

Abstract
Synapses are essential for transmitting, processing, and storing information, all of which decline in aging and Alzheimer’s disease (AD). Because synapse loss only partially accounts for the cognitive declines seen in aging and AD, we hypothesized that existing synapses might undergo molecular changes that reduce their functional capacity. Microarrays were used to evaluate expression profiles of 340 synaptic genes in aging (20–99 years) and AD across 4 brain regions from 81 cases. The analysis revealed an unexpectedly large number of significant expression changes in synapse-related genes in aging, with many undergoing progressive downregulation across aging and AD. Functional classification of the genes showing altered expression revealed that multiple aspects of synaptic function are affected, notably synaptic vesicle trafficking and release, neurotransmitter receptors and receptor trafficking, postsynaptic density scaffolding, cell adhesion regulating synaptic stability, and neuromodulatory systems. The widespread declines in synaptic gene expression in normal aging suggests that function of existing synapses might be impaired, and that a common set of synaptic genes are vulnerable to change in aging and AD.
Keywords: Microarray, Synaptic vesicle trafficking, Neurotransmitter receptors, Scaffolding molecules, Cortex, Limbic, Molecular reprogramming
1. Introduction
Aging is often accompanied by cognitive decline, in the absence of Alzheimer’s disease (AD) or other neurodegenerative diseases. Cognitive processes susceptible to age-related decline notably include working memory and other functions mediated by the dorsolateral prefrontal cortex, and declarative and episodic memory associated with hippocampal function. Elucidation of the multitude of factors that contribute to age-related cognitive decline is an area of active research, with many studies focusing efforts on defining how the structural integrity of the brain changes in normal aging and the extent to which structural changes correlate with cognitive declines. These studies have culminated in a growing appreciation that age-related cognitive decline is likely associated with alterations in synaptic connectivity and function, rather than neuron loss (Nicholson et al., 2004).
There is growing evidence that in aging, the brain undergoes synapse loss with minimal, if any, neuron loss. Interestingly, synapse loss occurs in a highly region-specific and synapse-type specific pattern. For example, the dorsolateral prefrontal cortex (in particular Brodmann area [BA] 46) undergoes substantial age-related synapse loss (30%–60%) that is not apparent in other regions of the prefrontal cortex, as demonstrated by electron microscopy studies in nonhuman primates (Dumitriu et al., 2010; Peters et al., 2008). In contrast to the synaptic loss reported in BA 46, the hippocampus (HC) appears to be characterized by more subtle and subregion-specific synaptic changes, for example, reduced postsynaptic density (PSD) size in the CA1 in the absence of synapse loss, and increased numbers of nonsynaptic boutons in the dentate gyrus (Hara et al., 2011; Nicholson et al., 2004). Taken together, the literature indicates that the nature of synaptic aging differs depending on brain region (Morrison and Baxter, 2012) and that subtle synaptic alterations can occur in the absence of overt synapse loss that correlate with age-related cognitive decline (Nicholson et al., 2004).
Age-related synaptic alterations that affect function have been most frequently documented at the structural and neurochemical levels (Hof and Morrison, 2004; Morrison and Baxter, 2012). It is likely that these structural and neurochemical changes are accompanied by reprogramming of the molecular machinery of the synapse, an idea that is consistent with evidence that the HC shows reduced PSD sizes in the CA1 and increased numbers of nonsynaptic boutons in the dentate gyrus with aging (Hara et al., 2011; Nicholson et al., 2004). Overall synaptic efficacy depends critically on the integrated function of multiple pre- and postsynaptic systems that include synaptic vesicle trafficking and release, neurotransmitter receptors, PSD scaffolding molecules, and adhesion molecules that regulate the stability of the synapse, among others. Deficits in 1 or more of these systems would be predicted to compromise synaptic function and impair cognition. The theme of this report is to examine the constellation of molecular changes in synapse-related genes that occur across the brain in aging and AD, and how these profiles differ across brain regions that undergo different structural and neurochemical alterations with age and neurodegeneration.
High throughput microarray technology is well suited to provide a global view at the molecular level of changes in the synaptic machinery, because gene expression profiles of hundreds of synapse-related genes can be simultaneously assessed. Although a number of studies have reported array data on global gene expression changes in the human brain in AD (e.g., Liang et al., 2008; Yao et al., 2003) or in aging (Berchtold et al., 2008; Colantuoni et al., 2011; Erraji-Benchekroun et al., 2005; Lu et al., 2004), no previous microarray study has specifically addressed, in normal aging and AD, synaptic gene expression changes in multiple brain regions from the same cases. In the present study, microarrays were used to comprehensively compare expression profiles of genes related to synaptic function in young, aged, and AD individuals. Four cortical regions were profiled: 3 that accumulate pathology and undergo functional decline in aging and AD (the HC, entorhinal cortex [EC], and superior frontal gyrus [SFG]) and a fourth region that is relatively unaffected in AD (the postcentral gyrus [PCG]) (Braak and Braak, 1991). These data thus build on our previous studies in which we analyzed aging-related changes in these regions using the same set of young and aged cases, across all genes represented on the microarray. Our previous studies revealed highly significant enrichment of synaptic and energy-related classes of genes downregulated with aging, while upregulated genes were highly enriched in genes related to immune function (Berchtold et al., 2008; Cribbs et al., 2012).
The study design permitted analysis of a number of hypotheses. First, we tested the possibility that synaptic genes undergo region-specific patterns of change in aging and AD, particularly with respect to brain regions that accumulate extensive pathology in AD. Second, we evaluated whether select synaptic components or the broad class of synaptic genes were altered in aging and AD. Third, we tested the hypothesis that synaptic changes that are characteristic of the AD brain are already present to some degree in the aged brain. Overall, the findings suggest a new perspective on the manner in which the molecular machinery of synapses in the brain is affected across the lifespan and in AD.
