

Abstract
Background
X-linked intellectual disability (XLID) is a group of genetically heterogeneous disorders characterized by substantial impairment in cognitive abilities, social and behavioral adaptive skills. Next generation sequencing technologies have become a powerful approach for identifying molecular gene mutations relevant for diagnosis.
Methods & objectives
Enrichment of X-chromosome specific exons and massively parallel sequencing was performed for identifying the causative mutations in 14 Finnish families, each of them having several males affected with intellectual disability of unknown cause.
Results
We found four novel mutations in known XLID genes. Two mutations; one previously reported missense mutation (c.1111C > T), and one novel frameshift mutation (c. 990_991insGCTGC) were identified in SLC16A2, a gene that has been linked to Allan-Herndon-Dudley syndrome (AHDS). One novel missense mutation (c.1888G > C) was found in GRIA3 and two novel splice donor site mutations (c.357 + 1G > C and c.985 + 1G > C) were identified in the DLG3 gene. One missense mutation (c.1321C > T) was identified in the candidate gene ZMYM3 in three affected males with a previously unrecognized syndrome characterized by unique facial features, aortic stenosis and hypospadia was detected. All of the identified mutations segregated in the corresponding families and were absent in > 100 Finnish controls and in the publicly available databases. In addition, a previously reported benign variant (c.877G > A) in SYP was identified in a large family with nine affected males in three generations, who have a syndromic phenotype.
Conclusions
All of the mutations found in this study are being reported for the first time in Finnish families with several affected male patients whose etiological diagnoses have remained unknown to us, in some families, for more than 30 years. This study illustrates the impact of X-exome sequencing to identify rare gene mutations and the challenges of interpreting the results. Further functional studies are required to confirm the cause of the syndromic phenotypes associated with ZMYM3 and SYP in this study.
Background
Intellectual disability (ID) can be defined as a significant impairment of cognitive and adaptive functions and has an estimated prevalence of 1.5-2% [1]. It is defined by an intelligence quotient (IQ) below 70 and an impairment in social and adaptive skills diagnosed before the age of 18 years [2]. ID can be caused by genetic as well as Anon-genetic factors. A consistent finding among individuals with ID has been the excess of males [1], indicating a role of defects on the X-chromosome. Mutations in X-linked genes contribute for 10%-15% of ID cases in males [3]. There is substantial evidence that X-linked ID (XLID) is genetically an extremely heterogeneous disorder [4].
The recent development of sequencing technologies has provided an effective tool to analyze the chromosomal distribution of mutations underlying ID [5-10]. Sequencing of the coding regions of the X-chromosome in families with multiple affected males has resulted in the identification of about 100 XLID genes to date [11]. Also in sporadic male patients, the possibility of a de novo mutation on the X-chromosome has to be taken into consideration [12].
The aim of this study was to identify the causative gene defects underlying XLID in Finnish families with two or more affected males.
Methods
Patients
Families with two or more affected males with ID were enrolled in this study (n = 14). Many of these patients have been under the care of experienced child neurologists and/or clinical geneticists since 1980. Inspite of detailed clinical investigations the diagnosis for these families has remained unknown. The Ethical Committee of the Helsinki University Central Hospital approved this study. Written informed consent was obtained from either the parents or the guardians of the patients. DNAs from Finnish blood donors were used as controls.
Exome sequencing
For enrichment of genomic DNA from 11 index patients we used the Agilent SureSelect Human X-Chromosome Exome kit (Agilent, California, USA) and for three index patients (D299, D301, D303) we performed droplet-based multiplex PCR (7,367 amplicons, 757 genes, 1.54 Mb) similarly to the previously described study [13]. DNAs were sequenced on the Illumina GAII and HiSeq2000 platforms, respectively. For all samples coverage of at least 10X was reached for >86% of the targeted regions. Average sequencing depth ranged between 149–583. The sequence files were then aligned against the hg19 reference genome by SOAP2.21 with the default settings. Variants in the form of SNVs, indels and CNVs were called by an in house software package (Medical Re-sequencing Analysis Pipeline (MERAP), developed at the Max Planck Institute for Molecular Genetics, Berlin. All sequence variants were screened against publicly available databases (dbSNP138, 1000 Genomes project, Exome Variant Server (ESP6500) and the in-house database of the Max Planck Institute, Berlin) for annotating likely non-pathogenic and previously reported neutral variants. In addition, the OMIM catalogue and the Human Gene Mutation Database (HGMD) were used as a filter to identify all previously described pathogenic changes.
Mutation analysis and Sanger sequencing
Validation of variants was performed using the standard Sanger sequencing protocol. First, primers (Oligomer Ltd, Helsinki, Finland) were designed to detect each individual mutation using their respective reference sequence. All PCR reactions were performed in 25 μl volume using Dynazyme polymerase (Finnzymes, Espoo, Finland) under standard conditions. PCR products were purified using Exo-SapIT (Affymetrix, Santa Clara, CA) according to the standard protocol. Sequencing reactions were performed using Applied Biosystems BigDye terminator v3.1 (Life Technologies, Carlsbad, CA) and products were analyzed on the ABI 3730 sequencer. Finally, the accurate genotype of each variant was confirmed by sequence analysis with the Sequencher software v4.8 (Life Technologies) in both the family members and the anonymous Finnish controls. Primer sequences are available on request.
