

Abstract
Background
Mutations in genes encoding components of the Brahma-associated factor (BAF) chromatin remodeling complex have recently been shown to contribute to multiple syndromes characterised by developmental delay and intellectual disability. ARID1B mutations have been identified as the predominant cause of Coffin-Siris syndrome and have also been shown to be a frequent cause of nonsyndromic intellectual disability. Here, we investigate the molecular basis of a patient with an overlapping but distinctive phenotype of intellectual disability, plantar fat pads and facial dysmorphism.
Methods/results
High density microarray analysis of the patient demonstrated a heterozygous deletion at 6q25.3, which resulted in the loss of four genes including AT Rich Interactive Domain 1B (ARID1B). Subsequent quantitative real-time PCR analysis revealed ARID1B haploinsufficiency in the patient. Analysis of both patient-derived and ARID1B knockdown fibroblasts after serum starvation demonstrated delayed cell cycle re-entry associated with reduced cell number in the S1 phase. Based on the patient’s distinctive phenotype, we ascertained four additional patients and identified heterozygous de novo ARID1B frameshift or nonsense mutations in all of them.
Conclusions
This study broadens the spectrum of ARID1B associated phenotypes by describing a distinctive phenotype including plantar fat pads but lacking the hypertrichosis or fifth nail hypoplasia associated with Coffin-Siris syndrome. We present the first direct evidence in patient-derived cells that alterations in cell cycle contribute to the underlying pathogenesis of syndromes associated with ARID1B haploinsufficiency.
Keywords: Intellectual disability, Chromatin remodelling, Coffin-Siris syndrome, ARID1B mutation, Cell cycle, Haploinsufficiency
Introduction
The control of gene expression is an intricately regulated process that requires many multiprotein complexes. Chromain remodeling regulates gene expression by modulating the access of transcription machinery proteins to the condensed genomic DNA via dynamic modification of the chromatin architecture. This modification is mediated by either covalent histone modifications via specific enzymes such as histone acetyltransferases or ATP-dependent alteration of DNA-nucleosome topology [1]. The latter mode of modification is mediated by a class of protein complexes called ATP-dependent chromatin-remodeling complexes, which are known to regulate gene expression in specific cellular contexts or at defined time points [2]. Recent studies have linked mutations in these complexes to both developmental disorders and cancer [3]. Mutations in ARID1B and several other genes encoding components of the Brahma-associated factor (BAF, also referred to as switching defective and sucrose nonfermenting SWI/SNF-α) chromatin remodeling complex, were recently shown to cause Coffin-Siris syndrome (CSS) [4,5]. Subsequent studies have demonstrated that mutations in ARID1B are the main cause of CSS [6-8]. CSS is characterised by intellectual disability, severe speech impairment, coarse facial features, microcephaly, developmental delay and hypoplastic nails on the fifth digits (MIM 135900). However, ARID1B mutations have also been identified in a broader cohort of patients, including nonsyndromic intellectual disability and deletions in individuals with intellectual disability, autism and agenesis of the corpus callosum [9-11]. It is unclear as to why patients with ARID1B mutations present such a broad phenotypic spectrum.
ARID1B encodes a DNA-binding protein component of the ubiquitous ATP-dependent chromatin remodelling BAF complex, which is known to regulate gene expression, including genes involved in proliferation and differentiation [12]. These complexes are made up of at least ten core proteins and have fifteen known interchangeable components to give rise to an array of functionally distinct and cell-type specific BAF complexes, such as neuronal progenitor BAF complex [12,13]. Thus, mutations in different BAF components are likely to perturb the function of BAF complexes to varied degrees in different cell types. In agreement with this notion, a striking feature of mutations in the BAF components is the phenotypic variability presented by the patients [6,7]. To date, the biological processes affected in patients with ARID1B mutations are largely unknown.
Here, we report ARID1B haploinsufficiency in five patients with moderate intellectual disability, absent speech and dysmorphic features with narrow palpebral fissures, long eyelashes, a thin upper lip and full lower lip. Alterations in cell cycle were observed in fibroblasts derived from a patient and ARID1B-knockdown control fibroblast cells. Our study broadens the phenotypic spectrum of ARID1B-mediated disorders and provides the first evidence that alterations in cell cycle contribute to the underlying pathogenesis of syndromes associated with ARID1B haploinsufficiency.
