

Abstract
Plasmonic gold nanostars offer a new platform for Surface-Enhanced Raman Scattering (SERS). However, due to the presence of organic surfactant on the nanoparticles, SERS characterization and application of nanostar ensembles in solution have been challenging. Here we applied our newly developed surfactant-free nanostars for SERS characterization and application. The SERS enhancement factors (EF) of silver spheres, gold spheres and nanostars of similar sizes and concentration were compared. Under 785 nm excitation, nanostars and silver spheres have similar EF, and both are much stronger than gold spheres. Having plasmon matching the incident energy and multiple “hot spots” on the branches bring forth strong SERS response without the need to aggregate. Intracellular detection of silica-coated SERS-encoded nanostars was also demonstrated in breast cancer cells. The non-aggregated field enhancement makes the gold nanostar ensemble a promising agent for SERS bioapplications.
Keywords: Gold nanoparticle, nanostars, SERS, in vitro, silica
INTRODUCTION
Surface-enhanced Raman scattering (SERS) spectroscopy, by enhancing the intrinsically inefficient Raman process, has become a promising tool for delineating the unique molecular “fingerprint” of the analyte. An enhancement up to 1013–1014 –fold allows for sensitive single-molecule detection.[1, 2] This enhancement has been proposed to originate from: 1) the localized surface plasmon resonance (LSPR) on the metal nanostructure; 2) the charge-transfer (CT) between the molecule and the metal conduction band.[3–5] It’s noteworthy that while the enhancement for a single isolated gold nanoparticle is less than four orders of magnitude,[6, 7] further enhancement can be obtained at “hot spots”, which are highly localized surface plasmons generated from sharp protrusions (e.g. rods, stars) or coupled configurations (e.g., dimers, aggregates, gaps).[4, 8, 9] The potential for engineering the geometry of nanoplatforms (i.e., tuning the plasmon and hot spots) enables optimization of the optical properties to achieve maximum SERS signal.[10–13]
Our group has been involved in the development and application of various SERS plasmonic platforms ranging from nanoparticles, to nanopost arrays, nanowires and nanochips.[13–16] Out of a wide variety of types of nanostructures, gold nanoparticles have been the most widely studied for many reasons. Gold nanoparticles are easy to synthesize in different sizes and are chemically inert and typically non-toxic.[17] In addition, the plasmon of unique-shaped gold nanoparticles can be tuned to the near infrared (NIR) region, which is preferable to the visible region for in vivo application due to superior tissue penetration.[18, 19] Therefore, gold nanoparticles are a potential candidate for application in biological systems.[20–22]
Amongst all NIR-tuned gold nanoparticles, star-shaped gold nanoparticles (nanostars) area promising new nanoplatform for bioapplications. Nanostars not only have plasmon bands tunable in the NIR tissue optical window, but also multiple sharp branches acting as “hot-spots” due to the “lightning rod” effect.[23, 24] According to several 2D simulation models, the plasmon resonant wavelength correlates with the branching.[25–27] The nanostar plasmon results from hybridization of the plasmon from each branch, while the plasmon peak intensity depends on the polarization angle. From a 3D polarization-averaged model, the plasmon peak position and intensity correlate with the branch aspect ratio and branch length/number, respectively.[24] Based on the two important attributes of nanostars (tunable NIR plasmon and multiple “hot spots” at the branches), their applications have been numerous in various biomedical arenas, including SERS,[23, 28–37] photodynamic therapy (PDT),[38] photothermal therapy (PTT),[39–41] photoacoustic imaging,[33] biosensing,[42] and magnetomotive imaging.[43]
In this article, we investigate the SERS enhancement factor (EF) of surfactant-free nanostars and apply the silica-coated SERS probe for intracellular detection. To date, previous studies have reported SERS EFs of 5–7 orders of magnitude on nanostars.[29, 44, 45] However, due to the presence of organic surfactant on most nanostars studied, the realistic enhancement from nanostar ensembles in solution requires the removal of these surfactants. Such washing processes predispose nanoparticles to aggregation, which could lead to further unwanted enhancement. Using our newly developed surfactant-free nanostars, no washing step is required before surface functionalization. We compare the SERS EF of nanostars for the first time against silver spheres (AgNP) and gold spheres (AuNP) of similar sizes and concentrations. Nanostars have EFs similar to AgNP but much higher than AuNP. The hot spots from the branching seem to play a significant role in addition to the LSPR on SER Senhancement from nanostars. The in vitro SERS detection was demonstrated by using silica-coated SERS-encoded NS incubated with BT549 cancer cells.
RESULTS AND DISCUSSION
The SERS EF is determined by multiple factors, including metal composition, LSPR wavelength, particle concentration, and number of hot spots (e.g. aggregation, sharp branch, gap, etc).[4] To properly compare the SERS EF of different nanoparticles, the effect from the dyes (charge, molecular structure, surface orientation, surface coverage, resonance) should be kept constant; thus the concentration of both dye and nanoparticle were controlled during the SERS measurement. Also, because a very small portion of aggregation can dominate the overall enhancement, we therefore added no NaCl and carefully monitored the aggregation state in this study.