X-inactivation analysis
X-inactivation study was performed in the families with symptomatic carrier females. 500 ng of DNA was divided into two fractions; one set was digested in a 20 μl volume of buffered solution with HpaII (New England Biolabs, UK) while the other set was digested with RsaI (Roche, Basel, Switzerland). Both aliquots were incubated at 37°C for 17 hours. Next, exon 1 of the androgen receptor gene (AR) was amplified from 2 μl of each of the HpaII and RsaI digested DNA samples using the forward 5'-labelled primer TCCAGAATCTGTTCCAGAGCGTGC and CTGGGACGCAACCTCTCTC reverse primer [14]. The amplification was performed for 35 cycles with an annealing temperature of 63.3°C. DNAs from healthy males were used as controls. Two μl of the PCR product was mixed with 10 μl of Hi-Di formamide (Life Technologies) and 0.025 μl of GeneScan 500 Liz size marker (Life Technologies). The samples were run on an ABI3730xl genetic analyzer. The peaks were analyzed using the GeneMarker software version1.4 (Life Technologies).
In silico analysis
Multiple programs, including SIFT, PolyPhen-2, PhD-SNP, PANTHER and SNPs&GO were used to predict the functional impact of the mutation on the protein function [15-19].
Multiple protein sequence alignment using orthologous species and human sequences was performed using ClustalX to verify the evolutionary conservation of the respective amino acid positions [20]. The HOPE server was used to predict the structural effects of the mutation [21]. In addition, we used the ConSeq server to analyze the evolutionary conservation of the structures of proteins [22].
Results
Computational analysis of the sequencing data revealed a total of 1,032 variants in coding and non-coding exons on chromosome X, or potentially affecting splice sites. Out of these, 405 protein changing mutations were either present in publicly available databases or were recurrent in our in-house dataset. The final set of 25 novel variants including those that were reported as heterozygous in publicly available databases, the recurrent pathogenic SLC16A2 mutation present in HGMD and the recurrent variants investigated for segregation in this study are listed in the Additional file 1: Table S1. In four families, D299, D174, D172 and D301 a novel mutation in an X-chromosomal gene was found to co-segregate with the clinical phenotype. In one family L107, a recurrent pathogenic mutation in an X chromosomal gene was identified. In one family D222, a novel missense mutation in a candidate XLID gene was identified. The clinical phenotypes and the identified mutations are described below. In the remaining eight families no functionally relevant candidate genes were found.
Clinical description of the patients and X-exome sequencing results
Family L107
Family L107 has three affected males (II-1, II-2 and II-9, Figure 1a), who presented with moderate to severe ID. Patients II-1 born in 1950 and II-2 born in 1954 never walked. They have spastic diplegia in the lower limbs, athetosis in the upper limbs, alalia and dysarthria. In contrast, the index patient (II-9) born in 1966 who is the half-brother could walk but lost his motor skills during early childhood. Patients II-2 and II-9 could speak a few words. No data was available from patient II-1 concerning speech. Clinical features are summarized in Table 1. Metabolic analyses, EEG, EMG and imaging were normal. The thyroid hormone levels for TSH and T4 from the index patient II-9 were within the normal range. Data about the thyroid hormone level for T3 was unavailable. The index patient (II-9) was previously analyzed for MECP2 aberration, as well as for mutations in ARX and CDKL5, all being normal. High-resolution, X-chromosome specific arrayCGH was also reported to be normal. X-exome sequencing of the index patient (II-9) identified a variant in SLC16A2, BCORL1 and GPR112, respectively. Variants in BC0RL1 and GPR112 were excluded as they were not clinically relevant. A missense mutation [NM_006517.4:c.1111C > T; p.Arg371Cys] in exon 4 of the SLC16A2 gene was identified and was compatible with the clinical phenotype. This mutation was also present in his half-brother (II-2) and his mother (I-2) was found to be carrier of this mutation. The oldest affected half-brother (II-1) did not participate in the mutation analysis study. According to PolyPhen-2, SIFT, PhD-SNP, SNPS&GO, and PANTHER, the mutation is predicted to be damaging. The ConSeq server also revealed that this residue is highly conserved with a score of 9. Based on HOPE [21], the altered amino acid has a change in charge and can disturb the ionic interactions with the other transmembrane helices. The mutant cysteine residue is more hydrophobic than the wild type arginine residue, possibly affecting the hydrophobic interactions within the core of the protein or with the membrane lipids and, as a result, disturbing the correct folding of the protein. The mutation was absent in 100 Finnish controls.
Figure 1.