Methods
Patients
Informed consents were obtained from patients’ parents for the publication of clinical, genetic and molecular analyses. After receiving institutional Ethics Committee (Royal Children’s Hospital, Melbourne, Australia) blood and tissue samples were obtained. Genomic DNA was extracted from whole blood using the BACC DNA extraction kit (GE Healthcare Life Sciences) according to the manufacturer’s instructions. Standard karyotype analysis was performed using G-banding and high density SNP array data (Affymetrix Human SNP array 6.0) was analysed for copy number variation using Karyo-studio (Illumina).
Generation of ARID1B knockdown human fibroblast
Primary fibroblast cultures were established using standard procedures and maintained in BME supplemented with 10% fetal bovine serum (FBS). MicroRNA-adapted shRNA (shRNAmir)-mediated knockdown of ARID1B was performed using lentivirus containing ARID1B-targeting shRNAmir (V3THS_306691 or V3THS_306692, Open Biosystems) or scrambled non-silencing control (RHS4743, Open Biosystem). The lentiviral particles were generated by transfecting HEK293FT with V3THS_306691, V3THS_306692 or RHS4743 plasmids, and SPAX2 (Addgene) and pMD2.G (Addgene) lentiviral packaging plasmids using Lipofectamine 2000 (Invitrogen) according to manufacturers’ instructions. The viral supernatants were harvested after 48 hours and concentrated at 70, 000 g, 4°C for 2 hours. Wildtype fibroblasts were transduced with both V3THS_306691 (MOI = 20) and V3THS_306692 (MOI = 20) or RHS4743 (MOI = 20) to generate ARID1B-knockdown and non-silencing control fibroblast lines respectively. Transduced cells were cultured in complete media containing 4 μg/ml puromycin for 2 weeks and transduction of the fibroblast lines was confirmed by expression of the co-expressed marker protein turboRFP.
Quantitative real-time PCR analysis
Fibroblast cells were cultured in 60 cm dish and RNA was extracted using SV Total RNA Isolation system (Promega) according to manufacturer’s instructions. 100 ng RNA was used to generate cDNA library for each sample using Transcriptor cDNA First Strand Synthesis kit (Roche) according to manufacturer’s instructions. Real-time PCR reactions were performed in triplicate with Light Cycler 480II instrument (Roche) using Taqman probes directed against ARID1B (Applied Biosystems, hs00368175) and the housekeeping control Human Large Ribosomal Protein (RPLO) (Applied Biosystems, 432631E-0904009) and Fast Start Taqman Probe Master Mix (Roche) according to manufacturer’s instructions. The relative ARID1B expression was analysed using Pfaffl method with respect to RPLO expression [14]. Standard deviations were calculated using Gaussian error propagation. T-Test was used to investigate the difference in relative ARID1B expression between samples. Each experiment was independently performed at least three times.
Serum starvation assay
Fibroblasts were cultured in 60 cm dishes and incubated in serum free media for seven days to induce cell cycle arrest. Subsequently, complete media (10% FBS) was added and cells were incubated for up to two days prior to FACS analysis to quantify the number of cells undergoing S1 phase. Cells were detached, fixed with 80% ethanol and incubated with propidium iodide/RNAse A staining solution (50 μg propidium iodide/ml, 0.2 mg RNAse A/ml, 0.05% Triton-X100 in PBS) for 2 hours prior to Fluorescence-Activated Cell Sorting (FACS) analysis. A minimum of 10,000 cells were counted for each sample using BD LSR II Flow Cytometer (BD science) with excitation laser set at 488 nm and emission filter set at 695 nm/40 and analysed using ModFit LT analysis software (Verity Software House). Each experiment was independently performed at least three times.
Sanger sequencing of ARID1B and whole Exome sequencing
Exons of ARID1B were amplified via PCR with primer sets listed in Additional file 1: Table S1. The resulting PCR products were purified and sequenced using BigDye Terminator kit v3.1 (Applied Biosystem) and ABI 3730 DNA Analyzer (Applied Biosystems). Exome sequencing of patient 5 was performed on an Illumina Hiseq 2000 platform with the NimbleGen VCRome 2.1 exome capture design for enrichment. The 125 bp paired-end reads were mapped to UCSC genome Browser Hg 19 and variants and indels were called using HGSC Mercury analysis pipeline [15]. High quality variant calls were filtered on nonsynonymous changes in the coding regions or changes affecting the canonical splice sites. Variants that were present with a frequency above 1% in dbSNP and the Nijmegen in-house database containing data of over 1,300 exomes, or in 200 exomes analysed using the same Illumina Hiseq 2000 platform were excluded.
