

Abstract
Background
Homozygous or compound heterozygous mutations in the PTEN-induced kinase 1 (PINK1) gene are causative of autosomal recessive, early onset PD. Single heterozygous mutations have been repeatedly detected in a subset of patients as well as in non-affected subjects, and their significance has long been debated. Several neurophysiological studies from non-manifesting PINK1 heterozygotes have shown the existence of neural plasticity abnormalities, indicating the presence of specific endophenotypic traits in the heterozygous state.
Methods
In the present study, we performed a functional analysis of corticostriatal synaptic plasticity in heterozygous PINK1 knock-out (PINK1+/−) mice by a multidisciplinary approach.
Results
We found that, despite a normal motor behavior, repetitive activation of cortical inputs to striatal neurons failed to induce long-term potentiation (LTP), whereas long-term depression (LTD) was normal. Although nigral dopaminergic neurons exhibited normal morphological and electrophysiological properties with normal responses to dopamine receptor activation, we measured a significantly lower dopamine release in the striatum of PINK1+/−, compared to control mice, suggesting that a decrease in stimulus-evoked dopamine overflow acts as a major determinant for the LTP deficit. Accordingly, pharmacological agents capable of increasing the availability of dopamine in the synaptic cleft restored a normal LTP in heterozygous mice. Moreover, MAO-B inhibitors rescued a physiological LTP and a normal dopamine release.
Conclusions
Our results provide novel evidence for striatal plasticity abnormalities even in the heterozygous disease state. These alterations might be considered an endophenotype to this monogenic form of PD, and a valid tool to characterize early disease stage and design possible disease-modifying therapies.
Keywords: PINK1, autosomal recessive Parkinson’s disease, heterozygous mutations, synaptic plasticity, striatum
Introduction
Gene mutations are causative of distinct inherited forms of Parkinson’s disease (PD), playing a relevant role in the etiology of the disease. Mendelian forms of PD account for a relatively small fraction of PD cases, as only 20% of patients with an early-onset and no more than 3–5% of those with a late onset have a monogenic etiology, with classical autosomal recessive or dominant inheritance [1]. Homozygous or compound heterozygous mutations in the PINK1 gene cause PD with Lewy body pathology, characterized by early onset often in a limb, slow progression and good response to levodopa [2–4]. Single heterozygous mutations or rare variants in the PINK1 gene have been detected in a subset of sporadic PD patients, who show a later onset and clinical features indistinguishable to idiopathic PD, as well as in a similar proportion of healthy controls [3].
The loss of dopaminergic inputs to the dorsal striatum is currently thought to be the major cause for the appearance of motor symptoms in PD [5,6], that are preceded by a prolonged, though yet undefined pre-symptomatic window, characterized by a 30–50% reduction of basal ganglia dopamine (DA) levels [7,8], suggesting a substantial ability to compensate for nigrostriatal cell loss. Functional MRI studies in clinically unaffected carriers of PINK1 single heterozygous mutations showed an increase of cortical motor-related activity during execution of self-initiated movements [9], interpreted as a reorganization of the motor system in the pre-symptomatic disease stage [9–11]. Moreover, PET studies demonstrated a reduced striatal dopaminergic F-dopa uptake [11,12], suggesting that the natural progression of subclinical motor signs may precede an overt parkinsonian syndrome. Finally, neurophysiological studies demonstrated impaired temporal discrimination thresholds in PINK1 heterozygous healthy carriers [13].
Based on such evidence, the condition of non-manifesting PINK1 heterozygous carrier provides a unique model to study the effects of a subclinical dopaminergic dysfunction on motor learning and plasticity. We performed recordings from heterozygous PINK1 knock-out mice (PINK1+/−) [14] slices and explored how the heterozygous condition may interfere with normal expression of synaptic plasticity at corticostriatal synapses, and nigral dopaminergic neurons excitability. Additionally, this electrophysiological study was paralleled by behavioral and neurochemical analyses.
Materials and Methods
PINK1 mice and genotyping
Animal experiments were carried out in accord with EC, Internal Institutional Review Committee, EU directive and Italian rules (86/609/EEC; D.Lvo 116/1992, 63/2100 EU, 153/2001A–IHM and 5/2010 UV). Mice were generated and characterized as reported [14]. Breeding colonies of homozygous (PINK−/−), heterozygous knock-out mice (PINK1+/−), and of their wild-type littermates (PINK1+/+) were established at our animal house. All experiments were performed blindly. For genotyping, DNA was isolated from mouse-tail using the Extract-N-Amp Tissue PCR Kit (XNAT2, Sigma-Aldrich). To amplify the 324 and 501 bp fragments, three specific primers were utilized (10 μM; PINK1-F: 5′ AGA GGA TGC TAG TCC CTG TGA AGG G 3′; PINK1-X: 5′ ACA CTC AGT CCT TGG GCA ATG CTA 3′; NeoA: 5′ ACC AAA GAA GGG AGC CGG TTG 3′). PCR reactions were carried out with the Extract-N-Amp PCR reaction mix (XNAT2 kit) in a My Cycler thermal cycler (Bio-Rad; 35 cycles, annealing temperature 62°C). The 324 and 501 bp sequences were identified via 1.5% agarose gel electrophoresis using 2% SYBR Safe. Representative images of PCR products separated on 1.5% agarose gel (Fig. 1A) show the differences among genotypes. Line 2 identifies heterozygous mice.
Figure 1. Characterization of mouse genotype and of dopaminergic nigral cells.

A. Analysis of PINK1+/+, PINK1+/− and PINK1−/− mice genotype. Representative gel images of PCR products are shown. Products from PINK1+/+, PINK1+/− and PINK1−/− mice appear in lanes 1, 2 and 3, respectively. Lane 1 shows a fragment of 324 bp, lane 3 a fragment of 501 bp, and lane 2 shows both fragments. B. Representative confocal microscope images (z-series projections) showing tyrosine hydroxylase (TH) immunostaining in substantia nigra pars compacta (SN) slices. Similar TH immunoreactivity was found in PINK1+/− compared to PINK1+/+ littermates. C. The plot shows both similar cellular distribution of TH-positive neurons and cell count in the two genotypes in a total area of 425×425 μm analyzed for each SN slice (scale bar: 20 μm). D,E. Sharp microelectrode recordings of dopaminergic neurons from nigral slices obtained from PINK1+/+ and PINK1+/− mice, respectively, showing typical spontaneous, rhythmic firing activity. In slices from both genotypes brief dopamine (DA) application (100 μM, 30 s) hyperpolarizes the cell membrane and blocks the firing activity. Upon DA washout, the membrane slowly recovers, and action potential discharge returns to control levels. The insets show normal amplitude and duration of single action potentials recorded from dopaminergic neurons in the two genotypes. F,G. Representative traces showing the effects of quinpirole (300 nM, 1 min) on nigral dopaminergic neurons. Bath-applied quinpirole hyperpolarizes the cell membrane and abolishes the spontaneous firing activity of neurons recorded from PINK1+/+ and PINK1+/− mice, respectively. Similar responses are recorded from the two groups of mice. Upon quinpirole washout, the membrane slowly returns to pre-drug level and firing activity resumes. H. The quinpirole effect is prevented by pretreatment of the slice with the D2R antagonist sulpiride (3 μM), confirming the specificity of the response.