Overview of the mutations reported in SLC16A2 gene in two Finnish families with Allan Herndon Dudley Syndrome. a) Family pedigree of L107 showing the inheritance of SLC16A2 mutation, open circles denote females; circles with a dot in the middle denote obligate carrier females, empty square denote males, the left half of the black squares denote affected males, the right half of the squares denote mutation positive males, crossed symbols denote deceased individuals, wt denote mutation negative subject, b) Sanger sequencing confirming the missense mutation c.1111C > T, c) Multiple species protein sequence alignment showing conservation of the mutated R371 residue, d) Family pedigree of D299 showing the inheritance of SLC16A2 mutation, e) Sanger sequencing confirming the frameshift insertion c.990_991insGCTGC.
Table 1.
Overview of the clinical features of the previous and present families with Allan-Herndon-Dudley syndrome
| Clinical findings | Clinical findings reported previously | D299 | L107 |
|---|---|---|---|
|
Short stature |
+ |
+ |
- |
|
Scoliosis |
+ |
+ |
+ |
|
Low weight |
+ |
+ |
+ |
|
Microcephaly |
+ |
+ |
- |
|
Muscle hypoplasia |
+ |
+ |
NA |
|
Hypotonia |
+ |
+ |
NA |
|
Contractures |
+ |
+ |
NA |
|
Dystonic movements |
+ |
+ |
+ |
|
Athetosis |
+ |
+ |
+ |
|
Intellectual disability |
+ |
+/S |
+/MO/S |
|
Absent speech |
+ |
+ |
+ |
|
Dysarthria/Limited speech |
+ |
+ |
+ |
|
Seizures |
+ |
+ |
NA |
|
Pectus excavatum |
+ |
+ |
NA |
|
Narrow long face |
+ |
- |
+ |
|
Round face |
+ |
+ |
- |
|
Valgus |
+ |
+ |
NA |
|
Hyperreflexia |
+ |
+ |
NA |
|
Simple ears |
+ |
+ |
- |
|
Cupped ears |
+ |
- |
+ |
| Spastic quadriplegia | + | + | - |
MO→Moderate, S→Severe, NA→No data available,+→clinical feature present, -→clinical feature absent.
Family D299
The index patient (III-3) is 16 years old and his two half-brothers (III-1, III-2) deceased at the ages of 21 years and 16 years respectively (Figure 1d). Clinical features are summarized in Table 1.
The three affected sons of family D299 had similar clinical features, characterized by severe psychomotor retardation, absent speech, seizures, hypotonia and contractures. The thyroid hormone profile of the index patient III-3 was T3-9 (Normal range: 2.6-5.7 pmol/L), T4-8.6 (Normal range: 9–19 pmol/L), TSH-2.16 (Normal range: 0.35-5 mU/L) and TGB-870 (Normal range: 233–490 nM/L). The patients have obtained thyroxin medication at the age of 2–3 years without any improvement.
X-exome sequencing of the index patient (III-3) revealed two novel variants, one in SLC16A2 and the other in ODZ1 (Additional file 1: Table S1). The 5 base-pair insertion identified in exon 3 of SLC16A2 leads to a frameshift and a premature stop codon [NM_006517.4:c.990_991insGCTGC; p.G334PfsX11] and segregated with the clinical phenotype. The mother (II-2) carries the mutation on one of her X-chromosomes. The mother's sister (II-5) was also tested and she was found not to be a carrier. This novel mutation in the SLC16A2 gene was absent in 100 Finnish controls.
Family D174
Family D174 has three affected males (III-7, III-8 and II-5, Figure 2a). The patients aged 36, 35 and 57 years respectively, are characterized by severe ID with autistic features, epilepsy, short stature and behavioral problems such as self injury and aggressive outbursts (Table 2).
Figure 2.

Overview of the missense variant reported in GRIA3 in a Finnish Family with XLID. a) Family pedigree showing the inheritance of the GRIA3 mutation, open circles denote females; circles with a dot in the middle denote obligate carrier females, empty square denote males, the left half of the black squares denote affected males, the right half of the squares denote mutation positive males, crossed symbols denote deceased individuals, wt denote mutation negative subject, b) Photographs of the affected males, c) Sanger sequencing confirming the mutation, d) Multiple species protein sequence alignment showing conservation of the mutated G630 residue.
Table 2.