Results
Identification of patient 1
Patient 1 presented with moderate intellectual disability, absent speech and dysmorphic features with narrow palpebral fissures, long eyelashes, a thin upper lip and full lower lip. Fetal finger and toe pads and plantar lipomas were present (Figure 1A-C). Karyotype analysis showed an apparently balanced de novo reciprocal translocation involving chromosomes 4 and 6 (46,XY,t(4;6)(q35;q25.3)) in all examined cells (n = 15). The chromosome 4 breakpoint was located within a 0.3 Mb region that did not encode any genes, whereas the chromosome 6 breakpoint was mapped to a ~0.4 Mb region that encoded several genes. Copy number analysis did not identify any variations associated with the chromosome 4 breakpoint. However, a heterozygous deletion of ~1.2 Mb was identified on 6q25.3 that resulted in single allele loss of the genes encoding sorting nexin-9 (SNX9), zinc finger DHHC-type containing 14 (ZDHHC14), transmembrane protein 242 (TMEM242) and AT rich interactive domain 1B (ARID1B) (Figure 2).
Figure 1.
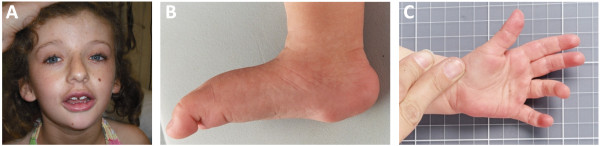
Clinical phenotype of patient 1. The facial photographs show dysmorphism including a broad face, narrow palpebral fissures, thin upper lip, full lower lip and low-set ears (A). The plantar fat pads anteromedial to the heel and fetal toe pads are prominent (B) and the hands show fetal finger pads and pillowing over the metacarpal heads (C).
Figure 2.

Molecular characterisation of patient 1. High density SNP chip array and CNV analysis identified a heterozygous de novo 4;6 reciprocal translocation that resulted in the deletion of four genes encoding sorting nexin-9 (SNX9), zinc finger DHHC-type containing 14 (ZDHHC14), transmembrane protein 242 (TMEM242) and AT rich interactive domain 1B (ARID1B) at 6q25.3.
ARID1B haploinsufficiency in patient 1
Given the documented role of ARID1B in disorders with a clinical presentation somewhat similar to our patient, we tested the effect of the deletion on ARID1B expression in patient-derived primary fibroblasts. Western blot analysis using multiple commercially available antibodies directed against ARID1B failed to robustly identify the predicted ~240 kDa protein in patient or control fibroblast cells (data not shown). However, real-time PCR analysis revealed that the transcription level of ARID1B in patient 1 was significantly reduced (35 ± 7%, mean ± SD, n = 5, P < 0.001) in comparison to the control fibroblasts (Figure 3A). In order to further characterise the effect of ARID1B haploinsufficiency, we utilised a knockdown approach. Wildtype fibroblasts co-transduced with ARID1B-targetting shRNAmir (V3THS_306691 and V3THS_306692) expressed ARID1B mRNA at approximately 60% of control levels (56 ± 12%, mean ± SD, n = 3, P < 0.01). In contrast, no effect on ARID1B mRNA level was observed when the non-silencing control was utilised (Figure 3A).
Figure 3.
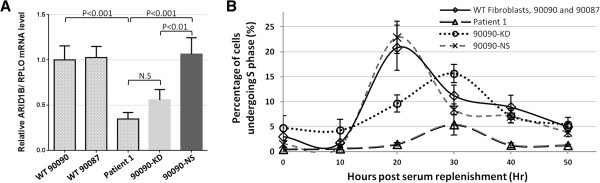
ARID1B haploinsufficiency results in delayed S1 Phase entry. Real Time Quantitative PCR analysis of patient-derived (Patient 1) and control (90087 and 90090) fibroblasts was performed using Taqman probes directed against ARID1B and the housekeeping control RPLO. Samples were normalised to control 90090, which was assigned a value of 1 and analysed by the Pfaffl relative quantification method. The expression of ARID1B was significantly reduced in both Patient 1 and the control 90090 after shRNAmir mediated knockdown of ARID1B (90090-KD), whereas the non-silencing control did not alter ARID1B expression (90090-NS) (A). Cell cycle dynamics of serum-starved cells undergoing S1 phase after serum replenishment was analysed using ModFit LT. The percentage of cells undergoing S1 phase at 20 hours post serum replenishment was significantly lower in both patient 1 (P = 0.0002) and 90090-KD (P = 0.005) compared to control (B).