To properly compare the SERS EF between gold and silver nanoparticles, they have to be prepared in similar sizes (Fig. 1, Fig. S1 (Supporting information)) and concentrations (adjusted to roughly 0.1 nM). Our newly developed surfactant-free seed-mediated synthesis method yielded monodisperse 50-nm nanostars, which can be easily labeled with SERS active dyes without the need for removal of surface polymer on the nanoparticles. Spherical silver and gold nanoparticles of roughly 50-nm in diameter were also prepared. For spherical silver nanoparticles (AgNPs), because both standard citrate and hydroxylamine reduction methods produced a wide size distribution, we utilized a seed-mediated methods to create monodisperse 50-nm AgNPs (Fig. 1, Fig. S2 (Supporting information)).[46] This AgNP, however, was sensitive to oxidation; SERS dyes addition to a 3-day old AgNP samplehas much less SERS intensity than the fresh-made AgNP. Because of this, all AgNP studied were prepared fresh. For spherical gold nanoparticles (AuNPs), the modified Turkevich citrate reduction method produced slightly oval-shaped, monodisperse 50-nm AuNPs. Both gold nanostars and spheres did not show significant oxidation effect.
Figure 1.
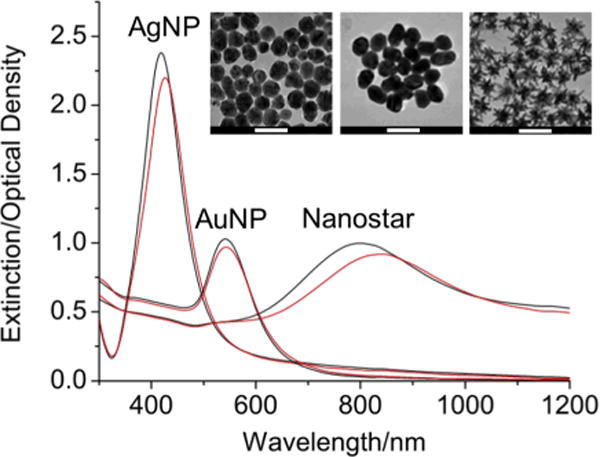
Extinction spectra of 0.1 nM AgNP, AuNP and nanostars before (black line) and after (red line) the addition of 4-MBA (final 1 μM) in MeOH (final 10 % v/v). A prominent plasmon red-shift was visible on nanostars but less on AgNP and AuNP. (inset) TEM images of AgNP, AuNP and nanostars (left to right). Scale bars are 100 nm.
The extinction spectrum maximum of the three different nanoparticles ranges from 400–800 nm based on their composition and geometry (Fig. 1). Previously, we have shown on simulation that nanostars’ plasmon peak and intensity are determined by the branch aspect ratio and branch number/length, respectively.[24] Because of the heterogeneous branch morphology, nanostar ensembles probably enablewider range of LSPR modes, which explains the broadening of the extinction spectra. Upon the addition of 1 μM 4-MBA, which has less than the full surface coverage (20~40%) on nanoparticles and does not induce apparent aggregation, a slight spectral red-shift was observed. The plasmon red-shifted more on AgNP (4 nm) than on AuNP (1 nm); this probably reflects the different thiol binding efficiency between silver (59 kcal/mol) and gold (45 kcal/mol). For nanostars, even though their branches contain a small amount of silver,[24] enhanced thiol binding does not explain the red-shift difference between nanostars (15 nm) and AgNP (4 nm). Trigari et al. found on simulation a remarkable plasmon red-shift on nanostars when the surrounding refractive index increases.[27] It is therefore very likely that the plasmon from the branches is more sensitive to the refractive index change than the plasmon from the spheres, hence a significant plasmon red-shift on nanostars.
Figure 2 shows the non-resonance SERS comparison of nanostars, AgNP and AuNP, with the most prominent peaks being the Stokes features appearing at 1013 cm–1 and 1078 cm−1, which are assigned to the C-O stretching mode of MeOH and ring breathing mode of the 4-MBA.[44] It is noteworthy that no NaCl was added to avoid nanoparticle aggregation. Although there was no strong evidence of aggregation on UVVIS, nanoparticle tracking analysis still found a small amount of larger particles on all samples after the addition of 4-MBA (Fig. S1 (Supporting information)). Because adding 4-MBA generated the smallest size change when compared to adding 4-aminothiophenol, thiophenol, and 4-methylbenzinethiol, we believe that nanoparticle ensembles labeled with 4-MBA were the closest to their non-aggregated state, which allows for our SERS measurement to reflect the realistic EM enhancement on nanoparticle ensembles. Although this is not a single particle SERS study, the ensemble-averaged signal still provides crucial information for SERS comparison between different compositions and geometries. At 785 nm, nanostars exhibit a slightly greater enhancement than AgNP but significantly outperform AuNP. The EF of isolated AuNP was not visible in our setup; an EF of lower than 4 orders of magnitude on isolated AuNP was previously reported.[6, 7, 47, 48] Nanostars, with the presence of multiple hot spots on the branches, have at least comparable EF to AgNP, which is typically 2 to 3 orders of magnitude stronger than AuNP.[49] Being able to achieve a similar EF to AgNP without the toxicity of silver, nanostars are particularly enticing due to their superior biocompatibility of gold. Moreover, because aggregation is generally hard to control and irreproducible, the advantage of having strong SERS without the need to form aggregates make gold nanostars an appealing candidate as SERS contrast agent.
Figure 2.