Overview of clinical features of previously published patients with GRIA3 deletion/duplication and present patients with GRIA3 mutation
|
Gecz et al. [1999] [31] Female patient |
Wu et al. [2007] [34] Male patients |
|
|
|
|
Bonnet et al. [2009] [36] Male/male patient |
Chiyonobu et al. [2007] [35] Male patient |
D174 Male patient (III-7) |
D174 Male patient (III-8) |
D174 Male patient (II-5) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 1 | n = 3 | n = 2 | n = 4 | n = 2 | n = 2 | n = 2 | n = 1 | ||||
|
Type of change |
Translocation t(X;12) (q24;q15) |
Deletion |
Missense variant p.G833R |
Missense variant p.R631S |
Missense variant p.M706T |
Missense variant p.R450Q |
Partial duplication of GRIA3 |
Partial duplication of GRIA3 |
Missense variant p.G630R |
Missense variant p.G630R |
Missense variant p.G630R |
|
Feature |
|
|
|
|
|
|
|
|
|
|
|
|
Intellectual disability |
+ |
+ |
+ |
+ |
+ |
- |
+/+ |
+ |
+ |
+ |
+ |
|
Mild |
- |
- |
- |
- |
+ |
- |
- |
- |
- |
- |
- |
|
Moderate |
+ |
+ |
+ |
+ |
+ |
- |
- |
- |
- |
- |
- |
|
Severe |
- |
- |
- |
- |
- |
- |
+/+ |
+ |
+ |
+ |
+ |
|
Autistic features |
ND |
ND |
+ |
ND |
ND |
ND |
+/ND |
+ |
+ |
- |
+ |
|
Behaviour troubles |
+ |
ND |
+ |
ND |
ND |
ND |
+/+ |
+ |
+ |
+ |
+ |
|
Self injury |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
+ |
|
Aggressive outbursts |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
- |
|
Dysmorphic features |
ND |
ND |
ND |
ND |
ND |
ND |
+/+ |
+ |
+ |
+ |
+ |
|
Brachycephaly |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
+ |
|
Macrocephaly |
ND |
ND |
+ |
ND |
ND |
ND |
ND |
ND |
- |
- |
- |
|
Deep set eyes |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
+ |
|
Aesthenic habitus |
ND |
ND |
ND |
+ |
+ |
ND |
ND |
ND |
- |
- |
- |
|
Myclonic jerks |
ND |
ND |
+ |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
|
Hyporeflexia |
ND |
+ |
ND |
ND |
+ |
ND |
ND |
ND |
ND |
ND |
ND |
|
Prominent supraorbital ridges |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
+ |
|
Short stature |
ND |
ND |
ND |
ND |
ND |
ND |
-/- |
+ |
+ |
+ |
+ |
|
Epilepsy |
+ |
ND |
ND |
ND |
ND |
ND |
-/- |
- |
+ |
+ |
+ |
|
Inguinal hernia |
- |
ND |
ND |
ND |
ND |
ND |
+/- |
+ |
- |
- |
- |
|
Brain MRI abnormalities |
ND |
ND |
ND |
ND |
ND |
ND |
-/+ |
- |
ND |
ND |
ND |
|
Bowel occlusions |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
- |
- |
+ |
|
Malposition of feet |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
+ |
- |
|
Hydronephrosis |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
+ |
ND |
ND |
| Ren arcuatus | ND | ND | ND | ND | ND | ND | ND | ND | + | ND | ND |
ND→No data, +→clinical trait present, -→clinical trait absent.
X-exome sequencing of the index patient III-8 revealed a novel missense mutation [NM_000828.4:c.1888G > C; p.Gly630Arg] in exon 12 of the GRIA3 gene that segregated in the family (Additional file 1: Table S1). The mutation was absent in 135 Finnish controls. Using PolyPhen-2, SIFT, PhD-SNP, SNPS&GO and PANTHER, the mutation was predicted to be probably damaging. The ConSeq server also revealed that this residue is highly conserved with a score of 9. Missense changes in OR13H1, GPR112 and F8 did not segregate with the phenotype. The variants identified in TAF1 and LRCH2 segregated in the family. However, both variants turned out to be common single nucleotide variations as they were present in 3/88 Finnish control samples.
The wild type residue glycine of the p.Gly630Arg is neutral and the mutant residue arginine is positively charged. HOPE [21] predicted that this can lead to disturbances in the ionic interactions with the other transmembrane helices. The mutant residue is larger than the wild type residue, which can disturb either the contacts with the other transmembrane domains or with the lipid membrane. This can also affect the hydrophobic interactions within the core of the protein or with the membrane lipids. The wild type residue, glycine, is considered to be the most flexible of all residues and this flexibility might be vital for this protein’s function. It is predicted that this glycine residue at this position is needed to make a special backbone conformation or to facilitate movement of the protein.
Family D172
This family has three affected males in two generations (III-5, III-8 and IV-3, Figure 3a). X-exome sequencing of the index patient (III-8) revealed two novel variants, one in DLG3 and the other in ZBTB33 (Additional file 1: Table S1). The ZBTB33 variant was excluded because it did not co-segregate with the phenotype. A donor splice site mutation [NM_021120.3:c.357 + 1G > C] in intron 1 of the DLG3 gene segregates in the family. The affected males who presented with mild to moderate ID, normal growth, narrow thorax, molar hypoplasia, short up-slanting palpebral fissures, a high vaulted palate, and hypotonia. Daytime wetting has been a problem until adult age (Table 3). In addition to the aforementioned features the index patient (III-8) had an external strabismus in his right eye. Chromosomes (300bands), FRAXA, vacuolated lymphocytes, urinary metabolic screen, ophthalmology and hearing examinations showed normal results. Females II-2, III-2 and III-12 are heterozyous carriers of this mutation. Female III-2 has passed regular basic education and completed vocational schooling. Her younger son (IV-5) has epilepsy but is of normal intelligence and does not have the DLG3 mutation. Female II-2 is 77 years old, who is also a mutation carrier and has no cognitive or memory problems. All females showed a normal X-inactivation pattern in their blood lymphocytes (data not shown).