2. Methods
Frozen unfixed tissue was obtained from 55 non-AD controls (age 20–99 years) and from 26 Alzheimer’s disease cases (age 74–95 years) (Table 1) from 7 well established National Institute on Aging Alzheimer’s disease brain banks. Inclusion criteria can be found in the Supplementary data (materials and methods). Total RNA was extracted from 4 brain regions (EC, HC, SFG [BA 9/46], PCG [BA 1, 2, and 3]) as described previously (Berchtold et al., 2008), and RNA quality was assessed using the Agilent BioAnalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA integrity numbers (RIN) are shown in Supplementary Tables 1A and B. There were no significant differences in RNA integrity numbers across comparison groups for any region (Supplementary Table 1B). Two hundred forty-two tissue samples were individually hybridized to Affymetrix HgU133 plus 2.0 arrays: SFG, 65 arrays; PCG, 66 arrays; HC, 59 arrays; and EC, 52 arrays (Supplementary Table 1). Samples were randomized across batch runs, and no batch effects were detected. Guanine-cytosine robust multi-assay analysis (GC-RMA) expression values were calculated, followed by per-chip and per-gene normalization, log-transformation of the geometric mean of expression using Gene-Spring 7.3.1 software (Agilent Technologies, Palo Alto, CA, USA). Five hundred sixty-two probe sets representing 340 synaptic related genes were investigated for statistical significance (p < 0.01) in aging, comparing young (n = 22,20–59 years, mean age 35.4 years) versus aged controls (n = 33, age 69–99, mean age 84.2 years) (Table 1). The cutoff separating young versus aged samples (e.g., age 60 years) was based on our previous analysis (Berchtold et al., 2008), and is supported by other microarray studies demonstrating that though gene expression is relatively constant through adulthood, many genes enter turning points at approximately age 60 (Colantuoni et al., 2011; Kang et al., 2011). AD-related gene expression changes were based on the comparison of AD cases (n = 26, ages 74–95 years, mean age 85.7) versus age-matched controls (age 69–99 years) (n = 33, age 69–99, mean age 84.2 years). At a threshold of p < 0.01, the expected number of false positives is 3.4 genes. A false discovery rate can be calculated as the ratio of ‘expected false positives’/‘significant genes’, such that a false discovery rate ≤0.05 corresponds to a significant gene list of n ≥ 68 genes. Aging-AD continuum genes were identified using a 2-step procedure. First, linear regression across all cases was used to model mean gene expression as a function of group (young, aged, AD), and genes expressing a significant (p < 0.01) trend across groups were identified for each region. Next, we identified genes that demonstrated a consistent pairwise relationship as defined by the upper and lower bounds of the corresponding confidence interval for the difference in means for each pairwise comparison (young vs. aged) and (aged vs. AD), using 85% confidence limits. The second step was taken to ensure that the genes expressing significant age trends in step 1 truly do exhibit a trend across all groups, and were not selected because of differences between only 2 of the 3 groups. A subset of synaptic genes was further analyzed by quantitative polymerase chain reaction (qPCR) in hippocampal tissue from 29 samples (young control, n = 9; aged control, n = 10; AD, n = 10) to validate the age- and AD-related changes derived from the gene chip studies. A subset of these samples (non-AD controls) has been used in a previous microarray study (Berchtold et al., 2008) for which data have been deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo) accession number GSE11882. More detailed methods can be found in the Supplementary data (materials and methods).
Table 1.
Total cases and cases/brain regions analyzed for each group
| Control (20–59 y)
|
Control (69–99 y)
|
AD (74–95 y)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Male, female | Av age ± SD | n | Male, female | Av age ± SD | n | Male, female | Av age ± SD | |
| All cases | 22 | 12, 10 | 35.4 ± 10.5 | 33 | 14, 19 | 84.4 ± 9.0 | 27 | 11, 16 | 85.7 ± 6.3 |
| SFG | 21 | 12, 9 | 35.0 ± 10.6 | 23 | 10, 13 | 84.1 ± 9.4 | 21 | 7, 14 | 87.2 ± 6.3 |
| PCG | 19 | 11, 8 | 34.7 ± 10.9 | 23 | 11, 12 | 82.9 ± 9.5 | 24 | 10, 14 | 86.1 ± 6.6 |
| HC | 18 | 10, 8 | 35.4 ± 10.9 | 23 | 12, 11 | 83.7 ± 9.0 | 18 | 9, 9 | 84.2 ± 6.8 |
| EC | 21 | 12, 9 | 35.4 ± 10.8 | 16 | 9, 7 | 82.8 ± 9.6 | 15 | 7, 8 | 86.5 ± 5.7 |
Key: AD, Alzheimer’s disease; EC, entorhinal cortex; HC, hippocampus; PCG, postcentral gyrus; SFG, superior frontal gyrus.
3. Results
3.1. Region-specific patterns of synaptic gene change in aging and AD
To evaluate the possibility that synapse-related genes undergo changes in messenger RNA (mRNA) availability in aging or AD, expression profiles of 562 probe sets representing 340 synaptic genes were assessed in the human brain, using 55 non-AD cases (aged 20–99 years) and 26 AD cases (aged 74–95 years) (Table 1, Supplementary Table 2). In addition, to determine if there are region-specific patterns of change in aging or AD, gene expression profiles were assessed in 4 brain regions of which 3 are vulnerable to functional decline in aging and AD (SFG, HC, EC). Gene expression profiles were additionally assessed in the PCG, a region that is relatively spared in AD, allowing us to test the prediction that more extensive synaptic gene change might occur in AD-vulnerable regions.
3.1.1. In aging, synaptic genes undergo extensive expression change in neocortical regions, and less in limbic regions
Gene expression in the HC, EC, SFG, and PCG was compared in young (20–59 years) versus aged (69–99 years, nondemented) controls, at a criterion level of p < 0.01. This analysis revealed an unexpectedly large number of significant expression changes in synapse-related genes in aging, with predominant declines in mRNA levels and region-specific response profiles (Fig. 1A). Contrary to our predictions, the most extensive aging-related changes in synaptic gene expression were observed in the neocortical SFG (198 genes) and PCG (168 genes), with surprisingly smaller responses in the limbic HC (58 genes) and EC (22 genes). Most synaptic genes underwent decreased expression with age, particularly in the neocortical regions, where approximately 80% of probe sets revealed reduced mRNA expression.
Fig. 1.
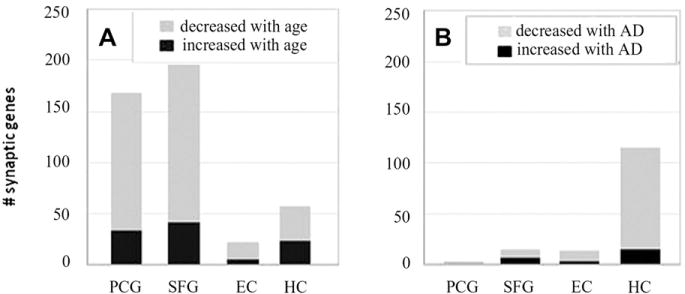
Aging and Alzheimer’s disease (AD) undergo unique region-specific patterns of synaptic gene responses, with the vast majority of genes downregulated. (A) In normal aging, neocortical regions (superior frontal gyrus [SFG], postcentral gyrus [PCG]) undergo extensive synaptic gene expression change, and limbic regions (hippocampus [HC], entorhinal cortex [EC]) show relatively little significant transcriptional change (p < 0.01, young vs. aged). (B) In AD, only the HC showed extensive synaptic gene expression changes, and relatively little transcriptional change was detected in the EC and SFG. Essentially no gene expression change was detected in the PCG (p < 0.01, aged vs. AD).