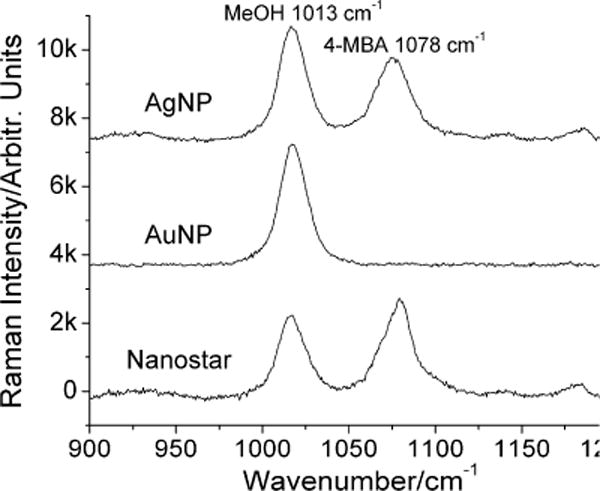
SERS spectra (baseline subtracted) of 1 μM 4-MBA in 0.1 nM AgNP, AuNP, and nanostar solutions examined through a Raman microscope under 785 nm excitation. Methanol (10% v/v) was used as an internal reference. Wavenumber 1013 and 1078 represents Raman signals from MeOH and 4-MBA, respectively.
Figure 3 illustrates a laser wavelength dependence of SERS EF on both AgNP and nanostars. Generally, maximal enhancement occurs at excitation near the LSPR maximum.[48–51] With AgNP, which has a plasmon maximum located near 405 nm, a 2-fold stronger EF was obtained from 785 nm than 633 nm excitation, suggesting a possible minute aggregation dominating the overall SERS response.[52] Such aggregation was barely detectable under UVVIS but was only slightly visible under Nanosight. On nanostars, more than 10-fold stronger EF was observed under excitation from 785 nm than from 633 nm. Both Rodríguez-Lorenzo et al. and Li et al. reported stronger SERS response under 785 nm than at shorter wavelength.[7, 53] Under 633 nm excitation, the ensemble-averaged nanostars SERS EF (0.3×105) is lower than reports from Nalbant Esenturk et al. and Jeong et al. (2–5.7×105),[29, 45] probably due to different dyes/nanostars concentrations or NaCl used. Under 785 nm excitation, the ensemble-averaged nanostars SERS EF is around 4×105, which is slightly higher than results from Li et al. (1×105) and Khoury et al. (2–5×103).[7, 44] Having sharper branches and no polymer coating on our nanostars might contribute the slight EF improvement. It is noteworthy that the SERS signal intensity increases with analyte concentration in the range investigated; an opposing non-linear trend was observed on the SERS EF (Fig. S3 (Supporting information)). When being excited under 785 nm excitation, which is in resonance with the branch plasmon,[24, 54] the EF was almost 10-fold greater at 0.1 nM than at 1 μM. It is likely that the non-linear presentation could be attributed to multiple factors, such as anisotropic EM field distribution, inhomogeneous molecular coverage/polarity on the hot spots, etc.[49, 55] However, the exact mechanism explaining these observations is beyond the scope of this paper and would be a topic of future comparative studies. In short, a combination of LSPR matching the excitation laser and presence of multiple hot spots on nanostars may explain their strong SERS enhancement.
Figure 3.
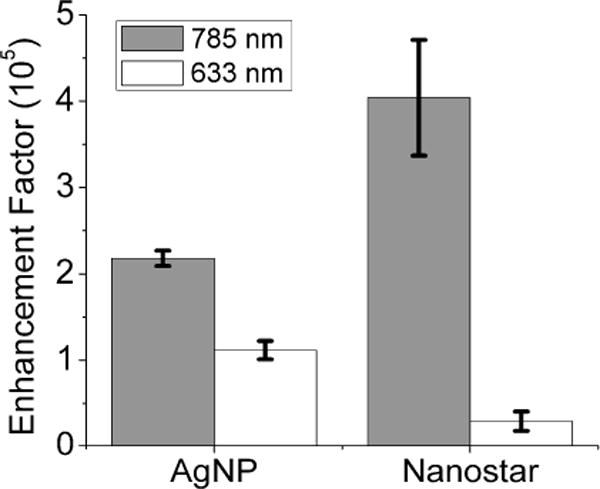
Enhancement factors of AgNP and nanostar under 785 nm (grey) and 633 nm (white) laser excitation. AuNP was omitted due to no 4-MBA SERS signal available for EF calculation. Error bar is 1 SD.
Although nanostars have slightly higher EF than gold nanorod ensembles(104–105), its EF is still lower than that from gold dimers (107–108) or clustered patterns (108–1010).[6, 30, 48, 56] With more branches per nanoparticle, it is reasonable that nanostars have several fold higher enhancement than nanorods. However, the EF from sharp protrusion may be less than from a coupled configuration.[28, 57] Dimers, despite their higher EF, remain difficult to fabricate uniformly. Film-based clustered patterns cannot be applied easily to biological systems as well. Although nanostars coupled to a gold film produced SERS EF of 1010,[30] we could not reach enhancement that high even upon NaCl induced aggregation in solution. It is noteworthy that the ensemble-averaged SERS increment upon aggregation was much less profound from nanostars than from spheres. Instead of a branch facing another branch, aggregated nanostars probably have branches collapsing into each other, hence breaking the hot spots on the branches. It is likely that nanostar aggregates produce much less EM enhancement than sphere aggregates due to the possible plasmon deactivation from interaction between tips with different geometry and orientation; this is in agreement with previous observation.[58] Although aggregates may produce strong SERS, keep in mind that the reproducible formation of, as well as access to, hotspots in nanoparticle dimers or aggregates is limited. Controlled aggregation in ensemble is appealing but still remains a difficult task with various yields.[46, 59] In contrast, nanostars possess a high EF as non-aggregated monodisperse entities, and therefore do not suffer from the non-linear enhancement effect due to aggregation. Until a more reliable method is developed for the formation of reproducible and accessible hot spots, nanostars may nonetheless be one of the most sensitive and controllable SERS platforms in solution.