Tissue slice preparation
All efforts were made to minimize the number of animals utilized and to reduce their suffering. Mice were sacrificed and corticostriatal parasagittal and nigral horizontal slices from substantia nigra pars compacta (SNpc) (290–350 μm) were prepared as described [15–18] in a Krebs’ solution bubbled with 95% O2 and 5% CO2. Individual slices were transferred into a recording chamber superfused with oxygenated Krebs’ medium, and maintained at 32–33°C. Nigral slices were kept at 34°–35°C.
Electrophysiology
Whole-cell patch-clamp were performed from medium spiny neurons (MSNs), visualized using infrared videomicroscopy, as described [14,18]. Recordings were made with an AxoPatch 200B amplifier coupled to pClamp 10.2 software (Molecular Devices), using borosilicate glass pipettes (resistance 2.5–5 MΩ). Membrane currents were continuously monitored and access resistance measured in voltage-clamp was in the range of 5–30 MΩ prior to electronic compensation (60–80% routinely used). Cells showing ≥ 20% change in series resistance during the experiment were discarded from the analysis. An internal solution with the following composition was used (in mM): 120 Cs-gluconate, 13.6 CsCl, 10 HEPES, 1.1 EGTA, 0.1 CaCl2, 2.5 Mg-ATP, and 0.3 Na-GTP, pH 7.3 [19,20].
For spontaneous glutamatergic excitatory post-synaptic currents (sEPSCs), MSNs were clamped at a holding potential of −60 mV [20], in the presence of picrotoxin (50 μM). sEPSCs were completely blocked by NMDA and AMPA receptor antagonists, MK-801 (30 μM) and CNQX (10 μM) respectively. Conversely, spontaneous GABAergic inhibitory post-synaptic currents (sIPSCs) were recorded at a holding potential of +10 mV, in the presence of MK-801 and CNQX. In these conditions sIPSCs were blocked by picrotoxin. To isolate both miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs, respectively) Tetrodotoxin (TTX, 1 μM), was added to the perfusing solution.
Sharp-electrode recordings of striatal and nigral neurons
Intracellular recording electrodes were filled with 2M KCl (30–60 MΩ). Signals were recorded with an Axoclamp 2B amplifier, displayed on an oscilloscope and stored on PC using Digidata 1322A and pClamp 10.2 (Molecular Device). A bipolar electrode was used to evoke corticostriatal excitatory postsynaptic potentials (EPSPs). Test stimuli were delivered at 0.1 Hz in picrotoxin. This electrophysiological approach was used to retain the fidelity of post-receptor signaling in synaptic plasticity experiments. For high frequency stimulation (HFS) protocol (three trains: 3s duration, 100 Hz frequency, 20s intervals) stimulus intensity was raised to suprathreshold levels. The amplitude of EPSPs was averaged and plotted as percentage of the control amplitude for 15 min before protocol induction. To optimize long-term potentiation (LTP) induction, magnesium was omitted from the external medium [17]. Nigral recordings were performed as described [15,21–24]. Dopaminergic neurons from the SNpc were identified by their electrophysiological properties. Traces were stored on pClamp10.2 running on a PC for off-line analysis.
Drug source and Statistical analysis
Drugs were from Tocris Cookson (UK), except for dopamine, nomifensine, L-Dopa, and tyramine (Sigma, Italy). Amphetamine was provided by Dr. N.B. Mercuri. Patch-clamp data were analyzed offline by Clampfit 10.2 (Molecular Devices), Origin 8 (Originlab Corp., Northampton, MA, USA) and Mini-Analysis 6.0 (Synaptosoft, Decatur, GA, USA) software. For analyzing spontaneous and miniature post-synaptic currents, the detection threshold (3–5 pA) was set to twice the noise after trace filtering (traces in Figures are not filtered). For data presented as mean ± SEM, statistical significance between groups was evaluated using a paired Student t-test, unpaired Student t-test or Wilcoxon rank-sum test. Percentage values are calculated for each single experiment and all data are presented as mean ± SEM for each condition. Student’s t-test and non-parametric Mann–Whitney test were used to compare means pre- and post-HFS/drug. Analysis of variance (ANOVA) test with a post-hoc Tukey test were performed among groups (P<0.05; alpha = 0.01). P was set at < 0.05.
Voltammetry
Amperometric detection of DA was obtained as described [23]. The tips of the carbon fiber electrodes (WPI) were gently positioned into corticostriatal slices. Constant potential amperometry was obtained with a WPI MicroC holding the electrodes at an oxidation potential of 0.55–0.60 V versus reference electrode Ag/AgCl [25,26]. Currents sampled at this oxidation peak potential were measured to provide profiles of DA concentration ([DA]o) versus time. Electrodes calibration was obtained by bath-perfusing DA (0.3–10 μM) in all experimental media. A supramaximal electrical stimulation of the striatum was obtained with a pulse of 1 mA, 80 μs. Data were analyzed by Student’s t test.
Histology and immunohistochemistry
Histological samples were obtained both from perfused animals and biocytin-loaded slices from both genotypes (Fig. 3B). Immediately after recording, slices containing biocytin-loaded cells were fixed in 4% paraformaldehyde in 0.1 M PB overnight (4°C). The slices were subsequently immersed in 30% PB/sucrose (4°C). They were then frozen with dry ice and cut into 35 μm-thick transverse sections. Sections derived from biocytin-loaded slices were incubated with Cy2- or Cy3-conjugated Streptavidin (1:200; Jackson Immunoresearch, USA) in PB 0.3% Triton X-100 for 1 h at RT, and images acquired as previously described [17].
Figure 3. Normal intrinsic electrophysiological properties of striatal medium spiny neurons (MSNs) from PINK1+/− mice.

A. Representative traces of current-clamp sharp microelectrode recordings obtained from both PINK1+/+ and PINK1+/− MSNs showing a tonic firing activity induced by a depolarizing current step (800 pA, 700 ms). The long depolarizing ramp to spike threshold, as well as the strong inward rectification during hyperpolarizing steps are peculiar features of MSNs. Note that similar voltage responses are observed in MSNs from both PINK1+/+ and PINK1+/− mice. B. Confocal microscope image of a biocytin-loaded MSN, recorded from a PINK1+/− slice. Note the branched dendrites densely embedded with spines. Scale bar: 20 μm. C. Whole-cell recordings of sEPSCs and mEPSCs (downward deflections) recorded from MSNs in the presence of picrotoxin (50 μM) or picrotoxin plus tetrodotoxin (TTX, 1μM), respectively, in PINK1+/+ and PINK1+/− mice. Holding potential: −60 mV. The plots show no significant changes in both mean frequency and amplitude in PINK1+/− mice compared to their wild-type littermates in sample neurons. The representative cumulative probability plot of inter-event intervals confirms no significant change in frequency of glutamate-mediated events in the two strains. D. Sample traces of sIPSCs and mIPSCs (upward deflections) recorded from MSNs in the presence of MK-801 (30 μM) and CNQX (10 μM) in slices from both genotypes. Holding potential: +10mV. mIPSCs were recorded in TTX. The graphs indicate no significant difference in both mean frequency and amplitude in PINK1+/− mice when compared to the PINK1+/+group. The representative cumulative probability plot of inter-event intervals reveals no significant difference in frequency of GABA-dependent currents in sample neurons. Data are expressed as means ± SEM.