Figure 3.

Overview of the two splice donor mutations identified in DLG3 in two Finnish families. a) Pedigree of family D172 showing the inheritance of the DLG3 splice donor mutation c.357 + 1G > C, open circles denote females; circles with a dot in the middle denote obligate carrier females, empty square show males, the left half of the black squares denote affected males, the right half of the squares denote mutation positive males, crossed symbols denote deceased individuals, wt denote mutation negative subject, b) Sanger sequencing result showing the mutation in one affected male, c) Pedigree of family D301 showing the inheritance of the DLG3 splice donor mutation c.985 + 1G > C, d) Sanger sequencing result showing the mutation in one affected male, e) Schematic representation of the DLG3 gene, along with overview of previously reported mutations and mutations reported in the current study.
Table 3.
Comparison of the clinical features of the patients of two families with DLG3 mutation
| Family | D172 | Family D301 | ||||||
|---|---|---|---|---|---|---|---|---|
| |
III-5 |
III-8 |
IV-3 |
III-5 |
III-9 |
IV-3 |
V-1 |
V-2 |
|
Male |
Male |
Male |
Male |
Male |
Male |
Male |
Male |
|
|
Clinical features |
|
|
|
|
|
|
|
|
|
Intellectual disability |
+/MO |
+/M |
+/MO |
+/S |
+/S |
+/MO |
+/MO |
+/MO |
|
Delayed motor development |
+ |
ND |
+ |
+ |
ND |
+ |
+ |
+ |
|
Delayed speech development |
+ |
ND |
+ |
+ |
+ |
+ |
+ |
+ |
|
Eneuresis |
+ |
- |
+ |
+ |
- |
- |
+ |
- |
|
Strabismus |
- |
- |
- |
+ |
- |
+ |
+ |
- |
|
ADHD |
- |
- |
- |
ND |
ND |
+ |
+ |
+ |
|
Seizures |
- |
- |
- |
+ |
- |
- |
- |
- |
| Behavioural problems | - | - | + | + | - | + | - | - |
ND→No data, M→Mild, MO→Moderate, S→Severe, +→clinical feature present, -→clinical feature absent.
Family D301
Family D301 has five affected males in three generations (III-5, III-9, IV-3, V-1 and V-2, Figure 3c). In the index patient, V-1, etiological investigations such as urine metabolic screening, karyotype analysis, Fragile-X testing, FISH on 22q11.2 locus, creatinine kinase, lactate, carnitine, very long fatty acids, EEG and brain MRI were normal.
The phenotype consists of delayed motor and language development. Three males had strabismus and attention deficit hyperactivity disorder (ADHD). The males do not have other dysmorphic features except a bifid uvula in patients V-1 and V-2. Only one patient had experienced seizures in childhood. The cognitive performance in the affected males varied from severe to moderate ID, with ID being less severe in the younger generations (Table 3). Female III-6 participated in the cognitive evaluation at the age of 60 years at the level of moderate ID; she had not been evaluated earlier. She had received her basic and vocational education in a special school. She has not been able to work but has lived independently and needs assistance in all paperwork and contacts with authorities. The mother (IV-2) of patients V-1 and V-2 and their grandmother (III-2) graduated from regular schools and obtained vocational education.