To fabricate a strong SERS probe for bioapplication, several dyes were investigated. Non-thiolated dyes (e.g. rhodamine, crystal violet) were found unable to remain on the nanoparticle surface during the silica coating process. Thiolated, thiocyanated, thionine dyes, such as 4-MBA, fluorescein isothiocyanate, methylene blue, all sustained the silica coating process but the resulting SERS signal under 785 nm excitation remains insufficient, requiring long integration time for adequate signal-to-noise ratio. One exception is 3,3′-diethylthiatricarbocyanine iodide (DTTC), which is a NIR dye that has two cyanine groups that will facilitate its anchoring onto the gold surface. DTTC is also resonant with the 785 nm excitation laser, creating a stronger resonance Raman for use in SERS detection.[60] Upon silica coating, the hydrodynamic size of the SERS probes used in this study were around 110 nm (Fig. 4B, Fig. S4 (Supporting information)).
Figure 4.
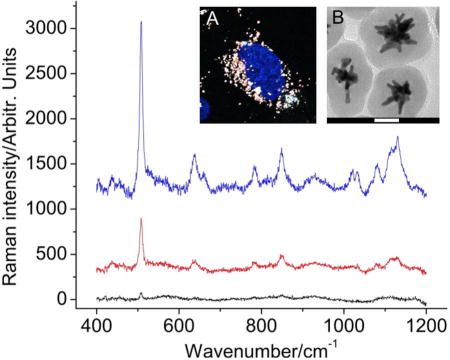
SERS spectra (baseline subtracted) of different intracellular regions (blue:cytoplasm, red: nucleus, black: glass) on BT549 cells incubated 24 hrs with silicacoated nanostars labeled with a SERS dye (DTTC). (A) Two-photon imaging confirming the presence of nanostars (white) locating primarily in the cytoplasm. Nucleus was stained blue. (B) TEM image of the silica-coated nanostars SERS probes. Scale bar is 50 nm.
Figure 4 shows the SERS spectra of silica-coated DTTC-encoded SERS probe in different compartments of cells with short integration time (10 sec). Similar strategies have been applied for SERS mapping in cells or tissues previously.[36, 37] In our study, two-photon microscopy disclosed that SERS probes were accumulated mostly in the cytoplasm with minimal intranuclear accumulation after 24 hours of probe incubation (Fig. 4A). SERS signals were found to be the greatest in the cytoplasm region but remained one-fifth as strong in the nuclear region. The discrepancy between the two-photon image (minimal intranuclear nanostars) and SERS spectra could be the fact that due to a low axial resolution (from the large axialoptical probe volume), the SERS spectra collected in the nuclear region maybe confounded by signals from the cytoplasm above or underneath the nucleus. Further use of multifunctional SERS probes by incorporating other modalities (e.g. photodynamic therapy, photothermal therapy, MRI contrast) may bring forth promises for molecular imaging and cancer therapy.[38, 61, 62]
CONCLUSION
In this paper we compare the ensemble-averaged SERS response on AgNP, AuNP and nanostars of similar sizes and concentrations under both 785 nm and 633 nm excitations. The surfactant-free nanostars allow direct dye labeling without washing cycles to remove the surfactant; doing soenables a more realistic and convenient SERS study. Nanostars, with the presence of multiple branchesas hot spots, generate a strong field enhancement, comparable to that from AgNP. With LSPR in the near IR, the enhancement from nanostars is 10-fold more prominent under 785 nm than 633 nm excitation. Furthermore, an intracellular detection of silica-encapsulated DTTC-encoded nanostars SERS probes was demonstrated in breast cancer cells. SERS probes accumulated primarily in the cytoplasm, displaying a high SERS signal upon examination. Having a high enhancement factor without the need for aggregation, nanostars thus have a strong potential in sensing and imaging applications.
EXPERIMENTAL
Chemicals
Gold(III) chloride trihydrate (HAuCl4), sodium citrate tribasic dihydrate (Na3Cit), L(+)-ascorbic acid (AA), silver nitrate (AgNO3), hydrochloric acid (HCl), O-[2-(3-mercaptopropionylamino)ethyl]-O′-methylpolyethylene glycol(MW 5,000; SHPEG5k), 4-mercaptobenzoic acid (4-MBA), ethanol (EtOH), methanol (MeOH), tetraethyl orthosilicate (TEOS), 3,3′-diethylthiatricarbocyanine iodide (DTTC) were purchased from Sigma-Aldrich (St. Louis, MO). All chemicals were used as received. Millipore Synergy ultrapure water (DI) of resistivity = 18.2 MΩ cm was used in all aqueous solutions. All glassware and stir bars were cleaned with aqua regia solution and oven-dried before use. (Caution: aqua regia is extremely dangerous. Please use it with extra caution.)