For immunostaining assay mice were deeply anesthetized (avertin, 0.002 ml/0.01 kg) and perfused with ice-cold 4% paraformaldehyde. The dissected brain were equilibrated with 30% sucrose three overnights, and 30 μm-thick coronal sections were cut. Slices were preincubated with 10% normal donkey serum solution (NDS) in PBS 0.25% Triton X-100 (TPBS) for 1 h at RT and incubated with the primary antibody rabbit polyclonal anti-tyrosine hydroxylase 1:1500 (Ab-Cam cod. ab112) at +4°C overnight in TPBS 1% NDS. After 3 washes (10 min each) with TPBS, the sections were incubated with Cy3-conjugated donkey anti-rabbit secondary antibody (Jackson ImmunoResearch cod. 711-165-152) 1:200 for 2 h at RT. Sections were washed three times, (last wash in Dapi 0.01mg/ml) mounted with Vectashield (Vector Labs) on plus polarized glass slides and coverslipped. To test for the specificity of the antisera, primary antibody was substituted with normal serum and, accordingly, the immunolabeling was absent (data not shown). Images were acquired with Zeiss LSM 700 confocal laser scanning microscope as described [27]. For quantitative analysis the area of 425×425 μm corresponding to the SNpc was selected to count manually TH+ cells in serial coronal sections of adult PINK1+/− and PINK1+/+ mice.
Behavior
Locomotor activity was assessed in PINK1+/+ (n=6) and PINK1+/− (n=6) mice. The apparatus comprised five gray opaque Plexiglas chambers (20×10 cm) placed inside a sound-attenuated room. Individual mice were introduced into each chamber and accustomed to the apparatus for 60 min. Then, all mice were removed and left undisturbed inside their home cages [28]. Subsequently, mice were placed individually in the same cages they had experienced and tested for 60 min. Behavioral data were collected and analyzed by the ‘EthoVision’ (Noldus, The Netherlands) fully automated video tracking system [29]. Briefly, a CCD video camera was used to record the experimental system. The signal was then digitized (by a hardware device called a frame grabber) and passed on to the computer’s memory. Then digital data were analyzed using EthoVision software to measure distance “distance moved” (in cm) as an estimate of locomotor activity.
Rotarod
Motor coordination and balance were assessed (n=5 per genotype) using the Accelerating Rotarod apparatus (U. Basile, mod. 7650; Italy). The test phase consisted of three trials, of maximum 300 sec, separated by 15 min inter-trial intervals [30]. The rod was initially rotating at a 4 rpm constant speed to allow positioning of all mice in their respective lanes. Once all mice were positioned, the trial was started and the rod accelerated from 4 rpm to 40 rpm in 5 min. Latency at which each mouse fell off the rod were measured. Data were analyzed using two-tailed non-parametric Mann–Whitney test.
Results
Cell-count and activity of nigral dopaminergic neurons are unaffected in PINK1+/− mice
Neurodegeneration of nigral neurons is a pathological hallmark of PD. However in prodromal and early disease stages no obvious depletion has been reported [31–33].
Previous evidence revealed the absence of significant neuronal loss in the SNpc of PINK1−/− mice at the ages of 2–3 and 8–9 months [14]. Similarly, our morphological characterization of SNpc of PINK1+/− mice did not reveal significant alterations of nigral dopaminergic neurons as compared to their controls. We performed immunofluorescence experiments with specific antibodies for tyrosine hydroxylase (TH) to detect nigral dopaminergic neurons. In serial coronal sections of both PINK1+/− and PINK1+/+ mice, no macroscopic alterations in morphology and density of dopaminergic cell bodies or terminals were detected (data not shown). TH-immunoreactive neurons count in the SNpc did not show significant difference in the number of nigral dopaminergic neurons of PINK1+/− mice compared to PINK+/+ (Fig. 1B,C; p>0.05; PINK1+/+ =115 ± 4.9, PINK1+/− =114.8± 2.9).
Recordings of nigral neurons showed no significant change in the firing activity from PINK1+/− and PINK1+/+ mice (Fig. 1D,E; PINK1+/+ 3.6 ± 1.6 Hz; n=17; PINK1+/− 3.52 ± 1.9 Hz; n=14; p> 0.05 Mann-Whitney). In slices from PINK1+/+ and PINK1+/− mice, bath-application of DA (100 μM, 30 s) inhibited cell firing and hyperpolarized the cell membrane (Fig. 1D,E; PINK1+/+ 5.4 ± 1.3 mV; PINK1+/− 4.9 ± 2 mV, n=9 per group). Upon washout, the resting membrane potential (RMP) gradually recovered, and action potential discharge returned to control levels (Fig. 1D,E). The inhibitory effect of DA on nigral neurons is mediated specifically by somatodendritic D2 autoreceptors (D2R), as it is completely blocked by D2R antagonists [34] and is absent in D2R knock-out mice [15]. Accordingly, the D2R agonist quinpirole (300 nM, 1 min) caused a longer-lasting membrane hyperpolarization and blockade of the firing discharge in dopaminergic neurons from PINK1+/+ and PINK1+/− mice, respectively (Fig. 1F,G; 8.82 ± 3.3 min; 8.82 ± 2.9 min; n=8 per genotype; p> 0.05;), an effect that was prevented by the application of D2R antagonist sulpiride (3μM) (Fig. 1H).
Behavioral analysis
We assessed the locomotor activity of PINK1+/− mice using a battery of behavioral test. Firstly, we analyzed spontaneous locomotor activity showing no statistical difference between the two genotypes in the total distance travelled (Fig. 2A,B; session 1: PINK1+/+ 7833.8 ± 589.8 cm; PINK1+/− 7457.9 ± 610.9 cm; n=6 per genotype, p>0.05 two tailed Mann-Whitney; session 2: PINK1+/+ 5897.6 ± 457.7 cm, PINK1+/− 6015.7 ± 568.4 cm; n=6 per genotype, p>0.05 two tailed Mann-Whitney). Next, motor ability was further assessed by means of an accelerated rotarod test. Of interest, PINK1+/− mice performed equally when compared to their respective control (Fig. 2C; mean latency to fall PINK1+/+ 270 ± 12.4 s, PINK1+/− 284.9 ± 15.1 s, n=5 per genotype, p>0.05 two tailed Mann-Whitney).