3.1.2. In AD, only the HC shows extensive changes in synaptic gene expression
Changes in synaptic gene expression were next assessed in AD cases relative to age-matched controls (p < 0.01), with the prediction that extensive change would be apparent in the HC, EC, and SFG and minimal in the PCG. Further, we predicted that more synaptic genes would undergo expression change in AD than in aging. Consistent with these predictions, a large number of synaptic genes in the HC underwent expression change in the AD brain (115 genes), a response that was more extensive than that seen in aging. However, in the EC and SFG, which are also pathologically vulnerable regions in AD, few synaptic genes were identified as showing significantly altered expression (13 and 15 genes, respectively), and, indeed, these regions underwent far less change in AD than was observed in aging (Fig. 1B). Finally, consistent with the fact that the PCG is minimally affected in AD, synaptic genes in the PCG showed essentially no AD-related change in expression (3 genes). Thus, this initial analysis revealed that in AD, extensive change in synaptic gene expression was only apparent in the HC, with minimal change detected in the PCG, SFG, and EC.
Because the EC and HC are early loci of pathology (Braak and Braak, 1991), undergo significant functional decline in AD (Sperling et al., 2010), and are highly interactive components of a functionally connected network (van Strien et al., 2009), we investigated if the 168 synaptic probe sets representing the 115 genes that were significantly changed in the HC in AD showed similar expression changes in the EC, using regression analysis of fold change. A strong correlation was found (r = 0.82; p < 0.0001), indicating that synaptic genes in these 2 interconnected, interactive structures might undergo similar patterns of change, although the gene responses appear to be more robust in the HC.
3.1.3. Synaptic genes undergo progressive change across aging and AD, especially in AD-vulnerable regions
A key unknown in aging and AD is whether expression changes in synaptic genes apparent in the AD brain are already initiated to some degree in aging. We therefore evaluated the extent to which synaptic genes in a given region show progressive increased or decreased expression across aging and AD. Linear regression across all cases was used to model mean gene expression as a function of group (young, aged, AD), followed by elimination of genes that did not show a consistent directional change across aging and AD, using 85% confidence intervals for the pair-wise comparison of ‘young versus aged’ and ‘aged versus AD.’ Genes meeting these criteria were designated “Aging-AD continuum” genes. This analysis revealed that a substantial number of synaptic genes underwent progressive expression change across Aging-AD (Fig. 2A). The strongest trend was apparent in the SFG and HC (113 and 104 genes, respectively) with relatively fewer continuum genes identified in the EC and PCG (60 and 64 genes, respectively) (Fig. 2A). The vast majority of Aging-AD continuum genes showed reduced expression, with 80%–95% of Aging-AD genes undergoing reduced expression in the EC, HC, and SFG. Overall, these data reveal that many of the gene expression changes in AD might have already been initiated to some degree in normal aging.
Fig. 2.
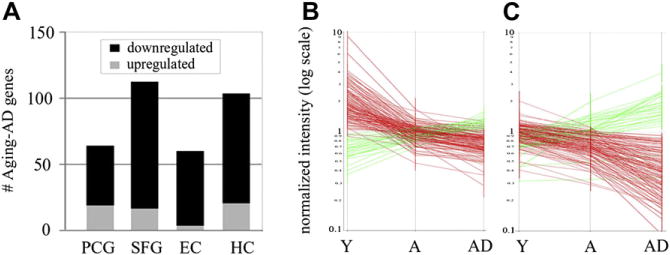
Numerous synaptic genes appear to undergo a progressive transcriptional change that is initiated in aging and continues in Alzheimer’s disease (AD). (A) Genes showing a progressive change across Aging-AD were most prevalent in the superior frontal gyrus (SFG) and hippocampus (HC), with relatively less change occurring in the entorhinal cortex (EC) and postcentral gyrus (PCG). (B) Signature profiles of Aging-AD genes in the SFG. (C) Signature profiles of Aging-AD genes in the HC. Abbreviations: A, aged; Y, young.
3.2. Functional classification of aging genes and Aging-AD genes
To gain insight into which elements of the synaptic machinery are most affected, we functionally categorized the synaptic genes showing altered expression with aging, AD, or across Aging-AD. Categorization by synaptic subfunction allowed the hypothesis to be tested that select synaptic components might be particularly susceptible to change, or that, alternatively, synaptic genes as a class might show broadly altered expression and affect multiple aspects of synaptic function.
This analysis revealed that multiple aspects of synaptic function appear to be broadly affected across Aging-AD, with genes predominantly showing decreased expression across all subclasses. These included many genes related to (1) synaptic vesicle trafficking and release; (2) neurotransmitter receptors and receptor trafficking; (3) PSD scaffolding and cell adhesion molecules regulating synaptic stability; and (4) neuromodulatory systems. These classes of synaptic genes showed extensive expression declines in cortical regions in the course of brain aging, and in AD these genes were primarily affected in the HC. A subset of synaptic genes showed progressive declines across aging and AD, particularly in the HC and SFG. Few genes in these synaptic subclasses showed increased expression in either aging or AD. These data are described in more detail in the next sections.
3.2.1. Synaptic vesicle trafficking and release
Many genes involved in synaptic vesicle trafficking and release were significantly changed with aging, AD, and across Aging-AD. Neurotransmitter release into the synapse is controlled by a complex and tightly regulated membrane fusion machinery consisting of a multitude of soluble/n-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) and sec/munc proteins. A set of core neuronal SNARE molecules (synaptobrevin2 (vesicle-associated membrane (VAMP) 2), SNAP-25, and syntaxin 1) directly mediate fusion of synaptic vesicles with the presynaptic membrane (Sollner et al., 1993), with many additional SNARE-related genes modulating synaptic vesicle assembly, function, and trafficking (e.g., synapsins, synaptotagmins, Rabs (one family of the Ras superfamily of small GTPases), synaptobrevins, dynamins, and syntaxins, among others).
Aging was characterized by widespread decreased gene expression of the machinery controlling synaptic vesicle trafficking and release, particularly in the SFG and PCG (Supplementary Table 2). In these neocortical regions, genes showing decreased expression included core SNAREs (synaptobrevin2/VAMP2, and SNAP-25), and multiple SNARE-related molecules including synaptotagmin I (the preferential Ca+ sensor of synaptic vesicles) and its homologue synaptotagmin V, the synaptobrevin homolog YKT6, SNAP29, VAMP4, synapsins I and II, synaptopodin, RABs 3A and 14, SEC22A and B, syntaxin 6, syntaxin binding proteins 5 and 6, synaptobrevin-like 1, dynamin 1, dynamin-like 1, α and β synucleins, piccolo, and bassoon (Supplementary Table 2). In contrast to the extensive decline in gene expression in the aging cortex, fewer genes of this class were affected in the HC and EC. In the aging HC, genes undergoing decreased expression included SNAP29, RABs (3A, 14), SEC22 (A, B) syntaxin 6, synaptobrevin-like 1, dynamin-1, and dynamin-like 1, and in the EC, only syntaxin 6, YKT6, and synaptopodin showed significant decreased gene expression. Interestingly, a subset of genes regulating synaptic vesicle trafficking and fusion showed increased gene expression in aging, primarily in the SFG, including SNAP23 and the vesicle associated proteins VAMP1 (synaptobrevin), VAMP3 (cellubrevin), VAMP5 (myobrevin), and VAMP8 (endobrevin) (Supplementary Table 2).