Gold seed synthesis
Gold seeds were made by adding 15 ml of 1% (w/v) Na3Cit solution into 100 ml of boiling 1 mM HAuCl4 solution under vigorous stirring. After 30 min, the solution was cooled, filtered by a 0.22 μm nitrocellulose membrane, and kept at 4 °C for long-term storage.
Gold spheres (50 nm) synthesis
Gold spheres were made by adding 0.8 ml of 1% (w/v) Na3Cit solution into 100 ml of boiling 0.25 mM HAuCl4 solution under vigorous stirring. After 30 min, the solution was cooled, filtered by a 0.22 μm nitrocellulose membrane and stored at 4 °C for long-term storage. Before use, the solution underwent centrifugal wash (3000 ×g for 15 min) once and was resuspended to 0.1 nM.
Silver spheres (50 nm) synthesis
Silver spheres were synthesized using a seed-mediated method modified from the nanostars synthesis method below. In 10 ml of 100 mM AgNO3 solution, 100 μl of the citrate gold seed solution was added under room temperature. Immediately afterwards, a mixture of 50 μl of 100 mM AA and 10 μl of HCl was administered. One hour later, the solution underwent centrifugal wash (3000 ×g for 15 min) once and was resuspended to 0.1 nM. The Ag spheres were used within a day to reduce the detrimental effect from surface oxidation.
Nanostarssynthesis
Gold nanostars were synthesized using a seed-mediated method. Detailed synthesis and characterization of the nanostars has been presented elsewhere.[24] Briefly, in 10 ml 0.25 mM HAuCl4 solution, 10 μl of 1N HCl and 12 nm citrate gold seeds 100 μl were added followed by the simultaneous addition of 100 μl AgNO3 and 50 μl 100 mM AA under stirring (700 rpm). The reaction was performed under room temperature, and the process is completed in less than a minute. Afterwards, the solution underwent centrifugation wash (3000 ×g 15 min) once, resuspended to 0.1 nM, and kept under 4 °C for long-term storage.
Structural and optical characterization
Transmission electronic microscopy (TEM; Fei Tecnai G2 Twin, 200 kV) was used for structural analysis. The particle hydrodynamic size distribution, concentration, and ζ-potential were determined by nanoparticle tracking analysis (NTA 2.1; build 0342) using NanoSight NS500 (Nanosight Ltd. UK). A UV-VIS spectrophotometer (Shimadzu UV-3600; Shimadzu corporation, Japan) was used to collect the extinction spectrum.
SERS measurements and instrumentation
100 μl of freshly prepared 4-MBA (10 μM in 10% MeOH) was mixed with 1 ml particles solutions for 10 min. All particle solutions were pre-diluted to 0.1 nM particle concentrations before adding the 4-MBA. Special care was taken to avoid particle aggregation. The 4-MBA Raman spectrum from each particle solution was collected using a Renishaw InVia Raman system (633 nm HeNe laser, 8 mW, 1800 gr/mm grating; Renishaw Inc. IL) or a Jobin Yvon Horiba LabRam ARAMIS (785 nm diode-laser, 40 mW, 1200 gr/mm grating; Horiba Scientific, NJ) (Fig. S1 (Supporting information)). Three samples were collected for each experimental condition.
Examining the enhancement factor
The SERS EF of a nanoparticle ensemble is determined by the ratio of SERS cross-section (σ4MBA:SERS) to normal Raman cross-section (σ4MBA:Raman) of 4-MBA in nanoparticle and normal solution, respectively .[63] 4-MBA was chosen because 1) it has a thiol group that strongly binds to the metal surface, 2) it does not fluoresce at 633 nm or 785 nm, and 3) the carboxyl groups maintain the negative surface charge at pH 7 in order to reduce particle aggregation. Also, ensembles in solution are less aggregated than dried solid phase. This allowed us to study the effect of particle geometry on SERS without the non-linear interference from aggregation.
Usually, the EF is calculated by . However, because these three types of nanoparticles have uneven surface area (e.g. oval shape, branches), the exact number of surface-bound 4-MBA (especially those located at the hot spot) cannot be simply estimated. Also, due to an overlapping absorption background to the Raman emission spectra, the SERS intensity needs to be normalized by an internal reference. To approach these issues, we selected a 4-MBA concentration of sub-total surface coverage per particle. Because all 4-MBA molecules would attach to the nanoparticle through the strong dative bond, the amount of 4-MBA added can be assumed as the amount of 4-MBA on the nanoparticle surface. Also, we added a small amount (5~10% v/v) of MeOH, which does not induce aggregation and is not enhanced by the nanoparticle, as an internal reference.[1] The next step is to obtain σ4MBA:SERS and σ4MBA:Raman.
The σ4MBA:SERS is calculated in reference to σMeOH:Raman using the equation , where C4MBA:SERS/CMeOH:SERS and I4MBA:SERS/IMeOH:SERS are the concentrations and SERS intensities of 4-MBA and MeOH in nanoparticle solution. The SERS intensity was defined by the area under curve, which was calculated by fitting the spectrum using the pseudo-Voigt function in OriginPro 8 (OriginLab Corporation, USA). Raman intensities of 4-MBA and MeOH were measured using the aromatic ring vibration (1078 cm–1) and the C-O stretch vibration(1016 cm–1), respectively.[64, 65] Because the two vibration bands were in close proximity, the background absorption from the solution would be similar on both bands. The same strategy can be applied to obtaining σ4MBA:Raman.
