this model represented the group 3 sPNET, the subtype of CNS PNET that often displayed metastasis at diagnosis.12 Utilizing a panel of CSC phenotypic markers, we showed that the CSC cells from this model were composed of a heterogeneous pool of tumor cells expressing CD133 and/or CD15 and that dual-positive (CD133+/CD15+) cells possessed the strongest self-renewal and tumorigenic capacity both in vitro and in vivo. We also showed that some dual-negative (CD133−/CD15−) cells were also tumorigenic at 1 000 cells/mouse, which justified the need of searching for new CSC markers.
Recent identification of molecular subtypes in pediatric brain tumors has significantly advanced our understanding of tumor biology and provided a novel therapeutic potential of targeting group-specific abnormalities. Our xenograft model of group 3 sPNET (IC-2664PNET) and the matching neurosphere line (BXD-2664PNET-NS) have thus provided a much-needed model system for both biological and preclinical studies. However, we do recognize that there may still exist intratumoral heterogeneities even among the group 3 sPNETs (as evidenced by the expression of primitive neural genes in IC-2664PNT) and that more models are desired. We are therefore actively recruiting new sPNET tumors, which are relatively rare, for the development of more orthotopic xenograft models.
Understanding the role of CSC represents another important aspect of sPNET biology. Despite the accumulating evidence that supports the CSC hypothesis, there are ongoing debates about the relative abundance of CSCs and the specificity of phenotypic markers.38,39,54 Surface markers provide obvious convenience for isolating CSCs57 and have been used extensively for the isolation of specific subpopulation of CSCs, (eg, CD133 and CD15 as mono-markers in MB and glioma and CD24low/CD44high for breast and prostate cancers).22,23,27,28,31,34 Although the intratumoral heterogeneity of CSCs has been hypothesized, comprehensive examination of this hypothesis in human solid tumors has rarely been reported. In the current study, we used a panel of markers that have been associated with stem cells, either normal or cancerous, to further dissect the subpopulations of CSCs in sPNET. With the sufficient supply of xenograft tumor tissues, which overcomes one of the major barriers of CSC study of human solid tumors, we identified tumor cells expressing different cell surface markers in the IC-2664PNET model. Since many efforts are being made to develop CSC-directed therapies, our demonstration that different CSC subpopulations co-exist in the same sPNET tumor suggests that targeting one or only a few CSC subtypes may have limited success and that all CSC cells, regardless of their phenotypic markers, must be eliminated to cure.
To facilitate the preclinical drug screening or functional studies of various new genetic pathways, it is often desirable to have an in vitro model system for CSCs. Our successful establishment of the neurosphere line BXD-2664PNET-NS has thus provided a matching in vitro line for the xenograft model (IC-2664PNET). Since most of the stem cell features, including the preservation of CD133+ and CD15+ cells and the expression of nestin, OTX2, were preserved during long term in vitro growth; and few cells expressed the markers of mature glial, neuronal and oligodendroglial markers, it is reasonable to believe that this neurosphere line can serve as a useful resource for sPNET stem cell studies. We do notice, however, that the levels of CD133+/CD15+ dual-positive cells in the neurosphere line were much lower than that in the orthotopic xenograft tumors, suggesting that the microenvironmental differences might have played a role.
In summary, we have successfully established a patient tumor-derived orthotopic xenograft mouse model that replicated the biology of group 3 childhood sPNET as well as a matching neurosphere line. Using this unique model system, we isolated and characterized CSCs for childhood sPNET. We confirmed the presence of mixed subpopulations of sPNET cells with strong self-renewal and tumor-initiating capacities and identified CD133 and CD15 (mono- or dual-positive) as major surface antigen markers of sPNET CSCs. Since some CD133−/CD15− cells also formed neurospheres in vitro and developed xenografts in vivo, although at low efficiency, future studies are needed to determine if they express new CSC markers. This model system has thus provided us with a unique opportunity to investigate the biology and test new therapies against group 3 sPNET tumors as well as to develop CSC-directed treatment.
Supplementary Material
Funding
This study was supported by Cancer Fighters of Houston (X.N.L) and the joint participation by Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the Eliminating Brain Tumor Stem Cell with an Oncolytic Picornavirus Program.
Conflict of interest statement. None declared.