Though in aging, genes regulating synaptic vesicle trafficking and release were extensively affected in the neocortex with less response in the HC and minimal change in the EC, a different region-specific pattern of response was apparent in AD. In AD, extensive response of genes regulating the synaptic vesicle release machinery was primarily apparent in the HC. In the HC, genes showing decreased expression included core SNAREs (synaptobrevin2/VAMP2 and SNAP-25), synapsins I and II, synaptotagmins I and V, dynamin 1, dynamin-like 1, synaptopodin, RAB 3A, SEC22B, α and β synucleins, syntaxin binding protein 5-like and 6, syntaxin 6, bassoon, and piccolo (Supplementary Table 2). Similar to the response in the aging neocortex, a few genes regulating synaptic vesicle trafficking and fusion showed increased mRNA levels in AD, including SNAP23 and synaptopodin 2 in the HC, and VAMP1 (synaptobrevin 1) in the HC and EC (Supplementary Table 2).
Finally, several genes in this functional class underwent a progressive change across Aging-AD, most notably in the HC and SFG, and to a lesser extent in the EC and PCG. Genes that underwent progressively decreased expression in the SFG and HC included synaptobrevin2/VAMP2, SNAP-25, YKT6, and synapsin I, with additional decreased expression in the HC of VAMP4, synaptotagmin I, and syntaxin binding proteins 5 and 6, and of piccolo in the SFG and EC. Several genes were consistently changed across Aging-AD in 3 or more regions, including decreased expression of synaptotagmin V, dynamin-1 like, and α synuclein, and progressively increased expression of SNAP23, in the SFG, PCG, and HC (Supplementary Table 2).
These data reveal an extensive decline in expression of genes regulating trafficking and release of synaptic vesicles, suggesting that the machinery for neurotransmission declines in functionality with age, especially in neocortical regions, and that the HC is additionally hard hit in AD. Many of the gene changes are initiated to some degree in normal aging and further progress in AD, notably in the HC and SFG. These include decreased expression of 2 of the 3 core neuronal SNAREs that directly mediate exocytosis (VAMP2 and SNAP25), which might be of particular consequence considering that the SNARE complex consisting of a 1:1:1 interaction of VAMP2, SNAP25, and syntaxin 1 (Sudhof and Rothman, 2009) governs neurotransmitter secretion at synapse.
3.2.2. Neurotransmitter receptors and glutamate receptor trafficking molecules
In parallel with the expression declines of the synaptic vesicle trafficking and release machinery in aging and AD, a similar pattern of reduced mRNA expression was apparent for nearly all classes of neurotransmitter receptors. Notably, neurotransmitter receptor genes showed extensive declines in expression in neocortical regions in the course of brain aging and in the HC in the AD brain, with many undergoing progressive declines across Aging-AD.
Genes for a vast array of glutamate and gamma-Aminobutyric (GAB) acid (GABA) receptor subtypes showed decreased expression in aging and AD, including the glutamatergic 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA), N-Methyl-D-aspartate (NMDA), kainic acid (KA), and metabotropic receptors (mGluRs), and multiple GABA-A, GABA-B, and GABA-G subunits (Supplementary Table 3). A subset of glutamate and GABA receptor subtypes underwent progressive declines across Aging-AD in multiple brain regions. In particular, multiple AMPA receptors (glutamate receptor, ionotropic, AMPA (GRIA): GRIA2, GRIA3, GRIA4), KA receptors (glutamate receptor, ionotropic, kainate: GRIK1, GRIK2), mGluR2, and GABA receptors (GABBR1, GABRA4, GABRB3, GABRD) underwent progressive expression declines in 3 or more brain regions. In parallel with decreased expression of glutamate receptors, multiple genes involved in glutamate receptor trafficking to the PSD showed decreased expression, particularly in cortical regions in aging, including neuronal pentraxin 1 and 2, islet cell autoantigen 1 (ICA1), Homer 1 and 2, and glutamate receptor interacting protein (GRIP) 1-associated protein (Supplementary Table 4).
In addition to the extensive declines in glutamate and GABA receptor gene expressions, many other classes of neurotransmitter receptors that modulate synaptic excitability and plasticity underwent declining gene expression in aging and AD, including multiple cholinergic, dopamine, glycine, noradrenergic, and serotonin receptor subtypes (Supplementary Table 3). Many of these genes underwent greater than 2-fold expression declines over the course of Aging-AD, including muscarinic cholinergic receptors 1 and 3, and serotonin receptors (HTR) 2A, and HTR2C. Interestingly, only 1 neurotransmitter-related gene was upregulated across aging-AD, specifically hippocampal expression of the adrenergic β 2 receptor.
Taken together with the data on synaptic vesicle machinery, these data reveal an extensive decline in expression of genes regulating neurotransmitter packaging and release along with multiple neurotransmitter receptors, suggesting that a broad decline in synaptic efficacy might be present in aging, particularly in the cortex. Many of these changes continue to evolve and progress in AD, particularly in the SFG and HC. These changes include decreased expression of receptor genes for the excitatory (glutamate) and inhibitory (GABA) neurotransmitter systems, and the neuromodulatory cholinergic and serotonin neurotransmitter systems.
3.2.3. PSD scaffolding and cell adhesion molecules regulating synaptic stability
In addition to the importance of neurotransmitter receptors and the synaptic vesicle release machinery, synaptic plasticity and brain function rely on proper formation, maintenance, and modification of connections between neurons. Critical components of the synaptic architecture include molecules involved in PSD scaffolding and cell adhesion, which regulate the stability and plasticity of the synaptic contact.
Our data reveal that many PSD scaffolding proteins and cell adhesion molecules undergo altered expression in aging and AD, including PSD93, PSD95, lin7B, Shank2, presynaptic neurexin and its postsynaptic partner neuroligin, contactin-associated proteins, multiple integrins, protocadherins, multiple ephrins, MintX11, and chapsyin 110 (Supplementary Tables 5 and 6). The most extensive response of this category of synaptic genes was seen in the neocortical regions in aging, with an intermediate response in the HC and minimal response in the EC. A number of genes underwent progressive expression change across Aging-AD, particularly in the SFG, PCG, and HC. Most genes related to PSD scaffolding and cell adhesion showed decreased expression, however, a subset showed increased expression, most prominently populated by integrins, neuroligin 1 and 2, and neurexin 3 (Supplementary Tables 5 and 6).