To obtain σ4MBA:Raman, 4-MBA was dissolved to 10–50 mM in NaOH 1 N and 2 % (v/v) MeOH. The average σ4MBA:Raman measured were calculated 9.23×10–30 and 2.75×10–30 cm2/molecule at 633 nm and 785 nm, respectively. The MeOH:Raman was calculated from multiple mixing ratios of MeOH and acetonitrile. MeOH’s C-O stretch vibration (1016 cm–1) and acetonitrile’s C-C stretch vibration (919 cm–1) were used for calculation. σMeOH:Raman at both 633 nm (0.693×10–30 cm2/molecule) and 785 nm (0.207×10–30 cm2/molecule) were determined in reference to acetonitrile’s cross-section extrapolated from the known value.[66]
The final step is .
SERS dye encoding and silica-encapsulation on nanostars
Freshly synthesized nanostars (10 mL) were capped with 5 μM SHPEG5k under gentle stirring for 15 min. The PEGylated particles were then centrifuged (10000 ×g) twice at 4 °C to remove excess PEG and redispersed in DI. DTTC (5 μM) in methanol was added to this solution and allowed to stir overnight. The DTTC-tagged particles were centrifuged (5000 ×g) twice at 4 °C to remove excess DTTC and resuspended in 2.3 mL of EtOH. A modified Stöber method was then used for formation of the silica shell.[38, 67] Under gentle stirring, 2.25 mL of the nanostars in ethanol was added to a solution containing 2.0 mL of water and 6.8 mL of EtOH followed by the addition of 160 μL of NH4OH. Silica coating was initiated by the addition of 30 μL of 10% TEOS in EtOH, and the reaction was allowed to proceed for 3 h. The nanoparticles were then centrifugally purified (3500 ×g) twice and redispersed into 5 mL DI.
Intracellular SERS detection
The BT549 breast cancer cells were a gift from Dr. Victoria Seewaldt. Cells were incubated in RPMI 1640 medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum, 25 mM HEPES, and 0.023 unit/mL insulin, in an incubator with a humidified atmosphere (37 °C and 5% CO2). Cells in exponential growth phase were used in the experiment. The nanoparticles were concentrated to □0.1nM in the growth media and incubated with BT549 for 24 hours. Cells were then washed, fixed, and stained by Hoescht 33342 (Invitrogen, Carlsbad, CA). For confirming the presence of intracellular nanostars, cells were examined under multiphoton microscopy (Olympus FV1000, Olympus America, Center Valley, PA). SERS spectra were taken using a 40x 0.8 NA water immersion objective under 785 nm, 4mW, 10 sec integration, and 2 accumulations. Laser spot was roughly focused to different cellular compartments (nucleus, cytoplasm, glass) based on the bright field images.
Supplementary Material
Acknowledgments
This work was sponsored by the National Institutes of Health (Grants R01 EB006201 and R01 ES014774). AMF is supported by a training grant from the National Institutes of Health (T32 EB001040).
Footnotes
SUPPORTING INFORMATION
All supporting information mentioned in the textis available in the online version of the journal.
References
- 1.Kneipp K, Wang Y, Kneipp H, Perelman L, Itzkan I, Dasari R, Feld M. Phys Rev Lett. 1997;78:1667. doi: 10.1103/PhysRevLett.76.2444. [DOI] [PubMed] [Google Scholar]
- 2.Nie S, Emery S. Science. 1997;275:1102. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 3.Moskovits M. J Raman Spectrosc. 2005;36:485. [Google Scholar]
- 4.Stiles PL, Dieringer JA, Shah NC, Van Duyne RP. Annu Rev Anal Chem. 2008;1:601. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 5.Lombardi JR, Birke RL. Acc Chem Res. 2009;42:734. doi: 10.1021/ar800249y. [DOI] [PubMed] [Google Scholar]
- 6.Wustholz KL, Henry AI, McMahon JM, Freeman RG, Valley N, Piotti ME, Natan MJ, Schatz GC, van Duyne RP. J Am Chem Soc. 2010;132:10903. doi: 10.1021/ja104174m. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Cushing SK, Zhang J, Lankford J, Aguilar ZP, Ma D, Wu N. Nanotechnology. 2012;23:115501. doi: 10.1088/0957-4484/23/11/115501. [DOI] [PubMed] [Google Scholar]
- 8.Xu H, Aizpurua J, Kall M, Apell P. Phys Rev E. 2000;62:4318. doi: 10.1103/physreve.62.4318. [DOI] [PubMed] [Google Scholar]