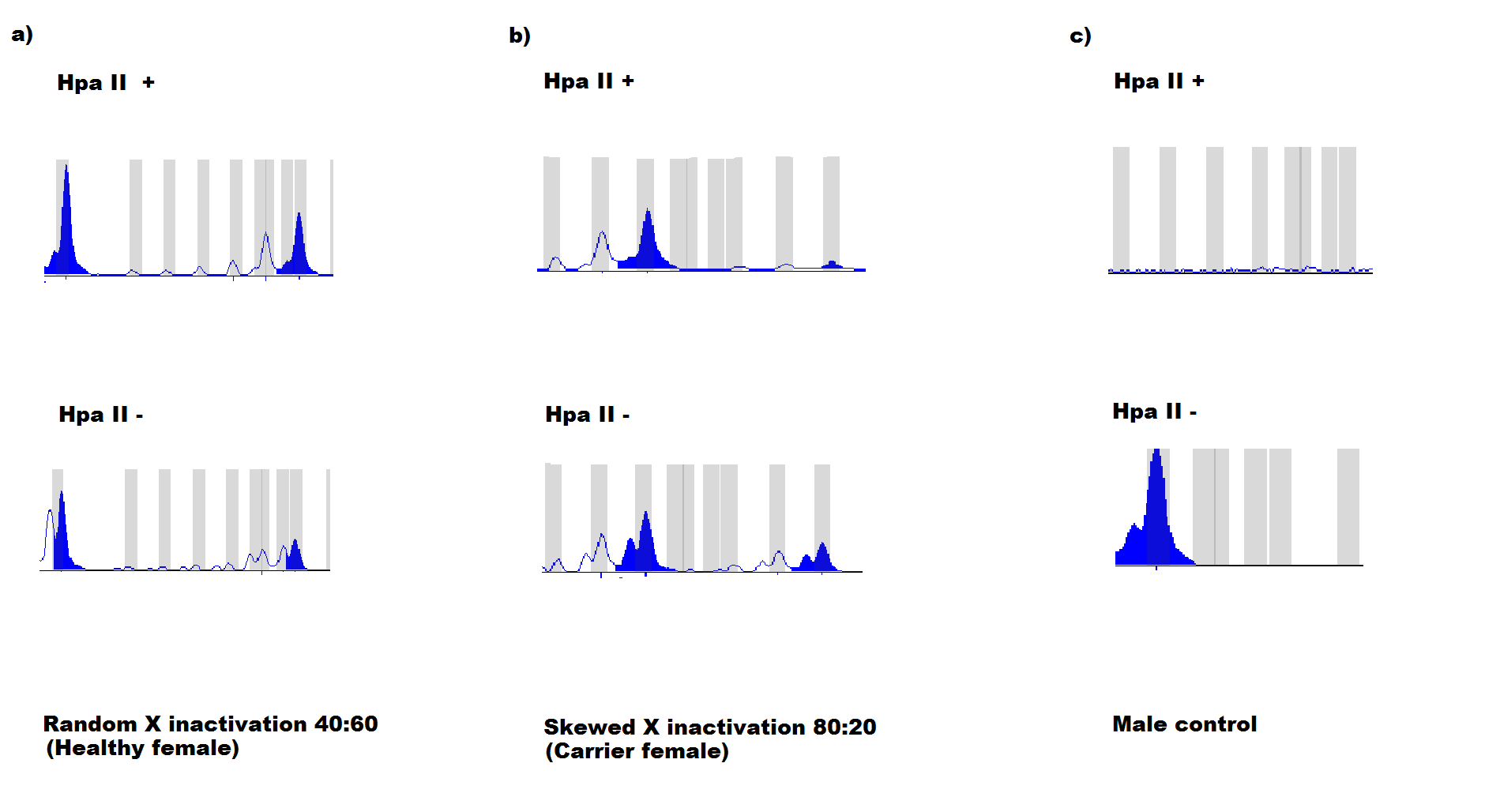
X-exome sequencing of the index patient (V-1) identified a novel donor splice site mutation [NM_021120.3:c.985 + 1G > C] in intron 6 of the DLG3 gene (Additional file 1: Table S1). One affected carrier female (III-6) had a skewed X-inactivation pattern of 80:20 (Additional file 2: Figure S1).
Family D222
Family D222 has three affected males who are half-brothers (III-1, III-2 and III-3, Figure 4a). The phenotype consists of small gestational age (SGA), hypospadia, mild aortic stenosis and leakage of the aortic valve, arcus aortae dexter, horseshoe kidney, slenderness and cup formed ear lobes. Their motor and language development were slightly delayed and they continue to have nocturnal enuresis up to school age. Two of them have ADHD. The cognitive performance is at the level of mild ID. The development of the youngest brother (III-4) is normal (Table 4).
Figure 4.

Clinical presentation of a novel syndrome in a Finnish family with XLID and segregation of the ZMYM3 missense mutation. a) Pedigree showing the inheritance of the ZMYM3 mutation in family D222, open circles show females, circles with a dot in the middle show obligate carrier females, empty square show males, the left half of the black squares show affected males, the right half of the squares show mutation positive males, crossed symbols denote deceased individuals, wt denote mutation negative subject, b) Photographs of the three affected males, c) Sanger sequencing confirming the mutation, d) Multiple species protein alignment showing conservation of the mutated R441 residue in ZMYM3.
Table 4.
Summary of clinical findings of patients with the ZMYM3 mutation
| Clinical trait | Patient III-1 | Patient III-2 | Patient III-3 |
|---|---|---|---|
|
Age |
15.5 y |
8.8 y |
7.3y |
|
Weight at birth |
2530 g |
2460 g |
3590 g |
|
Head circumference |
55.5 cm, −1 SD |
49.6 cm, −3 SD |
49.5 cm, −2.7 SD |
|
Hypospadia |
+ |
+ |
- |
|
Horseshoe kidney |
- |
+ |
- |
|
Enuresis nocturna |
+ |
+ |
rarely |
|
Large cupped formed ear lobes |
+ |
+ |
+ |
|
ADHD |
+ |
+ |
ND |
|
Arcus aortae dexter |
- |
+ |
- |
|
Bicuspid aortic valve |
+ |
+ |
- |
|
Sleep disorder |
+ |
+ |
+ |
| Intellectual disability | +/M | +/M | +/M |
M→Moderate, +→clinical trait present, -→clinical trait absent, SD→standard deviation.
X-exome sequencing of the index patient (III-1) identified novel variants in PTCHD1, RAB40A, ABCD1, COL4A5 and ZMYM3, respectively (Additional file 1: Table S1). PTCHD1 and RAB40A were excluded as a candidate gene because their variants did not segregate in the family (Additional file 1: Table S1). We suggest that the variant in COL4A5 is unlikely pathogenic because mutations in this gene have been previously associated with Alport syndrome [OMIM#301050].