3.2.4. Neuromodulatory systems
Many synaptic genes showing altered expression in aging and AD were related to various neuromodulatory systems, all of which underwent decreased expression. These included brain-derived neurotrophic factor (BDNF) and its receptor TrkB, corticotropin releasing hormone (CRH), somatostatin (SST) and the related neuropeptides cortistatin, histamine, and tachykinin (Supplementary Table 7).
Following the same region-specific pattern of change as for synaptic vesicle trafficking and neurotransmitter receptor genes, neuromodulatory genes showed decreased expression predominantly in neocortical regions in aging, with limbic regions minimally affected. In AD, expression of genes in this class of synaptic function was decreased in both the SFG and HC, with the most extensive declines across aging-AD apparent in the SFG. Many of these genes underwent greater than 2-fold declines across Aging-AD, including BDNF and its receptor TrkB, CRH, SST, and tachykinin. Indeed, in AD, gene expression for CRH underwent a dramatic decline to 8%–20% of young levels across all brain regions, SST similarly declined to 12%–30% of young levels, and BDNF declined to 20%–28% of young expression levels across all brain regions. The age-related decline in BDNF gene expression (EC, SFG, PCG) was paralleled by decreased TrkB gene expression (HC, SFG, PCG) with BDNF levels further declining in AD (HC, SFG). The pronounced declines in BDNF and TrkB might be of particularly functional consequence, because this neuromodulatory system is critical for facilitating synaptic plasticity, learning, and memory.
3.2.5. Other categories
Finally, several additional categories of genes important for synaptic function were notably affected in aging, AD, or across Aging-AD. These included glutamate and GABA transporters, multiple enzymes including those important for neurotransmitter synthesis, and many classes of voltage-gated ion channels (calcium, potassium, and sodium channels) (Supplementary Table 8). Most of these genes showed decreased expression with aging and AD. A few exceptions include the glial solute carrier family (SLC) high affinity aspartate/glutamate transporter (SLC1A6), the potassium intermediate/small conductance calcium-activated channel (KCN) subfamily N, member 1 (KCNN1), and dystrobrevin alpha, among others, which showed notably increased expression in multiple brain regions.
3.3. qPCR validation
To validate the age- and AD-related expression changes detected in the microarray analysis, qPCR was used to analyze gene expression patterns in hippocampal tissue for a subset of synaptic genes representing synaptic vesicle trafficking/release, neurotransmitter receptors, and neuromodulators, and expression patterns were compared with microarray data for the same samples. Genes assessed by qPCR included 3 genes critical for synaptic vesicle release (synaptobrevin 2/VAMP2, SNAP25, synaptophysin), several neurotransmitter receptors important for cognitive function (glutamate NMDA receptor 1, muscarinic cholinergic receptor 3, serotonin receptor 2a), and the neuromodulator SST, downregulation of which in AD is directly linked to poor cognitive function and memory impairment (Epelbaum et al., 2009). qPCR analysis confirmed the microarray data showing age-related decline in synaptic gene expression and further declines in AD, and comparison of relative expression changes derived from qPCR and microarray analysis for the same set of samples revealed a high correspondence between the 2 methods (Fig. 3). These data are consistent with our previous qPCR validation of a different gene subset of these array data, demonstrating high agreement between microarray and qPCR results (Berchtold et al., 2008; Cribbs et al., 2012).
Fig. 3.
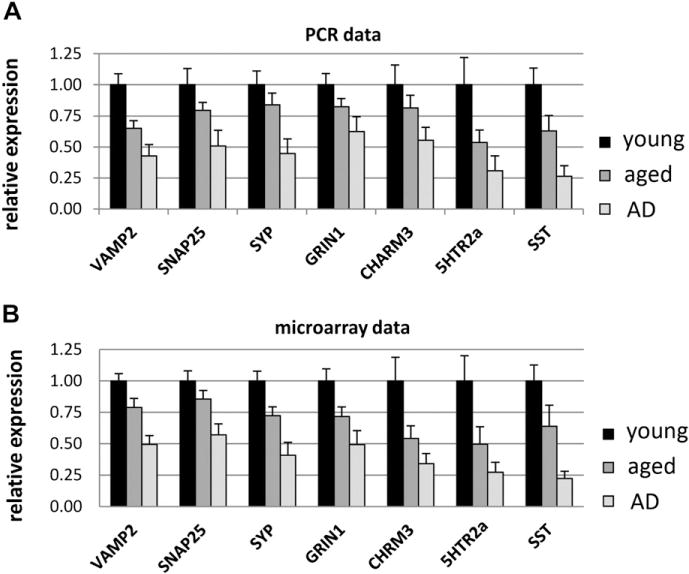
Quantitative polymerase chain reaction (qPCR) was undertaken in hippocampal tissue to validate the age- and Alzheimer’s disease (AD)-related changes derived from the gene chip studies for VAMP2 (synaptobrevin 2), SNAP25 (synaptosomal-associated protein-25), synaptophysin, GRIN1 (glutamate NMDA receptor 1), muscarinic cholinergic receptor 3 (CHARM3), serotonin receptor 2a (5HTR2a), and somatostatin (SST) using hippocampal tissue from 29 samples (young control, n = 9; aged control, n = 10; AD, n = 10). (A) qPCR data confirmed widespread downregulation of these synaptic genes in aging, with further downregulation in AD. Comparison of relative expression changes derived from qPCR and microarray analysis (B) for the same set of samples revealed a high correspondence between the 2 methods.
4. Discussion
Three major findings emerged from this study. First, many synaptic genes undergo pronounced declines in expression during normal aging, with a more extensive response in aging than in AD. Second, there is a region-specific pattern of response that is unique to aging or AD. Third, many synaptic genes showing decreased expression in normal aging undergo further declines in AD, revealing that many of the synaptic changes characteristic of the AD brain are already present to some degree in the aged brain.
Our data demonstrate that in aging (20–99 years), expression changes occur in a surprisingly high number of synaptic genes, with genes predominantly showing decreased expression. The extensive nature of synaptic gene expression change in the course of normal aging has not been previously reported, likely because previous studies have generally restricted investigation of narrower age ranges (e.g., approximately 60–100 years). Because the majority of age-related transcriptional changes in the human brain only emerge in the sixth to seventh decades (Berchtold et al., 2008; Colantuoni et al., 2011; Lu et al., 2004), studies that do not include younger adults likely miss detection of a large percentage of relevant genes.