Figure 2. Normal behavioral ability in PINK1+/− mice.
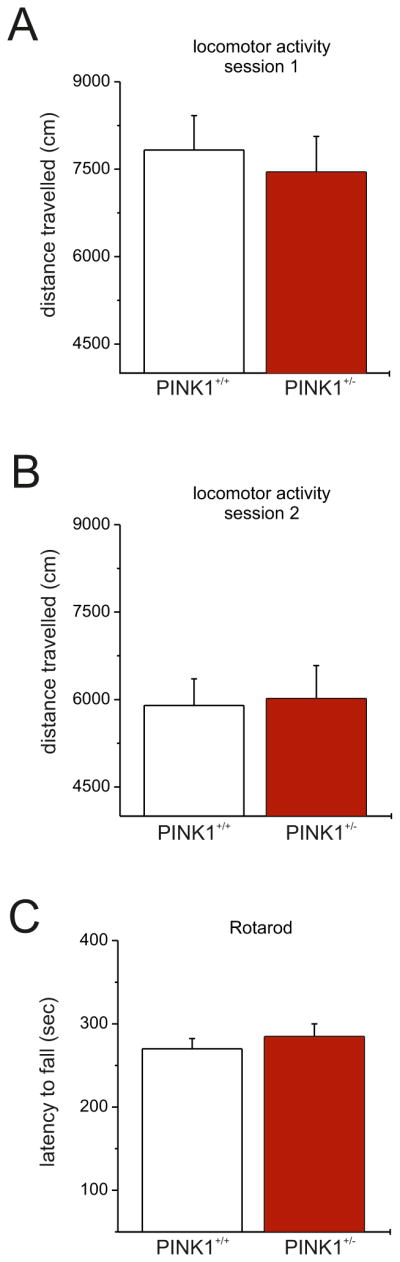
A,B. Spontaneous locomotor activity of PINK1+/− mice did not differ from their respective controls. There were no significant differences between the two genotype in the total distance traveled in both experimental sessions. C. PINK1+/− mice also demonstrated normal motor coordination and balance on the accelerated rotarod test. Values are expressed as mean ± SEM.
Electrophysiological properties of striatal medium spiny neurons are not altered in PINK1 heterozygous mice
Several genetic mouse models of PD exhibit dysregulation of striatal circuitry [14,35,36]. Recordings were obtained from striatal MNSs of PINK1+/− and PINK1+/+ slices. MSNs had similar resting membrane potential (RMP, −82 ± 3.15 mV for PINK1+/+, n=28; −83.4 ± 3.9 mV for PINK1+/− n=32; p>0.05, Mann-Whitney), were silent at rest; current pulses caused tonic action potential discharge and strong inward rectification that did not differ among genotypes (Fig. 3A). Analysis of short-term synaptic plasticity, EPSPs and paired-pulse ratio were unchanged in PINK1+/− mice (data not shown; p>0.05). Random labelling with biocytin confirmed the peculiar features of MSNs, with medium-sized soma (10–20 μm), and extensive spiny dendritic trees (Fig. 3B). To detect possible changes in glutamate release probability, sEPSCs were recorded in the presence of the GABAA receptor antagonist picrotoxin (50 μM). The frequency and amplitude of glutamate-dependent sEPSCs did not differ from PINK1+/+ (n=12) and PINK1+/− (n=14) MSNs (Fig. 3C; frequency: PINK1+/+ 5.34 ± 0.1 Hz; PINK1+/− 4.63 ± 0.2 Hz; amplitude: PINK1+/+ 9.44 ± 0.6 pA, PINK1+/− 9.62 ± 0.6 pA; p>0.05 T-test). Kinetic properties, such as rise time, decay time constant and half width were similar (not shown; p>0.05). Next, we recorded miniature EPSCs (mEPSCs), in the presence of TTX, to block action potential-dependent glutamate release. As expected, TTX homogenously reduced mean frequencies but not amplitude of spontaneous events in both PINK1+/+ (n=10) and PINK1+/− (n=8) MSNs (Fig. 3C; frequency: PINK1+/+ 3.41±0.57 Hz; PINK1+/− 2.99±0.14 Hz; amplitude: PINK1+/+ 9.32±0.7, pA; PINK1+/− 9.25±0.81; pA; p>0.05 t-test). Likewise, mean values of GABAergic sIPSCs amplitude and frequencies were comparable between genotypes (Fig. 3D; frequency: PINK1+/+ 2.24±0.27 Hz; PINK1+/− 1.46±0.3 Hz; amplitude: PINK1+/+ 15.26±0.42 pA, PINK1+/− 14.98±0.8 pA; n=9 for each genotype; p>0.05 Wilcoxon test). Bath-application of TTX resulted in a reduction of mean frequencies of mIPSCs but not of amplitude in both PINK1+/+ and PINK1 +/− MSNs (Fig. 3D; frequency: PINK1+/+ 1.49±0.2 Hz, PINK1+/− 0.62± 0.11 Hz; amplitude: PINK1+/+ 15.32±0.56 pA, PINK1+/− 15.09±0.6 pA; n=11 for each genotype; p>0.05 Wilcoxon). Kinetic properties were unaltered (not shown).
Selective DA-dependent LTP deficit in heterozygous PINK1+/− mice
We have previously demonstrated that PINK1 inactivation is responsible for a marked impairment of bidirectional striatal synaptic plasticity [14]. Therefore, in attempt to better characterize whether the heterozygous mutation of PINK1 gene might influence corticostriatal synaptic plasticity, we analyzed long-term depression (LTD) and long-term potentiation (LTP). HFS of glutamatergic afferents in presence of picrotoxin led to the induction of a robust LTD in MSNs in parasagittal slices from PINK1+/+ (Fig. 4A; 54.62 ± 4.48 % of control, measured 25 min post-HFS; n=25). Similarly, in MSNs from PINK1+/− mice, HFS was able to cause an LTD that was indistinguishable from that measured in wild-type mice (Fig. 4A; 57.43 ± 5.13 % of control, 25 min post-HFS; n=28, p>0.05 ANOVA).
Figure 4. Selective Long-term Potentiation impairment in PINK1+/− mice is associated with a reduced DA overflow.

A. Time-course of LTD in PINK1+/+ and PINK1+/− mice recorded from parasagittal slices. High-frequency stimulation (HFS, arrow) induces a robust LTD in both genotypes. Superimposed traces of EPSPs recorded before (pre) and 20 min after (post) HFS in PINK1+/+ and PINK1+/− MSNs. B. The LTP induction protocol causes a normal LTP in PINK1+/+ MSNs (gray circles), whereas the magnitude of LTP recorded in PINK1+/− mice is significantly lower (black circles). Sample traces of EPSPs recorded before and after HFS, showing that synaptic potentiation is significantly lower in magnitude in PINK1+/− MSNs. C. Amperometric measurement of striatal extracellular DA levels [DA] (expressed in μM) in PINK1+/− mice compared to PINK1+/+ mice. C. The graph shows [DA] levels in response to supramaximal stimulation (1 mA, 80 μs). In PINK1+/− mice striatal DA release is significant lower than in PINK1+/+ mice. Values are expressed as mean ± SEM.