Entry to S1 phase is delayed in patient 1
It was previously demonstrated that ARID1B deficiency delayed cell cycle re-entry after cell cycle arrest induced by serum starvation in mouse MC3T3 osteoblasts [16]. To investigate whether the molecular defect in patient 1 might lead to similar deficits, we analysed the kinetics of cell cycle re-entry after serum starvation [17]. Control fibroblasts showed a peak in cells undergoing S1 phase (21% ± 4.5, mean ± SD, n = 6) at 20 hours post serum replenishment (Figure 3B). In contrast, the percentage of cells undergoing S1 phase at 20 hours post serum replenishment was significantly lower in both patient 1 (1.5% ± 0.35, mean ± SD, n = 3, P = 0.0002) and 90090-KD (9.6% ± 1.7, mean ± SD, n = 3, P = 0.005) compared to control. The maximum number of patient 1 cells in S1 phase (5.4 ± 2.1%, mean ± SD, n = 3) was observed 30 hours post serum replenishment. Similarly, knockdown of ARID1B in control fibroblasts resulted in a delayed entry to S1 phase, with the peak also occurring at 30 hours post serum replenishment. The number of cells undergoing S1 phase in the knockdown fibroblasts at 30 hours post serum replenishment was greater than observed in patient 1. This may be because there was a trend to elevated ARID1B mRNA (and presumably protein) compared to patient 1, although it was not of statistical significance (Figure 3A). Both patient 1 and the knockdown cells demonstrated significantly reduced mRNA levels compared to the control fibroblasts and control fibroblasts transduced with the non-silencing ‘scramble’ control. While off-target effects were not specifically controlled for, the non-silencing control cells did not display alterations in either the number or timing of cells undergoing S1 phase. Moreover, both patient and ARID1B-knockdown cells displayed similar delayed entry to S1 phase, suggesting the effects were due to reduced ARID1B rather than a non-specific off target effect of the ARID1B-targetting shRNAmir used. Although we could not definitively demonstrate reduced endogenous ARID1B levels by western blot (antibodies tested were sc-32762 and NB100-57484), collectively these observations suggested that reduced ARID1B was associated with perturbed cell cycle regulation, which affected both the number and timing of cells entering S1 phase after serum starvation.
Identification of four additional patients with ARID1B mutations
The distinctive clinical features observed in patient 1 enabled us to ascertain four additional patients with a strikingly similar phenotype. All five patients, including patient 1 had intellectual disability, absent speech, plantar fat pads, fetal finger pads, and facial dysmorphism, consisting of narrow palpebral fissures, long eyelashes, a thin upper lip and full lower lip (Table 1). Direct sequencing of genomic DNA revealed that three additional patients encoded de novo single allelic ARID1B mutations; NM_020732.3(ARID1B):c.3208_3209delAA (p.(Lys1070Alafs*47) in patient 2, NM_020732.3(ARID1B):c.2306_2308delCCGinsTCCGCAGCCACTCC (p.(Pro769Leufs*17)) in patient 3 and NM_020732.3(ARID1B):c.4273dupT (p.(Tyr1425Leufs*34)) in patient 4. Whole exome sequencing of patient 5 identified 325 unique variants. Further filtering against an in-house database of genes previously implicated in intellectual disability identified a single candidate variant, which is predicted to be truncating. The de novo heterozygous ARID1B mutation NM_020732.3(ARID1B):c.2941C > T (p.(Gln981*)) was confirmed by Sanger sequencing (Figure 4). Two of these patients (patient 3 and patient 5) were previously published as having a phenotype consistent with Pierpont syndrome [18]. The molecular basis of Pierpont syndrome remains to be reported, but plantar fat pads are a key feature. The facial features in the five patients described here are distinct from those originally reported in Pierpont syndrome in that their palpebral fissures are not as narrow and their nasal tip not as broad [18,19]. Therefore we now believe their diagnosis should be of a BAF complex disorder due to ARID1B haploinsufficiency. In addition, we carried out mutation analysis of ARID1B (but not other members of the BAF complex) in four patients with classical features of Pierpont syndrome including patient 1 of the original case report [19] and did not identify any mutation (unpublished data).
Table 1.