Aging was characterized by a region-specific pattern of synaptic gene expression change that was extensive in neocortical regions but relatively mild in limbic regions, with the vast majority of genes showing decreased expression. The extensive declines in gene expression in the SFG is consistent with age-related synapse loss documented by electron microscopy in BA 46 in nonhuman primates (ranging from 30%–60% loss in layer 1 and 30%–35% loss in layer 3), with glutamatergic axospinous synapses accounting for the vast majority of lost synapses (Dumitriu et al., 2010; Peters et al., 2008). Consistent with the age-related loss of glutamatergic synapses in nonhuman primate BA 46, our data reveal extensive gene expression declines in multiple subtypes of AMPA, kainate, NMDA, and metabotropic glutamatergic receptors in the aged human SFG, with many genes exceeding 50% expression declines. Though a portion of the synaptic gene expression losses that we observe are undoubtedly beause of synapse loss, it is also likely that some of the gene expression changes reflect reprogramming of the molecular architecture of existing synapses. For example, in parallel with the declines in glutamatergic receptor gene expression, our data also reveal extensive declines in GABA-A receptors (α, β, and δ subtypes), which might represent a compensatory effort to counteract the loss of glutamatergic synaptic activity and maintain the excitatory/inhibitory balance in the SFG. Alternatively, the extensive declines in GABA might reflect that synapse loss in the aged SFG goes beyond loss of excitatory glutamatergic synapses, a possibility not yet supported by electron microscopy studies.
Though the gene expression declines in the SFG are consistent with aging studies documenting synaptic changes in the monkey BA 46, the similarly extensive declines in the PCG were unanticipated, because this is a primary somatosensory region not generally considered highly affected by aging. It is unlikely that the extensive changes in the PCG represent synapse loss, because several quantitative ultrastructural studies have documented a virtual absence of synaptic loss in this neocortical region (Adams, 1987; Scheff et al., 2001). However, because declines related to somatosensory function have been reported with aging, such as deterioration of proprioception (Kalisch et al., 2009), these extensive gene expression changes might reflect molecular reprogramming of the synaptic architecture that likely account for age-related changes in function involving this region.
In contrast to the extensive synaptic gene expression changes in neocortical regions, aging was characterized by only modest changes in the HC and minimal change in the EC. Though there is controversy regarding the extent to which synaptic loss occurs in the HC and EC in normal aging, it is generally thought that normal aging is characterized by general structural preservation in the medial temporal lobes, in contrast to AD which is associated with neuronal and synaptic loss, particularly in the HC (Morrison and Baxter, 2012). Nonetheless, recent rodent and monkey studies reveal that the aging HC undergoes a number of subtle synaptic changes indicating synaptic remodeling. For example, in the CA1, in the absence of synapse loss, there is a reduction in PSD size and fewer synaptic contacts per axonal bouton, and in the dentate gyrus, there is an increase in nonsynaptic boutons and preserved perforated synapse density (Hara et al., 2011; Nicholson et al., 2004). The synaptic gene expression changes that we observed in aging are likely related to these morphological changes and reflect molecular reprogramming of the synaptic architecture.
Finally, AD was characterized by minimal gene expression change in neocortical regions, but major declines in the HC, with correlated but less robust gene responses in the EC. The extensive declines in gene expression in limbic regions are consistent with the documented neuronal and synaptic loss in these regions in AD, most particularly in the HC (De Leon et al., 1997; Gomez-Isla et al., 1996), and with declining temporal lobe function characteristic of AD (Sperling et al., 2010). Though the gene responses were less marked in the EC than in the HC, it is possible that different cell layers in the EC undergo layer-specific gene response. In particular, layer II cells of the EC, which provide the major input to the HC and is the specific entorhinal circuitry most directly linked to the pathogenesis of AD, might be predicted to show a more pronounced change in synaptic gene expression than observed here for the whole EC. Ultimately, because the EC and HC are highly interactive components of a functionally connected network (van Strien et al., 2009), downstream effects of the extensive limbic synaptic gene downregulation in AD are likely compounded by the neocortical changes initiated in aging, ultimately leading to network deterioration and widespread cognitive and behavioral dysfunction in AD.
An important unresolved issue in the aging and AD fields is whether changes in gene expression levels that are apparent in the AD brain are initiated to some degree already in aging. In support of this idea, our data reveal that a remarkable number of synaptic genes undergo an apparent continuity of change across aging and AD, with predominantly decreased expression. Though the SFG and HC showed the greatest numbers of “continuum genes,” many were also present in the EC and PCG. Overall, our analysis supports the possibility that, within specific brain regions, AD might be part of a continuum with normal aging, with prominent, progressive downregulation of many genes regulating synaptic function. Classification of the synaptic genes undergoing progressive expression decline across aging and AD revealed that multiple aspects of synaptic function are affected. Genes associated with pre- and postsynaptic signaling mechanisms showed extensive declines in expression level, especially those regulating synaptic vesicle trafficking and release, neurotransmitter receptors, neuromodulatory systems, and synaptic formation and stability, as described in detail below.
Synaptic vesicle trafficking and neurotransmitter release into the synapse is tightly regulated by complex protein machinery. Central features of this machinery are SNARE proteins (Jahn and Scheller, 2006), particularly SNAP-25, synaptobrevin2/VAMP2, and syntaxin, which directly mediate fusion of synaptic vesicles with the presynaptic membrane, and are thus critical for regulating neurotransmitter release into the synapse. Working with SNAREs, a network of accessory molecules adds spatial and temporal specificity to neurotransmitter release and synaptic signaling. Our data reveal that numerous genes for SNAREs and SNARE accessory proteins undergo progressive gene expression declines across Aging-AD, particularly in the HC and SFG, with greater than 2-fold declines in SNAP-25, synaptobrevin2/VAMP2, synaptophysin, synapsins 1 and 2, and piccolo. Declines in the SFG are likely related to synaptic loss in this brain region, and the declines in the HC are more likely to be related to synaptic remodeling, because synaptic remodeling but not loss has been documented in the HC with aging. Rodent studies have demonstrated that downregulation of these vesicle trafficking genes can dramatically compromise synaptic and cognitive function. For example, targeted deletion of SNAP-25 or synaptobrevin2/VAMP2 impairs calcium-triggered neurotransmitter release (Bronk et al., 2007), while deletion of synapsin I or II results in fewer synaptic vesicles, synaptic depression on high-frequency stimulation, and age-dependent impairments in spatial and object recognition memory (Corradi et al., 2008), with similar cognitive impairments associated with synaptophysin knockout (Schmitt et al., 2009). Even modest reductions in gene expression can disrupt synaptic and cognitive function: antisense knockdown of piccolo by 30% in the mouse HC decreased K+ induced glutamate release and long-term potentiation (LTP) amplitude, with a corresponding impairment in spatial learning (Ibi et al., 2010). By comparison, our data reveal that piccolo is reduced by 65% (HC) and 52% (SFG) across Aging-AD. Overall, these data reveal that many genes regulating synaptic vesicle trafficking and neurotransmitter release undergo widespread and concurrent expression declines, with progress change across aging and AD.