The variant in ABCD1 was excluded as a causative mutation as mutations in ABCD1 previously have been linked to adrenoleukodystrophy [OMIM#300100]. The novel missense mutation [NM_201599.2:c.1321C > T; p.Arg441Trp] in exon 7 of the ZMYM3 gene was present in all three affected males and absent in their healthy brother. The mother (II-2) was found to be a carrier of this mutation. This mutation leads to substitution of the amino acid arginine with tryptophan (p.Arg441Trp) and was absent in 100 Finnish anonymous blood donors.
PolyPhen-2, SIFT, PhD-SNP, SNPS&GO and PANTHER predicted this mutation to be damaging. The ConSeq server also revealed that this residue is highly conserved with a score of 9. HOPE [21] predicted that the mutant residue is larger and more hydrophobic than the wild type residue. The amino acid substitution will lead to loss of hydrogen bonds in the core of the protein, thus preventing correct folding. The difference in the amino acid properties is likely to disturb a Zinc-finger domain known to bind DNA.
Family D175
This family has 9 affected males in three generations (II-1, II-13, III-2, III-5, III-11, III-13, III-14, IV-1 and IV-2, Figure 5a). The affected males are characterized by mild to moderate ID, normal growth, and dysmorphic facial features with prominent supraorbital ridges, deep set eyes, short philtrum, and prominent chin suggesting a novel syndrome (Figure 5b). One patient has epilepsy (III-5). Patients III-5, IV-1 and IV-2 have mild brain abnormalities in MRI (corpus callosum hypoplasia, mild cortical atrophy). Only limited clinical data is available for patients III-11, III-13 and III-14.
Figure 5.
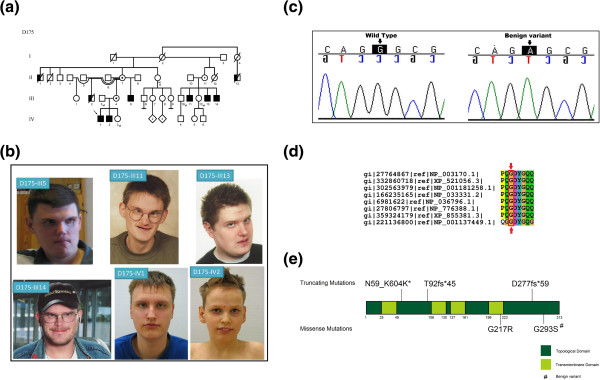
A clinical presentation of a novel syndrome in a Finnish family (D175) with XLID. a) Family pedigree showing the inheritance of the benign variant in SYP, open circles denote females, circles with a dot in the middle denote obligate carrier females, empty square denote males, the left half of the black squares denote affected males, the right half of the squares denote mutation positive males, crossed symbols denote deceased individuals, wt denote mutation negative subject, b) Photographs of the six affected males, c) Sanger sequencing confirming the benign variant, d) Multiple species protein alignment showing conservation of the mutated Gly293 residue in SYP, e) Schematic presentation of the SYP protein domains, location of the published mutations and the polymorphism identified in this study.
X-exome sequencing of the index patient (IV-1) identified a recurrent missense variant in exon 6 of the SYP gene [NM_003179.2:c.877G > A; p.Gly293Ser]. This missense variant was found in all the affected males. Variants in DMD, FAM47A, USP9X variants were not found in all of the affected males (Additional file 1: Table S1). Variant in FAM47B was not analyzed.
Discussion
In six families we identified five pathogenic mutations in three known XLID genes (SLC16A2, GRIA3 and DLG3) and one in the candidate XLID gene ZMYM3. In addition, two novel syndromes were identified, one with a novel missense mutation in a candidate XLID gene, ZMYM3[23] and the other with a previously reported benign variant in SYP in a large family (D175).
We identified two pathogenic mutations in SLC16A2. Mutations in this gene are known to cause Allan-Herndon-Dudley syndrome (AHDS; OMIM #300523). AHDS is typically characterized by developmental delay, poor head control, poor or no speech and muscle hypoplasia [24]. The clinical features of the patients investigated in this study are characteristic of AHDS. Previously, mutations in the SLC16A2 gene have been reported in more than 45 families across the world and one de novo translocation t(X;9)(q13.2;p24) has been reported in a female patient [24-29]. SLC16A2 belongs to the SLC16 gene family [30] and encodes the monocarboxylate transporter 8 (MCT8) protein, which is expressed in brain, liver, heart, intestine, placenta, kidney and thyroid. In the central nervous system MCT8 is present in neurons and astrocytes and is an essential thyroid hormone transporter, involved in the transport of thyroid hormone (TH) across the blood–brain barrier. The SLC16A2 gene consists of six exons and encodes a protein of 539 amino acids [24]. MCT8 contains twelve transmembrane domains (TMD) with intracellular amino- and carboxy-terminal domains [24]. In vitro studies showed that mutations in this gene lead to a reduced or absent supply of triiodothyronine (T3) to neurons [30]. The novel frameshift mutation p.G334PfsX11 identified in family D299 is located in the seventh TMD and the recurrent p.Arg371Cys amino acid substitution present in family L107 lies in the eighth TMD. The same missense mutation has previously been identified in a sporadic case (annotated as c.1333C > T, p.Arg445Cys using a different SLC16A2 isoform [29]. It has been previously shown that in a moderately affected individual with c.1333C > T missense mutation resulted in a more severe decrease in T3 uptake [29]. In the article by Capri et al. [29], the affected patient, aged 13 years, is able to walk with aid and can speak a few words. In family L107 the phenotype is variable as the two oldest patients (II-1 and II-2) never walked whereas the youngest II-9 could walk until early childhood. The patients in family L107 could speak some words. Based on the available data p.Arg445Cys mutation seems to be associated with variable phenotypes. To our knowledge, these are the first families with AHDS in the Finnish population.