Supplementary Material
References
- 1.Larouche V, Capra M, Huang A, et al. Supratentorial primitive neuroectodermal tumors in young children. J Clin Oncol. 2006;24:5609–5610. doi: 10.1200/JCO.2006.09.1033. [DOI] [PubMed] [Google Scholar]
- 2.Fangusaro J, Finlay J, Sposto R, et al. Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the Head Start I and II experience. Pediatr Blood Cancer. 2008;50:312–318. doi: 10.1002/pbc.21307. [DOI] [PubMed] [Google Scholar]
- 3.Johnston DL, Keene DL, Lafay-Cousin L, et al. Supratentorial primitive neuroectodermal tumors: a Canadian pediatric brain tumor consortium report. J Neurooncol. 2008;86:101–108. doi: 10.1007/s11060-007-9440-1. [DOI] [PubMed] [Google Scholar]
- 4.Friedrich C, von Bueren AO, von HK, et al. Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol. 2013;15:224–234. doi: 10.1093/neuonc/nos292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chintagumpala M, Hassall T, Palmer S, et al. A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro Oncol. 2009;11:33–40. doi: 10.1215/15228517-2008-079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBride SM, Daganzo SM, Banerjee A, et al. Radiation is an important component of multimodality therapy for pediatric non-pineal supratentorial primitive neuroectodermal tumors. Int J Radiat Oncol Biol Phys. 2008;72:1319–1323. doi: 10.1016/j.ijrobp.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 7.Bavishi S, Wong K, Delgardo T, et al. Successful radiation therapy for supratentorial primitive neuroectodermal tumor and epidermolysis bullosa simplex. Pediatr Blood Cancer. 2010;54:170–172. doi: 10.1002/pbc.22281. [DOI] [PubMed] [Google Scholar]
- 8.Inda MM, Munoz J, Coullin P, et al. High promoter hypermethylation frequency of p14/ARF in supratentorial PNET but not in medulloblastoma. Histopathology. 2006;48:579–587. doi: 10.1111/j.1365-2559.2006.02374.x. [DOI] [PubMed] [Google Scholar]
- 9.Miller S, Rogers HA, Lyon P, et al. Genome-wide molecular characterization of central nervous system primitive neuroectodermal tumor and pineoblastoma. Neuro Oncol. 2011;13:866–879. doi: 10.1093/neuonc/nor070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Lee KF, Lu Y, et al. Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell. 2009;16:533–546. doi: 10.1016/j.ccr.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Picard D, Miller S, Hawkins CE, et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol. 2012;13:838–848. doi: 10.1016/S1470-2045(12)70257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea--a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 14.Galmozzi E, Facchetti F, La Porta CA. Cancer stem cells and therapeutic perspectives. Curr Med Chem. 2006;13:603–607. doi: 10.2174/092986706776055661. [DOI] [PubMed] [Google Scholar]
- 15.Sutter R, Shakhova O, Bhagat H, et al. Cerebellar stem cells act as medulloblastoma-initiating cells in a mouse model and a neural stem cell signature characterizes a subset of human medulloblastomas. Oncogene. 2010;29:1845–1856. doi: 10.1038/onc.2009.472. [DOI] [PubMed] [Google Scholar]
- 16.Fan X, Eberhart CG. Medulloblastoma stem cells. J Clin Oncol. 2008;26:2821–2827. doi: 10.1200/JCO.2007.15.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietrich J, Diamond EL, Kesari S. Glioma stem cell signaling: therapeutic opportunities and challenges. Expert Rev Anticancer Ther. 2010;10:709–722. doi: 10.1586/era.09.190. [DOI] [PubMed] [Google Scholar]
- 18.Corsini NS, Martin-Villalba A. Integrin alpha 6: anchors away for glioma stem cells. Cell Stem Cell. 2010;6:403–404. doi: 10.1016/j.stem.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan X, Khaki L, Zhu TS, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeppernick F, Ahmadi R, Campos B, et al. Stem Cell Marker CD133 Affects Clinical Outcome in Glioma Patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 22.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh SK, Clarke ID, Hide T, et al. Cancer stem cells in nervous system tumors. Oncogene. 2004;23:7267–7273. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 24.Ward RJ, Lee L, Graham K, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009;69:4682–4690. doi: 10.1158/0008-5472.CAN-09-0342. [DOI] [PubMed] [Google Scholar]