In parallel, numerous genes related to neurotransmitter receptors and receptor trafficking similarly showed progressively decreased expression across Aging-AD. These included serotonin receptors, dopamine receptors, various GABA-A subunits, and many glutamate subtypes. Greater than 2-fold declines were apparent in the HC for GluR1 and multiple mGluRs, glutamate receptor subtypes critical for working memory in rodents (Bannerman, 2009). In addition, gene expression of multiple GABA-A subunits underwent more than 2-fold declines in multiple brain regions, with the HC notably affected. A corresponding change in GABA receptors at the protein level would be predicted to alter network excitability and might be related to the increased seizure susceptibility reported in AD (Larner, 2010). The extensive declines in expression of glutamate and GABA-A receptors, along with receptors for serotonin, dopamine, and acetylcholine (most prominently changed in the HC), suggests that the balance of synaptic excitability and inhibition might be reset during aging and AD, altering overall network signaling. These data are relevant to the accruing documentation from both the animal and human literature that aberrant increases in network excitability and compensatory inhibitory mechanisms might be major contributors to neural network dysfunction (Palop et al., 2007), with disrupted excitatory/inhibitory balance emerging as a key early signature linking aging and AD. This might be particularly relevant to HC function, in that elevated HC activation is observed in mild cognitive impairment, and studies in relevant animal models indicate that overactivity in selective HC circuits contributes to cognitive impairment (Bakker et al., 2012).
Decreased gene expression of neuromodulatory genes likely compounds the effects of transcriptional declines in genes regulating synaptic vesicle release and neurotransmitter receptors. Several neuromodulatory systems showed greater than 2-fold declines in mRNA availability across aging-AD. For example, striking decline was observed for SST, a neuromodulator implicated in the progression of AD (Epelbaum et al., 2009), which decreased in aging to 15%–50% of young levels, and further declined in AD to 12%–14% of young levels in the HC, PCG, and SFG. Decreased SST levels are consistently found in AD and are directly linked to poor cognitive function and memory impairment (Epelbaum et al., 2009). The prominent SST declines in the aging HC, PCG, and SFG are consistent with aging data from the frontal pole in humans (Lu et al., 2004), and aging-related declines in nonhuman primates identified in the HC, PCG, frontal cortex, temporal cortex, motor cortex, and visual cortex (Hayashi et al., 1997).
Further amplifying the altered synaptic function, our data reveal that many genes that direct synaptic maturation and stability undergo altered expression in aging, AD, and across Aging-AD, most notably in the HC, PCG, and SFG. These included decreased expression of numerous ephrins and ephrin receptors (important in modulating synaptic function and long-term changes in synaptic strength in the mature central nervous system; Hruska and Dalva, 2012), PSD95 (critical for clustering NMDA and AMPARs to the excitatory postsynaptic complex; Ehrlich and Malinow, 2004), and presynaptic neurexins paired with upregulation of their postsynaptic partners neuroligins. There is emerging evidence that synaptic plasticity and cognitive function can be strongly affected by shifts in the relative abundance of the various synaptic scaffolding and stabilization molecules (Dahlhaus et al., 2010; Sudhof, 2008). With this in mind, altered synaptic stability, concurrent with declines in presynaptic release mechanisms, neurotransmitter receptor, and neuromodulatory systems, likely converge to compromise network function, synaptic efficacy, and cognitive function.
The AD literature has identified a number of synaptic genes that show altered expression in the HC and frontal cortex in AD (Ginsberg et al., 2012; Gomez Ravetti et al., 2010; Miller et al., 2008; Tan et al., 2010). Our analysis of aging-AD continuum genes reveals that in addition to changing in AD, many of these synaptic genes show altered expression changes already in the course of aging. Previously identified AD genes that emerged as aging-AD genes in our analysis are summarized in Supplementary Table 9, and include downregulation of multiple synaptic vesicle and trafficking molecules, neurotransmitter receptors and synthesis, and neuromodulators including BDNF. In addition to confirming the previous AD literature, our data reveal that these genes already undergo AD-like changes in the course of aging in a region-specific manner. Importantly, our analysis identifies an extensive number of additional synaptic genes that undergo progressive change across aging-AD, thus suggesting new target genes that might be particularly important for preserving cognitive health before AD.
A number of studies suggest that the many of the same neurons and circuits that are affected during aging subsequently die in AD (Hof and Morrison, 2004; Masdeu et al., 2012; Morrison and Baxter, 2012; Neill, 2012). Our data demonstrating that the synaptic gene expression decline in aging is amplified with the transition to AD might provide support for this idea, because it is likely that the expression decline in synaptic markers is because of remodeling of synaptic connections of intact neurons and loss of neurons and synapses, particularly in the HC and SFG. However, our data from the PCG, which underwent prominent change in aging and revealed a number of aging-AD continuum genes in the apparent absence of synapse or neuron loss in aging and AD, suggest that synaptic gene change in this brain region might be primarily because of synaptic remodeling. Taken together, synaptic gene expression changes in the course of aging likely increase the vulnerability to neuron loss in AD, but in a region-specific manner. Further studies that undertake detailed morphologic analysis in combination with microarray data from the same cases will be indispensible to understand the manner in which the gene expression changes translate to neuron loss.
The results presented here are, of course, subject to limitations of microarray-based approaches. First, although the gene list was based on synaptic Gene Ontology categories, it is possible that some of the gene changes are not unequivocally of neuronal origin. Second, it is likely that some of the vesicle trafficking genes are not exclusive to synaptic vesicles, because numerous SNAREs are shared among systems (Jahn and Scheller, 2006). Third, some transcriptional changes might not have been detected because of sensitivity limitations of the microarray. Finally, mRNA and protein levels do not necessarily show a direct relationship. However, our data documenting extensive synaptic gene downregulation in human brain aging is consistent with protein data in rats demonstrating widespread alterations in the synaptoproteome with age (VanGuilder et al., 2010).
In summary, the current study provides a comprehensive dataset that defines the signature for gene expression at the synapse, how it evolves across aging and AD, and how different brain regions are affected. Our data revealing broad changes in synaptic gene expression across multiple functional classes suggests that an intervention to restore youthful levels of gene expression in the brain must have broad effectiveness across multiple synaptic classes. Animal studies indicate that 1 of the most effective interventions for exerting broad effects on brain gene expression patterns is exercise (Stranahan et al., 2010; Tong et al., 2001), and it will be important for future studies to examine the effectiveness of such lifestyle interventions to counteract gene expression patterns associated with aging and cognitive decline in humans.
Supplementary Material
Acknowledgments
Funding for this work was provided by National Institute on Aging grants R01 AG23173 (CWC), AG34667 (CWC), P50 AG16573 (CWC), AG00538 (CWC and DHC), RO1 AG36400 (PDC), AG7367 (JR), and by the Alzheimer Association grant IIRG 91822 (DHC).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.neurobiolaging.2012.11.024.
Footnotes
Disclosure statement
The authors have no actual or potential conflicts of interest.