Removal of magnesium from the perfusing solution reveals an NMDA-mediated component of EPSPs and optimizes LTP induction [37]. Accordingly, HFS protocol invariably induced LTP in PINK1+/+ mice (Fig. 4B; 172.48 ± 4.45 % of control, 25 min post-HFS; n=23; Mann–Whitney, p< 0.05). Unexpectedly, LTP amplitude in PINK1+/− MSNs was significantly lower (Fig. 4B; 137.7 ± 6.4 %; 25 min post-HFS; n=32, p<0.05 t-test). The impairment of bidirectional plasticity could be observed also in PINK1−/− mice, as previously reported (data not shown).
To support the hypothesis that an impairment of DA release could play a major role in the LTP deficit, we performed amperometric recordings in striatal slices. Indeed, in response to electrical stimulation, striatal extracellular DA release evoked in PINK1+/− slices was significantly lower when compared to PINK1+/+ mice (Fig. 4C).
Increasing DA release restores corticostriatal LTP
It is well established that D1Rs play a crucial role in the control of the striatal LTP [38,39]. Thus we pretreated striatal slices with the selective D1R agonist SKF38393 (10 μM, 20 min), which failed to restore LTP (Fig. 4A; 141 ± 4.2%; n=9, p<0.05 Mann–Whitney). In an attempt to increase the available DA amount for release upon stimulation, we pretreated striatal slices with the DA precursor L-Dopa (100 μM; 30–45 min before the LTP induction protocol). In this condition, we recorded a partial LTP rescue in PINK1+/− MSNs (Fig. 5A; 151.7 ± 7.3%, n=8; p< 0.05 t-test). Next, to distinguish between a potential failure of reuptake process and a deficient DA release, slices were pretreated with nomifensine (30 min, 100 μM), a DA-reuptake inhibitor, or with the DA releaser amphetamine (30 min, 100 μM). Nomifensine was ineffective in rescuing LTP in PINK1+/− mice (Fig. 5B; 135.5 ± 8.3%, n=6; p>0.05 t-test). Conversely, amphetamine induced a complete recovery of striatal LTP in PINK1+/− MSNs (Fig. 5B; 172.3 ± 7.9%, n=11; p<0.05 Mann–Whitney) to levels comparable to those measured in PINK1+/+ mice.
Figure 5. DA-dependence of LTP deficit in PINK1+/−mice.

A. Pretreatment of the slices with the D1R receptor agonist SKF38393 (10 μM, 20 min) does not restore LTP (grey squares). Conversely, a partial rescue is observed after slice preincubation with L-Dopa (100 μM, 30–45 min) (blue squares). B. Preincubation with 10 μM nomifensine (30 min), a DA-reuptake inhibitor, is unable to rescue LTP (grey squares), whereas amphetamine (100 μM, 30 min) restores LTP in PINK1+/− MSNs to control levels (blue squares). Superimposed EPSPs show the recovery of LTP by amphetamine pretreatment (post), as compared to the lack of effect by nomifensine. Each data point represents the mean ± SEM of >12 independent observations.
The finding that amphetamine could restore LTP, led us to test a number of pharmacological agents capable of increasing synaptic DA levels. In a first set of experiments, we tested the “trace amine” tyramine, whose action relies on the ability to release cathecolamines in an amphetamine-like manner, through the displacement from synaptic vesicles and promoting reversal of plasma membrane transporters [40]. Pretreatment of striatal slices with tyramine (10 μM, 30–45 min) was followed by a complete rescue of striatal LTP in PINK1+/− mice (Fig. 6A; 163±11.8%; n=8, p<0.01 t-test).
Figure 6. Increasing DA availability restores both LTP deficits and striatal DA content.

A. Preincubation of slices with the trace amine tyramine (10 μM, 30–45 min) is able to restore the LTP deficit recorded in PINK1+/− mice. B. Rasagiline (10 μM, 30–45 min) fails to rescue LTP, whereas selegiline (C, 30 μM, 2 hrs slice preincubation), restores LTP in PINK1+/− MSNs. Each data point represents the mean ± SEM of >12 independent observations. D. Measurements of striatal extracellular dopamine (DA) concentration ([DA] expressed in μM) in PINK1+/− mice show that both selegiline and rasagiline are able to restore a physiologic DA levels in response to supramaximal stimulation. Values are expressed as mean ± SEM.
Selective inhibition of brain MAO-B results in extension of the availability and activity of endogenous DA in the striatum through blockade of its degradation [15,41]. Firstly, we tested rasagiline (10 μM, 30–45 min), which did not cause, per sè, a significant change in EPSP amplitude, but failed to rescue LTP (Fig. 6B; 121.6 ± 11.3 %, n=9; p>0.05 Mann-Whitney). Selegiline, besides blocking MAO-B in an irreversible manner, acts also on neurotransmitter vesicular release through its amphetamine-like metabolite [42,43]. Indeed, slice preincubation with selegiline (30 μM, 2 hrs before HFS), resulted in a full rescue of striatal LTP (Fig. 6C, 164.7 ± 10.1%, n=11; p<0.01 Mann-Whitney). Our electrophysiological findings were paralleled by a set of amperometric recordings, which showed that pretreatment with selegiline (30 μM, n=18; p<0.05) completely rescued striatal DA content, whereas application of rasagiline (10 μM, n=14; p<0.05) only partially restored evoked DA release (Fig. 6D).
DISCUSSION
Compelling evidence obtained from animal models of monogenic forms of PD demonstrates remarkable abnormalities of DA-mediated neurotransmission and corticostriatal synaptic plasticity, indicative of a fundamental distortion of network function within the basal ganglia [14,16, for revs. see 44,45]. Recent neuroimaging studies of non-manifesting carriers of heterozygous mutations in the Parkin or PINK1 genes have unequivocally shown that a single mutant allele is associated with a dopaminergic nigrostriatal dysfunction conveying an increased risk to develop PD throughout lifetime [46–49].
Our findings provide novel evidence for a plausible cellular and synaptic correlate of the subclinical motor impairment observed in non-manifesting PINK1 heterozygotes, and have relevant pathophysiological implications for the role of PINK1 heterozygous mutations on DA-dependent synaptic plasticity. We show that, in heterozygous knockout mice, morphology and basic physiological cellular activity of both nigral and striatal neurons are preserved, as well as behavioral analyses. However, a significant decrease in evoked DA release paralleled a subtle alteration of LTP, whereas LTD expression was normal. More importantly, we report that drugs able to promote DA release, in an amphetamine-like manner, are able to restore plasticity deficits.