Comparison of patient clinical features
Patient 1
|
Patient 2
|
Patient 3 |
Patient 4
|
Patient 5
|
|
|---|---|---|---|---|---|
| Previously published |
- |
- |
Patient 5 in Wright et al., 2011 [18] |
- |
Patient 6 in Wright et al., 2011 [18] |
| Age at assessment |
8y |
2y |
6y 10m |
5y |
8y 2m |
| Height centile |
50th |
10-25th |
25th |
3rd |
2nd |
| Weight centile |
50-75th |
25th |
50-75th |
25th |
2nd |
| Head circumference |
50-98th |
25th |
50-98th |
25th |
50th |
| Heel Fat pads |
+ |
+ |
+ |
+ |
+ |
| Fetal finger and toe pads |
+ |
+ |
+ |
NR |
+ |
| Fifth nail hypoplasia |
- |
- |
- |
- |
- |
| Hirsutism |
- |
- |
- |
- |
+ |
| ID |
Mod |
Mod |
Mild |
Mod |
Mod |
| Speech |
Absent |
Absent |
Delayed |
Absent |
Delayed |
| Seizures |
- |
+ |
- |
+ |
- |
| Drooling |
+ |
+ |
+ |
- |
+ |
| Feeding difficulty |
+ |
- |
Mild |
- |
- |
| Scoliosis |
- |
- |
- |
- |
+ |
| Inguinal hernia |
- |
- |
- |
- |
- |
| Neuroimaging | Mega cisterna magna on brain MRI | Normal brain MRI | Hypoplastic posterior elements of corpus callosum on MRI | Head CT in first year of life, normal | Normal |
Figure 4.

Analysis of ARID1B in the patient cohort. The protein domain organisation of ARID1B and the position of the four mutations identified by sequencing are shown. The forward and reverse sequencing traces demonstrate the NM_020732.3(ARID1B):c.3208_3209delAA (p.(Lys1070Alafs*47)) mutation in patient 2 (A), the NM_020732.3(ARID1B):c.2306_2308delCCGinsTCCGCAGCCACTCC (p.(Pro769Leufs*17)) mutation in patient 3 (B), the NM_020732.3(ARID1B):c.4273dupT (p.(Tyr1425Leufs*34)) mutation in patient 4 (C) and the NM_020732.3(ARID1B):c.2941C > T (p.(Gln981*)) mutation in patient 5 (D). Where available, the forward sequencing trace for the parents is also shown. Mutation co-ordinates are derived from refseq NM_020732.3.
Discussion
Recent reports have demonstrated that mutations in ARID1B can cause both nonsyndromic intellectual disability and CSS. The phenotypes associated with ARID1B haploinsufficiency are variable but common features of the syndromic and nonsyndromic cases include intellectual disability and speech impairment. While the facial dysmorphism and intellectual disability present in our cohort is similar to previously published patients with ARID1B mutations [4-7], this study broadens the spectrum of ARID1B-associated phenotypes because all our patients presented with plantar fat pads and fetal finger/toe pads and none of our patients have the fifth nail hypoplasia hallmark of CSS. As some of the previous cohorts with ARID1B mutations were ascertained by a clinical diagnosis of CSS, there is a selection bias in the phenotypic features they will have. Although none of the point mutations in ARID1B are common between CSS and our cohort, three CSS-like patients with de novo single allelic deletion at chromosome 6 similar to patient 1 were identified by Santen et al. in a large cohort of patients with intellectual disability [4]. The phenotypes of these patients share some features but apparently represent distinct syndromic manifestations of ARID1B haploinsufficiency. Thus, our cohort demonstrates that the ARID1B phenotypic spectrum is broader than previously defined and includes the novel clinical features of plantar fat pads and fetal digit pads. The constant features across cohorts appear to be the facial gestalt and the presence of intellectual disability particularly affecting speech, in contrast to the marked variability in the presence and type of manifestations in the hands and feet.
ARID1B is a DNA-binding component of BAF complexes that are involved in regulating many biological pathways [12]. However, the repertoire of pathways regulated by these BAF complexes and the impact of mutations in BAF components on these pathways are still poorly understood. Here, we present the first direct evidence in patient-derived cells that alterations in cell cycle may contribute to the pathogenesis of syndromes associated with ARID1B haploinsufficiency. Given that the BAF complex comprises over 25 core and interchangeable protein subunits that give rise to functionally distinct and cell-type specific complexes [12], it is likely that additional variation in these components contributes to the observed phenotypic variability in ARID1B-mediated disorders.