All case identifiers have been removed and replaced with a unique case number by the providing tissue repositories, in compliance with protection of subjects and institutional review board (IRB) requirements.
References
- Adams I. Comparison of synaptic changes in the precentral and postcentral cerebral cortex of aging humans: a quantitative ultrastructural study. Neurobiol Aging. 1987;8:203–212. doi: 10.1016/0197-4580(87)90003-0. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DM. Fractionating spatial memory with glutamate receptor subunit-knockout mice. Biochem Soc Trans. 2009;37:1323–1327. doi: 10.1042/BST0371323. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, Beach T, Miller C, Troncoso J, Trojanowski JQ, Zielke HR, Cotman CW. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bronk P, Deak F, Wilson MC, Liu X, Sudhof TC, Kavalali ET. Differential effects of SNAP-25 deletion on Ca2+ -dependent and Ca2+ -independent neurotransmission. J Neurophysiol. 2007;98:794–806. doi: 10.1152/jn.00226.2007. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, Kleinman JE. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi A, Zanardi A, Giacomini C, Onofri F, Valtorta F, Zoli M, Benfenati F. Synapsin-I- and synapsin-II-null mice display an increased age-dependent cognitive impairment. J Cell Sci. 2008;121:3042–3051. doi: 10.1242/jcs.035063. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Berchtold NC, Perreau VM, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE, Christie BR, El-Husseini A. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity byaltering the ratio of excitation to inhibition in the hippocampus. Hippocampus. 2010;20:305–322. doi: 10.1002/hipo.20630. [DOI] [PubMed] [Google Scholar]
- De Leon MJ, George AE, Golomb J, Tarshish C, Convit A, Kluger A, De Santi S, McRae T, Ferris SH, Reisberg B, Ince C, Rusinek H, Bobinski M, Quinn B, Miller DC, Wisniewski HM. Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging. 1997;18:1–11. doi: 10.1016/s0197-4580(96)00213-8. [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, Rapp PR, Morrison JH. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30:7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelbaum J, Guillou JL, Gastambide F, Hoyer D, Duron E, Viollet C. Somatostatin, Alzheimer’s disease and cognition: an old story coming of age? Prog Neurobiol. 2009;89:153–161. doi: 10.1016/j.pneurobio.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Erraji-Benchekroun L, Underwood MD, Arango V, Galfalvy H, Pavlidis P, Smyrniotopoulos P, Mann JJ, Sibille E. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Che S. Gene expression levels assessed by CA1 pyramidal neuron and regional hippocampal dissections in Alzheimer’s disease. Neurobiol Dis. 2012;45:99–107. doi: 10.1016/j.nbd.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Ravetti M, Rosso OA, Berretta R, Moscato P. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS One. 2010;5:e10153. doi: 10.1371/journal.pone.0010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Park CS, Janssen WG, Punsoni M, Rapp PR, Morrison JH. Synaptic characteristics of dentate gyrus axonal boutons and their relationships with aging, menopause, and memory in female rhesus monkeys. J Neurosci. 2011;31:7737–7744. doi: 10.1523/JNEUROSCI.0822-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Yamashita A, Shimizu K. Somatostatin and brain-derived neurotrophic factor mRNA expression in the primate brain: decreased levels of mRNAs during aging. Brain Res. 1997;749:283–289. doi: 10.1016/S0006-8993(96)01317-0. [DOI] [PubMed] [Google Scholar]
- Hof PR, Morrison JH. The aging brain: morphomolecular senescence of cortical circuits. Trends Neurosci. 2004;27:607–613. doi: 10.1016/j.tins.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Hruska M, Dalva MB. Ephrin regulation of synapse formation, function and plasticity. Mol Cell Neurosci. 2012;50:35–44. doi: 10.1016/j.mcn.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Nitta A, Ishige K, Cen X, Ohtakara T, Nabeshima T, Ito Y. Piccolo knockdown-induced impairments of spatial learning and long-term potentiation in the hippocampal CA1 region. Neurochem Int. 2010;56:77–83. doi: 10.1016/j.neuint.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Kalisch T, Ragert P, Schwenkreis P, Dinse HR, Tegenthoff M. Impaired tactile acuity in old age is accompanied by enlarged hand representations in somatosensory cortex. Cereb Cortex. 2009;19:1530–1538. doi: 10.1093/cercor/bhn190. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Z, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, Mane S, Hyde TM, Huttner A, Reimers M, Kleinman JE, Sestan N. Spatiotemporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–77. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics. 2008;33:240–256. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Masdeu JC, Kreisl WC, Berman KF. The neurobiology of Alzheimer disease defined by neuroimaging. Curr Opin Neurol. 2012;25:410–420. doi: 10.1097/WCO.0b013e3283557b36. [DOI] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill D. Should Alzheimer’s disease be equated with human brain ageing? A maladaptive interaction between brain evolution and senescence. Ageing Res Rev. 2012;11:104–122. doi: 10.1016/j.arr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Nicholson DA, Yoshida R, Berry RW, Gallagher M, Geinisman Y. Reduction in size of perforated postsynaptic densities in hippocampal axospinous synapses and age-related spatial learning impairments. J Neurosci. 2004;24:7648–7653. doi: 10.1523/JNEUROSCI.1725-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Sethares C, Luebke JI. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience. 2008;152:970–981. doi: 10.1016/j.neuroscience.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Sparks DL. Quantitative assessment of possible age-related change in synaptic numbers in the human frontal cortex. Neurobiol Aging. 2001;22:355–365. doi: 10.1016/s0197-4580(01)00222-6. [DOI] [PubMed] [Google Scholar]
- Schmitt U, Tanimoto N, Seeliger M, Schaeffel F, Leube RE. Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience. 2009;162:234–243. doi: 10.1016/j.neuroscience.2009.04.046. [DOI] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol Aging. 2010;31:1937–1949. doi: 10.1016/j.neurobiolaging.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MG, Chua WT, Esiri MM, Smith AD, Vinters HV, Lai MK. Genome wide profiling of altered gene expression in the neocortex of Alzheimer’s disease. J Neurosci Res. 2010;88:1157–1169. doi: 10.1002/jnr.22290. [DOI] [PubMed] [Google Scholar]
- Tong L, Shen H, Perreau V, Balazs R, Cotman CW. Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis. 2001;8:1046–1056. doi: 10.1006/nbdi.2001.0427. [DOI] [PubMed] [Google Scholar]
- VanGuilder HD, Yan H, Farley JA, Sonntag WE, Freeman WM. Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J Neurochem. 2010;113:1577–1588. doi: 10.1111/j.1471-4159.2010.06719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Strien NM, Cappaert NL, Witter MP. The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. Nat Rev Neurosci. 2009;10:272–282. doi: 10.1038/nrn2614. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, Coleman PD. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer’s disease. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.