This study represents the first evidence that PINK1 haploinsufficiency differentially affects the expression of two distinct forms of corticostriatal synaptic plasticity. In fact, our data show a normal LTD and selective alteration of LTP in heterozygous PINK1 mice, whereas in homozygous PINK1 mice both forms of corticostriatal synaptic plasticity were impaired [14]. The precise process leading to such specific deficit of synaptic plasticity in the heterozygous condition remains to be established. We can hypothesize that the two distinct forms of synaptic plasticity may reflect the different stage of the disease course. In heterozygotes, when only one wild type PINK1 allele is normally functioning, the LTD is normally expressed, whereas the disruption of both PINK1 alleles, as it is seen in presence of homozygous or compound heterozygous mutations, represent both necessary and sufficient condition to determine the complete disruption of bidirectional corticostriatal synaptic plasticity. In support of this hypothesis, recent data emerging from a study conducted on a rodent model of early PD suggest that distinct degrees of DA denervation differentially affect the induction and maintenance of both corticostriatal LTP and LTD [50]. It is well accepted that the maintenance of dopaminergic tone governing striatal output is essential to preserve motor control [51,52]. Hence, the presence of a single mutant PINK1 allele may reflect an initial breakdown of the striatal DA homeostasis, which in turn influences the normal expression of striatal synaptic plasticity. These findings support the hypothesis that nigrostriatal dopaminergic dysfunction gives rise to an early dysfunction of basal ganglia circuits during the pre-symptomatic phase of the disease, which appears in agreement with our evidence of a normal motor behavior. A latent dopaminergic nigrostriatal dysfunction has been found also in non-manifesting individuals carrying heterozygous mutations in other autosomal recessive PD genes, such as Parkin [53–55]. Functional studies have demonstrated that a compensatory mechanism characterized by the recruitment of cortical motor areas may counteract the preclinical nigrostriatal dysfunction to ensure a normal level of performance during a motor sequence task [9,10,53,54].
At cellular level, available experimental evidence suggests that the complete expression of LTP requires a large amount of energy to enable the mobilization of synaptic vesicles from the reserve pool (RP), which is recruited only following HFS [56,57]. PINK1 plays an important role for cellular energy maintenance under increased demand, and PINK1 deficiency is related to mitochondrial respiratory defects [58,59]. Indeed, several reports indicate that mitochondria are essential for synaptic vesicle release under conditions of intense neuronal activity through vesicle mobilization from the RP, a process that requires numerous ATP-consuming steps [60–62]. Accordingly, it is plausible that a mitochondrial dysfunction may distinctively impact the maintenance of sufficient cellular ATP, affecting synaptic vesicle cycling required for synaptic plasticity induction. Studies from Drosophila reveal that deficiency or inactivation of PINK1 is associated with an impairment of mobilization of the RP during intense stimulation, affecting synaptic function [59]. More recently, studies from rodent models focused on the role of PINK1 in mitochondrial respiration, suggesting that loss of PINK1 causes defects in mitochondrial transmembrane potential through dysregulation of the mitochondrial permeability transition pore opening [58]. It is thus conceivable that changes in the mitochondrial ability to regenerate ATP may affect the magnitude of synaptic vesicle at presynaptic terminals, primarily via ATP synthesis.
A clear finding emerging from our work is the DA-dependence of striatal plasticity which has been matter of intense investigation [63,64]. Indeed, our data indicate a significant decrement in synaptic DA release at high frequencies of stimulation. Accordingly, displacement of neurotransmitter from storage areas to the synaptic cleft is able to restore LTP deficit, as observed with amphetamine, a DA releaser, but not with nomifensine, an uptake blocker. Notably, drugs exhibiting a similar, amphetamine-like pharmacodynamic profile restored LTP, such as tyramine and selegiline. Trace amines are found at low concentrations in neuronal tissues, where they are packed and released along with traditional amine compounds [65]. Trace amines have been shown to displace active biogenic amines and to act on transporters in an amphetamine-like manner, thereby promoting DA release and new DA synthesis [66,67].
Currently, strategies in PD treatment consider MAO-B inhibitors as they increase DA levels by blocking its degradation [15,41,68,69]. Indeed, DA levels are finely tuned by MAO enzymes, which exist in two functional isoforms, MAO-A and MAO-B. In line with current literature, our amperometric measurements revealed that, although to a different extent, both rasagiline and selegiline are able to increase DA striatal levels in PINK1+/− mice compared to controls. However, we found that only selegiline treatment was able to restore normal LTP expression. This is not surprising, as pharmacological properties of these two MAO-B inhibitors may account for such difference. In fact, selegiline and rasagiline exhibit distinct pharmacokinetic properties. Selegiline is metabolized in vivo to 1-amphetamine and to 1-methamphetamine [68] and has amphetamine-like properties [69], whilst rasagiline is metabolized to aminoindan, which is devoid of amphetamine-like actions [70]. It is therefore conceivable that the LTP rescue in PINK1 heterozygous mice is determined by the ability to force release from synaptic vesicles. The abnormalities of synaptic plasticity described in heterozygous mice might be considered an endophenotype to this monogenic form of PD, and a valid tool to design potentially disease-modifying therapies. This appears an intriguing working hypothesis, although results obtained from mouse models need to be interpreted with caution and further refined with additional analyses.
Acknowledgments
Support grants: Italian Ministry of Health (Giovani Ricercatori 2009), the Italian Ministry of Instruction, University and Research (MIUR, PRIN 2011), INAIL to AP; Italian Ministry of Health (Ricerca Corrente 2013, Ricerca Finalizzata Malattie Rare 2008, Giovani Ricercatori 2009), the Italian Telethon Foundation (GGP10140) and MIUR (FIRB Accordi di Programma 2010) to EMV; NIH NS041779 to JS.
We thank Mr. M. Tolu and V. Batocchi for their excellent technical assistance.
Footnotes
Conflict of interest: nothing to report.
Author roles: AP, NBM, PB: designed and wrote the paper. GM (Madeo), TS, GM (Martella), FP, MF, ECL performed the experiments and analyzed the data. GM (Madeo), GM (Martella), FP prepared illustrations. EMV, JS, SPA: reviewed critically the manuscript providing comments and revisions.
Financial disclosures: AP, NBM are employees at the University of Rome “Tor Vergata”, Italy, EMV at the University of Salerno, Italy; SPA at the Sapienza University of Rome; MF and PB are employees at Fondazione Santa Lucia; other authors: none. AP, EMV, NBM, JS, hold grants that are not related to the subject of the present study.