- 9.Banholzer MJ, Millstone JE, Qin L, Mirkin CA. Chem Soc Rev. 2008;37:885. doi: 10.1039/b710915f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lal S, Link S, Halas NJ. Nat Photonics. 2007;1:641. [Google Scholar]
- 11.Sau TK, Rogach AL, Jäckel F, Klar TA, Feldmann J. Adv Mater (Weinheim, Ger) 2010;22:1805. doi: 10.1002/adma.200902557. [DOI] [PubMed] [Google Scholar]
- 12.Pazos-Perez N, Ni W, Schweikart A, Alvarez-Puebla RA, Fery A, Liz-Marzán LM. Chem Sci. 2010;1:174. [Google Scholar]
- 13.Vo-Dinh T, Dhawan A, Norton SJ, Khoury CG, Wang HN, Misra V, Gerhold MD. J Phys Chem C. 2010;114:7480. doi: 10.1021/jp911355q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vo-Dinh T, Hiromoto M, Begun G, Moody R. Anal Chem. 1984;56:1667. [Google Scholar]
- 15.Vo-Dinh T, Meier M, Wokaun A. Anal Chim Acta. 1986;181:139. [Google Scholar]
- 16.Vo-Dinh T. TrAC, Trends Anal Chem. 1998;17:557. [Google Scholar]
- 17.Baker G, Moore D. Anal Bioanal Chem. 2005;382:1751. doi: 10.1007/s00216-005-3353-7. [DOI] [PubMed] [Google Scholar]
- 18.Gobin AM, Lee MH, Halas NJ, James WD, Drezek RA, West JL. Nano Lett. 2007;7:1929. doi: 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Neretina S, El-Sayed MA. Adv Mater (Weinheim, Ger) 2009;21:4880. doi: 10.1002/adma.200802789. [DOI] [PubMed] [Google Scholar]
- 20.Sperling RA, Rivera Gil P, Zhang F, Zanella M, Parak WJ. Chem Soc Rev. 2008;37:1896. doi: 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]
- 21.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 22.Lévy R, Shaheen U, Cesbron Y. Nano Rev. 2010;1:4889. doi: 10.3402/nano.v1i0.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abalde-Cela S, Aldeanueva-Potel P, Mateo-Mateo C, Rodríguez-Lorenzo L, Alvarez-Puebla RA, Liz-Marzán LM. J R Soc Interface. 2010;7:S435. doi: 10.1098/rsif.2010.0125.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan H, Khoury (co-first author) CG, Hwang H, Wilson CM, Grant GA, Vo-Dinh T. Nanotechnology. 2012;23:075102. doi: 10.1088/0957-4484/23/7/075102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao F, Nehl CL, Hafner JH, Nordlander P. Nano Lett. 2007;7:729. doi: 10.1021/nl062969c. [DOI] [PubMed] [Google Scholar]
- 26.Senthil Kumar P, Pastoriza-Santos I, Rodríguez-González B, Garcia de Abajo FJ, Liz-Marzán LM. Nanotechnology. 2008;19:015606. doi: 10.1088/0957-4484/19/01/015606. [DOI] [PubMed] [Google Scholar]
- 27.Trigari S, Rindi A, Margheri G, Sottini S, Dellepiane G, Giorgetti E. J Mater Chem. 2011;21:6531. [Google Scholar]
- 28.Hrelescu C, Sau TK, Rogach AL, Jackel F, Feldmann J. Appl Phys Lett. 2009;94:153113. [Google Scholar]
- 29.Nalbant Esenturk E, Hight Walker A. J Raman Spectrosc. 2009;40:86. [Google Scholar]
- 30.Rodriguez-Lorenzo L, Alvarez-Puebla RA, Pastoriza-Santos I, Mazzucco S, Stephan O, Kociak M, Liz-Marzán LM, Garcia de Abajo FJ. J Am Chem Soc. 2009;131:4616. doi: 10.1021/ja809418t. [DOI] [PubMed] [Google Scholar]
- 31.Barbosa S, Agrawal A, Rodriguez-Lorenzo L, Pastoriza-Santos I, Alvarez-Puebla RA, Kornowski A, Weller H, Liz-Marzan LM. Langmuir. 2010;26:14943. doi: 10.1021/la102559e. [DOI] [PubMed] [Google Scholar]
- 32.Lu W, Singh AK, Khan SA, Senapati D, Yu H, Ray PC. J Am Chem Soc. 2010;132:18103. doi: 10.1021/ja104924b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim C, Song HM, Cai X, Yao J, Wei A, Wang LV. J Mater Chem. 2011;21:2841. doi: 10.1039/C0JM04194G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerrero-Martínez A, Barbosa S, Pastoriza-Santos I, Liz-Marzán LM. Curr Opin Colloid Interface Sci. 2011;16:118. [Google Scholar]
- 35.Su Q, Ma X, Dong J, Jiang C, Qian W. ACS Appl Mater Interfaces. 2011;3:1873. doi: 10.1021/am200057f. [DOI] [PubMed] [Google Scholar]
- 36.Schütz M, Steinigeweg D, Salehi M, Kömpe K, Schlücker S. Chem Comm. 2011;47:4216. doi: 10.1039/c0cc05229a. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Lorenzo L, Krpetic Z, Barbosa S, Alvarez-Puebla RA, Liz-Marzán LM, Prior IA, Brust M. Integr Biol. 2011;3:922. doi: 10.1039/c1ib00029b. [DOI] [PubMed] [Google Scholar]