It is possible that the developmental features observed in patients with ARID1B haploinsufficiency are the result of abnormal regulation of cell cycle re-entry of developmentally arrested cells. This would impair developmental processes by upsetting the initiation of progenitor cell proliferation. In support of this notion, homozygous knockout of arid1b in mouse is embryonic lethal but the ES cells demonstrated a reduced proliferation rate and perturbation of differentiation and cell cycle [20]. An analogous observation is the presence of microcephaly in a minority of patients with ARID1B mutations and more broadly in Coffin-Siris syndrome [6,8]. Besides regulating cell cycle, chromatin-remodeling events and BAF complexes have been shown to be critically important during neural development and dendrite formation [13,21,22]. Although a specific role for ARID1B in early brain development remains to be demonstrated, the gene is predominantly expressed in neural tissues in the developing mouse embryo, suggesting that it is important for development of the brain when multipotent neuroepithelial cells are actively proliferating [9,23]. Future studies should investigate if impaired neural development contributes to the intellectual disability and speech impairment that are consistently observed in patients with ARID1B haploinsufficiency.
Conclusion
Our study demonstrates that the ARID1B phenotypic spectrum is broader than previously defined and includes the novel clinical features of plantar fat pads and fetal digit pads. These features can manifest in the absence of the hypertrichosis or fifth nail hypoplasia associated with Coffin-Siris syndrome. In addition, we present the first direct evidence in patient-derived cells that alterations in cell cycle may contribute to the underlying pathogenesis of syndromes associated with ARID1B haploinsufficiency.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JS performed molecular analysis and co-wrote the manuscript. SW provided and interpreted the clinical data and co-wrote the manuscript. EF, GW, GG, HM, SJ and DM performed molecular analysis and interpreted data. KP performed patient recruitment and read/contributed to the manuscript. PT, ATVvS, SM, RJL, MBD and DJA provided and interpreted the clinical data and revised the manuscript. PJL collected and analysed the data, wrote the manuscript and is responsible for overall content as guarantor and corresponding author.
Supplementary Material
Details of primer sequences. Primers were designed to amplify DNA encoding ARID1B (NM_020732.3) for direct sequence analysis.
Contributor Information
Joe C H Sim, Email: joe.sim@mcri.edu.au.
Susan M White, Email: sue.white@vcgs.org.au.
Elizabeth Fitzpatrick, Email: libby_fitzpatrick@yahoo.com.au.
Gabrielle R Wilson, Email: gabrielle.wilson@mcri.edu.au.
Greta Gillies, Email: greta.gillies@mcri.edu.au.
Kate Pope, Email: kate.pope@mcri.edu.au.
Hayley S Mountford, Email: hayley.mountford@mcri.edu.au.
Pernille M Torring, Email: pernille.toerring@ouh.regionsyddanmark.dk.
Shane McKee, Email: shane.mckee@belfasttrust.hscni.net.
Anneke T Vulto-van Silfhout, Email: a.silfhout@antrg.umcn.nl.
Shalini N Jhangiani, Email: jhangian@bcm.edu.
Donna M Muzny, Email: donnam@bcm.edu.
Richard J Leventer, Email: richard.leventer@mcri.edu.au.
Martin B Delatycki, Email: martin.delatycki@vcgs.org.au.
David J Amor, Email: david.amor@vcgs.org.au.
Paul J Lockhart, Email: paul.lockhart@mcri.edu.au.
Acknowledgements
We thank the families for their participation in this study and the generous support of the Lefroy and Handbury families. We thank Professor Han Brunner, Professor Di Donnai, Professor Raoul Hennekam, Dr Marlies Kempers, Dr Maria Kibaek, Dr Sahar Mansour, Dr Ann Olney, Dr Mary Ella Pierpont and Dr Emma Wright for their contribution of patients for this study. We also thank Professors Richard A. Gibbs and James R. Lupski for input into the manuscript.