- 25.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 27.Son MJ, Woolard K, Nam DH, et al. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Read TA, Fogarty MP, Markant SL, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–147. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao XG, Zhang X, Xue XY, et al. Brain Tumor Stem-Like Cells Identified by Neural Stem Cell Marker CD15. Transl Oncol. 2009;2:247–257. doi: 10.1593/tlo.09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright MH, Calcagno AM, Salcido CD, et al. Brca1 breast tumors contain distinct CD44+/ Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fillmore C, Kuperwasser C. Human breast cancer stem cell markers CD44 and CD24: enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007;9:303. doi: 10.1186/bcr1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 33.Simon RA, di Sant'Agnese PA, Huang LS, et al. CD44 expression is a feature of prostatic small cell carcinoma and distinguishes it from its mimickers. Hum Pathol. 2009;40:252–258. doi: 10.1016/j.humpath.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Hurt EM, Kawasaki BT, Klarmann GJ, et al. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98:756–765. doi: 10.1038/sj.bjc.6604242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, et al. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67:6796–6805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 36.Filip S, Mokry J, Vavrova J, et al. Homing of lin(-)/CD117(+) hematopoietic stem cells. Transfus Apher Sci. 2009;41:183–190. doi: 10.1016/j.transci.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 37.Adhikari AS, Agarwal N, Wood BM, et al. CD117 and Stro-1 Identify Osteosarcoma Tumor-Initiating Cells Associated with Metastasis and Drug Resistance. Cancer Res. 2010;70:4602–4612. doi: 10.1158/0008-5472.CAN-09-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintana E, Shackleton M, Sabel MS, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clement V, Dutoit V, Marino D, et al. Limits of CD133 as a marker of glioma self-renewing cells. Int J Cancer. 2009;125:244–248. doi: 10.1002/ijc.24352. [DOI] [PubMed] [Google Scholar]
- 40.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 41.Shu Q, Wong KK, Su JM, et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells. 2008;26:1414–1424. doi: 10.1634/stemcells.2007-1009. [DOI] [PubMed] [Google Scholar]
- 42.Yu L, Baxter PA, Voicu H, et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol. 2010;12:580–594. doi: 10.1093/neuonc/nop056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li XN, Shu Q, Su JM, et al. Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol Cancer Ther. 2005;4:1912–1922. doi: 10.1158/1535-7163.MCT-05-0184. [DOI] [PubMed] [Google Scholar]
- 44.Li XN, Parikh S, Shu Q, et al. Phenylbutyrate and phenylacetate induce differentiation and inhibit proliferation of human medulloblastoma cells. Clin Cancer Res. 2004;10:1150–1159. doi: 10.1158/1078-0432.ccr-0747-3. [DOI] [PubMed] [Google Scholar]
- 45.Simon R, Lam A, Li MC, et al. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 46.Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 47.Saeed AI, Bhagabati NK, Braisted JC, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 48.Shu Q, Antalffy B, Su JM, et al. Valproic Acid prolongs survival time of severe combined immunodeficient mice bearing intracerebellar orthotopic medulloblastoma xenografts. Clin Cancer Res. 2006;12:4687–4694. doi: 10.1158/1078-0432.CCR-05-2849. [DOI] [PubMed] [Google Scholar]
- 49.Bonnefoix T, Bonnefoix P, Verdiel P, et al. Fitting limiting dilution experiments with generalized linear models results in a test of the single-hit Poisson assumption. J Immunol Methods. 1996;194:113–119. doi: 10.1016/0022-1759(96)00077-4. [DOI] [PubMed] [Google Scholar]
- 50.Shackleton M. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 51.Chaichana K, Zamora-Berridi G, Camara-Quintana J, et al. Neurosphere assays: growth factors and hormone differences in tumor and nontumor studies. Stem Cells. 2006;24:2851–2857. doi: 10.1634/stemcells.2006-0399. [DOI] [PubMed] [Google Scholar]
- 52.Mangiola A, Lama G, Giannitelli C, et al. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: possible prognostic implications. Clin Cancer Res. 2007;13:6970–6977. doi: 10.1158/1078-0432.CCR-07-1229. [DOI] [PubMed] [Google Scholar]
- 53.Sun Y, Kong W, Falk A, et al. CD133 (Prominin) negative human neural stem cells are clonogenic and tripotent. PLoS ONE. 2009;4:e5498. doi: 10.1371/journal.pone.0005498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Sakariassen PO, Tsinkalovsky O, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122:761–768. doi: 10.1002/ijc.23130. [DOI] [PubMed] [Google Scholar]
- 55.Joo KM, Kim SY, Jin X, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 56.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–1757. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 57.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.