- 38.Fales AM, Yuan (co-first author) H, Vo-Dinh T. Langmuir. 2011;27:12186. doi: 10.1021/la202602q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van de Broek B, Devoogdt N, D’Hollander A, Gijs HL, Jans K, Lagae L, Muyldermans S, Maes G, Borghs G. ACS Nano. 2011;5:4319. doi: 10.1021/nn1023363. [DOI] [PubMed] [Google Scholar]
- 40.Yuan H, Khoury CG, Wilson CM, Grant GA, Bennett AJ, Vo-Dinh T. Nanomedicine: NBM. 2012 doi: 10.1016/j.nano.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan H, Fales AM, Vo-Dinh T. J Am Chem Soc. 2012;134:11358. doi: 10.1021/ja304180y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dondapati SK, Sau TK, Hrelescu C, Klar TA, Stefani FD, Feldmann J. ACS Nano. 2010;4:6318. doi: 10.1021/nn100760f. [DOI] [PubMed] [Google Scholar]
- 43.Song HM, Wei Q, Ong QK, Wei A. ACS Nano. 2010;4:5163. doi: 10.1021/nn101202h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khoury C, Vo-Dinh T. J Phys Chem C. 2008;112:18849. doi: 10.1021/jp8054747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong G, Lee Y, Kim M, Han S. J Colloid Interface Sci. 2009;329:97. doi: 10.1016/j.jcis.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Chen G, Wang Y, Yang M, Xu J, Goh SJ, Pan M, Chen H. J Am Chem Soc. 2010;132:3644. doi: 10.1021/ja9090885. [DOI] [PubMed] [Google Scholar]
- 47.Metiu H, Das P. Annu Rev Phys Chem. 1984;35:507. [Google Scholar]
- 48.Orendorff C, Gearheart L, Jana N, Murphy C. Phys Chem Chem Phys. 2006;8:165. doi: 10.1039/b512573a. [DOI] [PubMed] [Google Scholar]
- 49.Alvarez-Puebla RA. J Phys Chem Lett. 2012;3:857. doi: 10.1021/jz201625j. [DOI] [PubMed] [Google Scholar]
- 50.McFarland AD, Young MA, Dieringer JA, van Duyne RP. J Phys Chem B. 2005;109:11279. doi: 10.1021/jp050508u. [DOI] [PubMed] [Google Scholar]
- 51.Guo H, Ruan F, Lu L, Hu J, Pan J, Yang Z, Ren B. J Phys Chem C. 2009;113:10459. [Google Scholar]
- 52.Rivas L, Sanchez-Cortes S, Garcia-Ramos J, Morcillo G. Langmuir. 2000;16:9722. [Google Scholar]
- 53.Rodriguez-Lorenzo L, Alvarez-Puebla RA, Javier Garcia de Abajo F, Liz-Marzán LM. J Phys Chem C. 2010;114:7336. [Google Scholar]
- 54.Cialla D, März A, Böhme R, Theil F, Weber K, Schmitt M, Popp J. Anal Bioanal Chem. 2012;403:27. doi: 10.1007/s00216-011-5631-x. [DOI] [PubMed] [Google Scholar]
- 55.Pierre MCS, Mackie PM, Roca M, Haes AJ. J Phys Chem C. 2011;115:18511. [Google Scholar]
- 56.Yap FL, Thoniyot P, Krishnan S, Krishnamoorthy S. ACS Nano. 2012;6:2056. doi: 10.1021/nn203661n. [DOI] [PubMed] [Google Scholar]
- 57.Khoury CG, Norton SJ, Vo-Dinh T. ACS Nano. 2009;3:2776. doi: 10.1021/nn900664j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aldeanueva-Potel P, Carbó-Argibay E, Pazos-Perez N, Barbosa S, Pastoriza-Santos I, Alvarez-Puebla RA, Liz-Marzán LM. ChemPhysChem. 2012;13:2561. doi: 10.1002/cphc.201101014. [DOI] [PubMed] [Google Scholar]
- 59.Freitag I, Neugebauer U, Csaki A, Fritzsche W, Krafft C, Popp J. Vib Spectrosc. 2012;60:79. [Google Scholar]
- 60.Mohs AM, Mancini MC, Singhal S, Provenzale JM, Leyland-Jones B, Wang MD, Nie S. Anal Chem. 2010;82:9058. doi: 10.1021/ac102058k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Maltzahn G, Centrone A, Park JH, Ramanathan R, Sailor MJ, Hatton TA, Bhatia SN. Adv Mater (Weinheim, Ger) 2009;21:3175. doi: 10.1002/adma.200803464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kircher MF, de la Zerda A, Jokerst JV, Zavaleta CL, Kempen PJ, Mittra E, Pitter K, Huang R, Campos C, Habte F, Sinclair R, Brennan CW, Mellinghoff IK, Holland EC, Gambhir SS. Nat Med. 2012;18:829. doi: 10.1038/nm.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le Ru EC, Blackie E, Meyer M, Etchegoin PG. J Phys Chem C. 2007;111:13794. [Google Scholar]
- 64.Mammone J, Sharma S, Nicol M. J Phys Chem. 1980;84:3130. [Google Scholar]
- 65.Orendorff C, Gole A, Sau T, Murphy C. Anal Chem. 2005;77:3261. doi: 10.1021/ac048176x. [DOI] [PubMed] [Google Scholar]
- 66.Biswas N, Umapathy S. Appl Spectrosc. 1998;52:496. [Google Scholar]
- 67.Fernandez-Lopez C, Mateo-Mateo C, Alvarez-Puebla RA, Perez-Juste J, Pastoriza-Santos I, Liz-Marzan LM. Langmuir. 2009;25:13894. doi: 10.1021/la9016454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.