Funding
This work was funded in part by National Health and Medical Research Council Australia Program Grant 490037 to DJA. MBD is supported by an NHMRC Practitioner Fellowship (546452) and PJL is supported by an NHMRC Career Development Fellowship (APP1032364). This study was accomplished in part through the Centers for Mendelian Genomics research effort funded by the National Institutes of Health and supported by the National Human Genome Research Institute grant U54HG006542 to the Baylor-Hopkins Center for Mendelian Genomics. This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
References
- Teif VB, Rippe K. Predicting nucleosome positions on the DNA: combining intrinsic sequence preferences and remodeler activities. Nucleic Acids Res. 2009;37:5641–5655. doi: 10.1093/nar/gkp610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Balliano A, Hayes JJ. Mechanism(s) of SWI/SNF-induced nucleosome mobilization. Chembiochem. 2011;12:196–204. doi: 10.1002/cbic.201000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N. et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- Wieczorek D, Bogershausen N, Beleggia F, Steiner-Haldenstatt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmuller J, Alanay Y, Kayserili H, Klein-Hitpass L, Böhringer S, Wollstein A, Albrecht B, Boduroglu K, Caliebe A, Chrzanowska K, Cogulu O, Cristofoli F, Czeschik JC, Devriendt K, Dotti MT, Elcioglu N, Gener B, Goecke TO, Krajewska-Walasek M, Guillén-Navarro E, Hayek J. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22(25):5121–5135. doi: 10.1093/hmg/ddt366. [DOI] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, van der Wielen MJ, Vollebregt MJ, Breuning MH, Kriek M, van Haeringen A, den Dunnen JT, Hoischen A, Clayton-Smith J, de Vries BB, Hennekam RC, van Belzen MJ. Coffin-Siris consortium. Coffin-Siris Syndrome and the BAF Complex: Genotype-Phenotype Study in 63 Patients. Hum Mutat. 2013;34:1519–1528. doi: 10.1002/humu.22394. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Dalal AB, Al-Kindy A, Liebelt J, Mowat D, Nakashima M, Saitsu H, Miyake N, Matsumoto N. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2013. DOI:10.1111/cge.12225. [DOI] [PubMed]
- Backx L, Seuntjens E, Devriendt K, Vermeesch J, Van Esch H. A balanced translocation t(6;14)(q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet Genome Res. 2011;132:135–143. doi: 10.1159/000321577. [DOI] [PubMed] [Google Scholar]
- Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, Nielsen M, den Hollander NS, Ruivenkamp CA, Isidor B, Le Caignec C, Zannolli R, Mucciolo M, Renieri A, Mari F, Anderlid BM, Andrieux J, Dieux A, Tommerup N, Bache I. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clinical genetics. 2012;82:248–255. doi: 10.1111/j.1399-0004.2011.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Göhring I, Zink AM, Rappold G, Schröck E, Wieczorek D, Riess O, Engels H, Rauch A, Reis A. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, Kovar CL, Wang M, Hawes AC, Reid JG, Eng C, Muzny DM, Gibbs RA. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome medicine. 2013;5:57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagl NG Jr, Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007;26:752–763. doi: 10.1038/sj.emboj.7601541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PK, Ho A, Dowdy SF. Biological methods for cell-cycle synchronization of mammalian cells. BioTechniques. 2001;30:1322–1326. doi: 10.2144/01306rv01. 1328, 1330–1321. [DOI] [PubMed] [Google Scholar]
- Wright EM, Suri M, White SM, de Leeuw N, Vulto-van Silfhout AT, Stewart F, McKee S, Mansour S, Connell FC, Chopra M, Kirk EP, Devriendt K, Reardon W, Brunner H, Donnai D. Pierpont syndrome: a collaborative study. Am J Med Genet A. 2011;155A:2203–2211. doi: 10.1002/ajmg.a.34147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierpont ME, Stewart FJ, Gorlin RJ. Plantar lipomatosis, unusual facial phenotype and developmental delay: a new MCA/MR syndrome. Am J Med Genet. 1998;75:18–21. doi: 10.1002/(SICI)1096-8628(19980106)75:1<18::AID-AJMG5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Yan Z, Wang Z, Sharova L, Sharov AA, Ling C, Piao Y, Aiba K, Matoba R, Wang W, Ko MS. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem cells. 2008;26:1155–1165. doi: 10.1634/stemcells.2007-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron. 2007;56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Alcantar A, Gonzalez-Sandoval A, Escalante-Alcalde D, Lomeli H. Dynamics of expression of ARID1A and ARID1B subunits in mouse embryos and in cells during the cell cycle. Cell Tissue Res. 2011;345:137–148. doi: 10.1007/s00441-011-1182-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of primer sequences. Primers were designed to amplify DNA encoding ARID1B (NM_020732.3) for direct sequence analysis.