References
- 1.Puschmann A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Rel Dis. 2013;19:407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Samaranch L, Lorenzo-Betancor O, Arbelo JM, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133:1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- 3.Marongiu R, Ferraris A, Ialongo T, et al. PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20719. [DOI] [PubMed] [Google Scholar]
- 4.Ibanez P, Lesage S, Lohmann E, et al. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain. 2006;129:686–694. doi: 10.1093/brain/awl005. [DOI] [PubMed] [Google Scholar]
- 5.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 6.Calabresi P, Di Filippo M, Gallina A, et al. New synaptic and molecular targets for neuroprotection in Parkinson’s disease. Mov Disord. 2013;28(1):51–60. doi: 10.1002/mds.25096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrish PK, Sawle GV, Brooks DJ. Regional changes in [18F]dopa metabolism in the striatum in Parkinson’s disease. Brain. 1996;119 (Pt 6):2097–103. doi: 10.1093/brain/119.6.2097. [DOI] [PubMed] [Google Scholar]
- 8.Hilker R, Schweitzer K, Coburger S, et al. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol. 2005;62:378–82. doi: 10.1001/archneur.62.3.378. [DOI] [PubMed] [Google Scholar]
- 9.van Nuenen BF, Weiss MM, Bloem BR, et al. Heterozygous carriers of a Parkin or PINK1 mutation share a common functional endophenotype. Neurology. 2009;72:1041–7. doi: 10.1212/01.wnl.0000338699.56379.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Nuenen BF, van Eimeren T, van der Vegt JP, et al. Mapping preclinical compensation in Parkinson’s disease: an imaging genomics approach. Mov Disord. 2009;24 (Suppl 2):S703–10. doi: 10.1002/mds.22635. [DOI] [PubMed] [Google Scholar]
- 11.Eggers C, Schmidt A, Hagenah J, et al. Progression of subtle motor signs in PINK1 mutation carriers with mild dopaminergic deficit. Neurology. 2010;74:1798–1805. doi: 10.1212/WNL.0b013e3181e0f79c. [DOI] [PubMed] [Google Scholar]
- 12.Khan NL, Valente EM, Bentivoglio AR, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol. 2002;52:849–53. doi: 10.1002/ana.10417. [DOI] [PubMed] [Google Scholar]
- 13.Fiorio M, Valente EM, Gambarin M, et al. Subclinical sensory abnormalities in unaffected PINK1 heterozygotes. J Neurol. 2008;255:1372–7. doi: 10.1007/s00415-008-0923-6. [DOI] [PubMed] [Google Scholar]
- 14.Kitada T, Pisani A, Porter DR, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007;104(27):11441–6. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercuri NB, Scarponi M, Bonci A, Siniscalchi A, Bernardi G. Monoamine oxidase inhibition causes a long-term prolongation of the dopamine-induced responses in rat midbrain dopaminergic cells. J Neurosci. 1997;17(7):2267–72. doi: 10.1523/JNEUROSCI.17-07-02267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldberg MS, Pisani A, Haburcak M, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45(4):489–96. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 17.Martella G, Tassone A, Sciamanna G, et al. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132(Pt 9):2336–49. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28(25):6483–92. doi: 10.1523/JNEUROSCI.0435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cossart R, Hirsch JC, Cannon RC, et al. Distribution of spontaneous currents along the somato-dendritic axis of rat hip- pocampal CA1 pyramidal neurons. Neuroscience. 2000;99:593–603. doi: 10.1016/s0306-4522(00)00231-1. [DOI] [PubMed] [Google Scholar]
- 20.Cuomo D, Martella G, Barabino E, et al. Metabotropic glutamate receptor subtype 4 selectively modulates both glutamate and GABA transmission in the striatum: implications for Parkinson’s disease treatment. J Neurochem. 2009;109(4):1096–105. doi: 10.1111/j.1471-4159.2009.06036.x. [DOI] [PubMed] [Google Scholar]
- 21.Mercuri NB, Stratta F, Calabresi P, Bonci A, Bernardi G. Activation of metabotropic glutamate receptors induces an inward current in rat dopamine mesencephalic neurons. Neuroscience. 1993;56(2):399–407. doi: 10.1016/0306-4522(93)90341-c. [DOI] [PubMed] [Google Scholar]
- 22.Mercuri NB, Bonci A, Johnson SW, Stratta F, Calabresi P, Bernardi G. Effects of anoxia on rat midbrain dopamine neurons. J Neurophysiol. 1994;71(3):1165–73. doi: 10.1152/jn.1994.71.3.1165. [DOI] [PubMed] [Google Scholar]
- 23.Napolitano F, Bonito-Oliva A, Federici M, et al. Role of aberrant striatal dopamine D1 receptor/cAMP/protein kinase A/DARPP32 signaling in the paradoxical calming effect of amphetamine. J Neurosci. 2010;30(33):11043–56. doi: 10.1523/JNEUROSCI.1682-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercuri NB, Bonci A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. Eur J Neurosci. 1995 Mar 1;7(3):462–9. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 25.Kawagoe KT, Wightman RM. Characterization of amperometry for in vivo measurement of dopamine dynamics in the rat brain. Talanta. 1994;41:865– 874. doi: 10.1016/0039-9140(94)e0064-x. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 auto-receptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sciamanna G, Tassone A, Mandolesi G, et al. Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J Neurosci. 2012;32:11991–2004. doi: 10.1523/JNEUROSCI.0041-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ventura R, Alcaro A, Cabib S, Conversi D, Mandolesi L, Puglisi-Allegra S. Dopamine in the medial prefrontal cortex controls genotype-dependent effects of amphetamine on mesoaccumbens dopamine release and locomotion. Neuropsychopharmacology. 2004;29(1):72–80. doi: 10.1038/sj.npp.1300300. [DOI] [PubMed] [Google Scholar]
- 29.Spink AJ, Tegelenbosch RA, Buma MO, Noldus LP. The EthoVision video tracking systemFa tool for behavioral phenotyping of transgenic mice. Physiol Behav. 2001;73:731–744. doi: 10.1016/s0031-9384(01)00530-3. [DOI] [PubMed] [Google Scholar]
- 30.Glasl L, Kloos K, Giesert F, et al. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp Neurol. 2012;235 (1):214–27. doi: 10.1016/j.expneurol.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Grinberg LT, Rueb U, Alho AT, Heinsen H. Brainstem pathology and non-motor symptoms in PD. J Neurol Sci. 2010;289(1–2):81–8. doi: 10.1016/j.jns.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 32.Jellinger KA. Synuclein deposition and non-motor symptoms in Parkinson disease. J Neurol Sci. 2011;310(1–2):107–11. doi: 10.1016/j.jns.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Smith GA, Isacson O, Dunnett SB. The search for genetic mouse models of prodromal Parkinson’s disease. Exp Neurol. 2012;237(2):267–73. doi: 10.1016/j.expneurol.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 34.Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitada T, Pisani A, Karouani M, et al. Impaired dopamine release and synaptic plasticity in the striatum of Parkin −/− mice. J Neurochem. 2009;(110):613–621. doi: 10.1111/j.1471-4159.2009.06152.x. [DOI] [PubMed] [Google Scholar]
- 36.Tozzi A, de Iure A, Di Filippo M, et al. The Distinct Role of Medium Spiny Neurons and Cholinergic Interneurons in the D2/A2A Receptor Interaction in the Striatum: Implications for Parkinson’s disease. J Neurosci. 2011;31(5):1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term Potentiation in the Striatum is unmasked by removing the Voltage-dependent Magnesium Block of NMDA Receptor Channels. Eur J Neurosci. 1992;4(10):929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 38.Centonze D, Gubellini P, Picconi B, et al. An abnormal striatal synaptic plasticity may account for the selective neuronal vulnerability in Huntington’s disease. Neurol Sci. 2001;22(1):61–2. doi: 10.1007/s100720170047. [DOI] [PubMed] [Google Scholar]
- 39.Kerr JN, Wickens JR. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. J Neurophysiol. 2001;85(1):117–24. doi: 10.1152/jn.2001.85.1.117. [DOI] [PubMed] [Google Scholar]
- 40.Ledonne A, Berretta N, Davoli A, Rizzo GR, Bernardi G, Mercuri NB. Electrophysiological effects of trace amines on mesencephalic dopaminergic neurons. Front Syst Neurosci. 2011;5:56. doi: 10.3389/fnsys.2011.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dimpfel W, Hoffmann JA. Electropharmacograms of rasagiline, its metabolite aminoindan and selegiline in the freely moving rat. Neuropsychobiology. 2010;62:213–20. doi: 10.1159/000319947. [DOI] [PubMed] [Google Scholar]
- 42.Schapira AHV. Monoamine oxidase B inhibitors for the treatment of Parkinson’s disease. CNS Drugs. 2011;25:1061–71. doi: 10.2165/11596310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 43.Engberg G, Elebring T, Nissbrandt H. Deprenyl (selegiline), a selective MAO-B inhibitor with active metabolites; effects on locomotor activity, dopaminergic neurotransmission and firing rate of nigral dopamine neurons. J Pharmacol Exp Ther. 1991;259(2):841–7. [PubMed] [Google Scholar]
- 44.Madeo G, Martella G, Schirinzi T, Ponterio G, Shen J, Bonsi P, Pisani A. Aberrant striatal synaptic plasticity in monogenic parkinsonisms. Neuroscience. 2011 doi: 10.1016/j.neuroscience.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 45.Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson’s disease. Adv Exp Med Biol. 2012;970:553–72. doi: 10.1007/978-3-7091-0932-8_24. [DOI] [PubMed] [Google Scholar]
- 46.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- 47.Hilker R, Klein C, Ghaemi M, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367–376. [PubMed] [Google Scholar]
- 48.Khan NL, Scherfler C, Graham E, et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–136. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- 49.Walter U, Klein C, Hilker R, Benecke R, Pramstaller PP, Dressler D. Brain parenchyma sonography detects preclinical parkinsonism. Mov Disord. 2004;1912:1445–1449. doi: 10.1002/mds.20232. [DOI] [PubMed] [Google Scholar]
- 50.Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, Viscomi MT, Giampà C, Fusco FR, Gardoni F, Bernardi G, Greengard P, Di Luca M, Calabresi P. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci. 2010;30(42):14182–93. doi: 10.1523/JNEUROSCI.2149-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agid Y. Parkinson’s disease: pathophysiology. Lancet. 1991;337:1321–1324. doi: 10.1016/0140-6736(91)92989-f. [DOI] [PubMed] [Google Scholar]
- 52.Bezard E, Dovero S, Prunier C, et al. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J Neurosci. 2001;21:6853–6861. doi: 10.1523/JNEUROSCI.21-17-06853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buhmann C, Binkofski F, Klein C, et al. Motor reorganization in asymptomatic carriers of a single mutant Parkin allele: a human model for presymptomatic parkinsonism. Brain. 2005;128:2281–90. doi: 10.1093/brain/awh572. [DOI] [PubMed] [Google Scholar]
- 54.Schneider SA, Talelli P, Cheeran BJ, et al. Motor cortical physiology in patients and asymptomatic carriers of parkin gene mutations. Mov Disord. 2008;23:1812–9. doi: 10.1002/mds.22025. [DOI] [PubMed] [Google Scholar]
- 55.Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a Parkin kindred: an [18F]dopa PET and clinical study. Brain. 2002;125:2248– 2256. doi: 10.1093/brain/awf237. [DOI] [PubMed] [Google Scholar]
- 56.Kuromi H, Kidokoro Y. Exocytosis and endocytosis of synaptic vesicles and functional roles of vesicle pools: lessons from the Drosophila neuromuscular junction. Neuroscientist. 2005;11(2):138–47. doi: 10.1177/1073858404271679. [DOI] [PubMed] [Google Scholar]
- 57.Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6(1):57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- 58.Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress 2008. Proc Natl Acad Sci U S A. 2008;105(32):11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morais VA, Verstreken P, Roethig A, et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. 2009 EMBO Mol Med. 1(2):99–111. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;22(2):139. doi: 10.3389/fnsyn.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mochida S. Activity-dependent regulation of synaptic vesicle exocytosis and presynaptic short-term plasticity. Neurosci Res. 2011;70(1):16–23. doi: 10.1016/j.neures.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Ivannikov MV, Sugimori M, Llinás RR. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci. 2013;49(1):223–30. doi: 10.1007/s12031-012-9848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30(5):211–9. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 64.Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 65.Parker EM, Cubeddu LX. Comparative effects of amphetamine, phenylethylamine and related drugs on dopamine efflux, dopamine uptake and mazindol binding. J Pharmacol Exp Ther. 1988;245(1):199–210. [PubMed] [Google Scholar]
- 66.Bailey BA, Philips SR, Boulton AA. In vivo release of endogenous dopamine, 5-hydroxytryptamine and some of their metabolites from rat caudate nucleus by phenylethylamine. Neurochem Res. 1987;12(2):173–8. doi: 10.1007/BF00979534. [DOI] [PubMed] [Google Scholar]
- 67.Geracitano R, Federici M, Prisco S, Bernardi G, Mercuri NB. Inhibitory effects of trace amines on rat midbrain dopaminergic neurons. Neuropharmacology. 2004;46(6):807–14. doi: 10.1016/j.neuropharm.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 68.Reynolds GP, Elsworth JD, Blau K, Sandler M, Lees AJ, Stern GM. Deprenyl is metabolized to methamphetamine and amphetamine in man. Br J Clin Pharmacol. 1978;6:542–544. doi: 10.1111/j.1365-2125.1978.tb00883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simpson LL. Evidence that deprenyl, A type B monoamine oxidase inhibitor, is an indirectly acting sympathomimetic amine. Biochem Pharmacol. 1978;27(11):1591–5. doi: 10.1016/0006-2952(78)90490-2. [DOI] [PubMed] [Google Scholar]
- 70.Youdim MB, Gross A, Finberg JP. Rasagiline [N-propargyl-1R(fl)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol. 2001;132:500–506. doi: 10.1038/sj.bjp.0703826. [DOI] [PMC free article] [PubMed] [Google Scholar]