We identified a missense mutation c.1888G > C (p.Gly630Arg) (RefSeq NM_000828.4) in the glutamate receptor, ionotropic, AMPA 3 (GRIA3) gene. GRIA3 belongs to a class of an alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)-sensitive glutamate receptor that operates as a ligand-gated ion channel in the central nervous system and has an essential role in excitatory synaptic transmission [31]. These receptors contain three functional domains – transmembrane, ligand binding, and receptor channel core [32]. Ionotropic glutamate receptors (iGluRs) are related to learning and memory [33]. In vitro functional studies on GRIA3 missense variants have shown that mutations in functional domains of GRIA3 are associated with kinetic changes in AMPA receptor function leading to significant reduction in iGluR3 channel function which is found to be linked with moderate ID [34,35]. Previously, four missense mutations, one whole gene deletion and three duplications have been reported in GRIA3[34-38]. One of the missense mutations (p.Arg631Ser) [34] affected the amino acid residue adjacent to the Gly630 mutated in the family described here. The patients of these families have facial dysmorphic features in common and these are different from the other reported patients with a mutation in GRIA3. The affected individuals of family D174 exhibit a severe phenotype. They are all severely intellectually disabled with behavioral disturbances. Three of the four previously reported patients with a GRIA3 mutation were moderately intellectually disabled, whereas one patient with a missense change (p.Met706Thr) had mild ID [34]. We conclude that there is wide variation in the severity of ID among patients with mutations in the GRIA3 gene. Further phenotype studies are needed to confirm the syndromic features underlying GRIA3 mutations.
The DLG3 gene, located at Xq13.1, encodes the synapse-associated protein SAP102 [39]. SAP102 is a member of the membrane-associated guanylate kinase (MAGUK) protein family [39,40]. SAP102 is highly expressed in both young and mature neurons and localizes to the postsynaptic density of excitatory synapses [41]. It is the first XLID gene to be associated to glutamate receptor-mediated postsynaptic signaling, a process which is crucial for the regulation of synaptic formation and plasticity in brain development [40]. Mutated DLG3 has been identified as a rare cause of XLID with, so far, only five families diagnosed, with a total of 17 affected males presenting with moderate to severe ID [41,42]. Of eleven mutation carrier women, one has mild ID and in another family a female carrier has mild ID and a history of seizures. These expressing females, however, do not have a skewed X-inactivation pattern [41,42]. Our families expand the phenotype spectrum of DLG3 mutations to mild ID.
The missense mutation c.1321C > T (p.Arg441Trp) (RefSeq NM_201599.2) is the first variant in the coding region of ZMYM3 reported to date. Previously, a chromosomal breakpoint in the 5’ UTR of ZMYM3 has been reported in an intellectually disabled female. In addition to ID, the patient had scoliosis and spotty hyperpigmentation of the skin. She also had slight facial asymmetry and clinodactyly [23]. However, the patients described in this study do not have any of the reported clinical features except for mild ID. Recently, a missense variant of unknown significance (c.356A > G (p.Gln119Arg)) in ZMYM3 was reported in the index patient of a family with XLID with four affected males in two generations. However, the patient also carries a missense change in the established XLID gene HCFC1[43].
The ZMYM3 gene is also referred to as ZNF261 and DXS6673E. ZMYM3 belongs to the MYM family. This gene contains 5 tandem repeats of a Cys-X2-Cys-X19–22-Cys-X3-Cys-X3-Cys-X13–19-Cys-X2-Cys-X19–25-Cys-X3-Cys motif [44]. ZMYM3 encodes a protein that is an integral component of histone deacetylase-containing multiprotein complexes. Further clarification is needed regarding the role of ZMYM3 as a XLID gene [45]. In addition, a previously reported benign polymorphism in SYP was identified in six affected males with a syndromic phenotype (Additional file 1: Table S1). Pathogenic mutations in SYP, including three truncating and one missense change (OMIM #300802) have previously been reported [11]. The patients in our study presented with mild to moderate ID, epilepsy, and common dysmorphic features. The missense benign variant c.877G > A (p.Gly293Ser) (RefSeq NM_003179.2) has been reported in the Exome Variant Server with a MAF frequency of T = 0.001/1 suggesting that SYP is not explaining the syndromic phenotype in this large family.
The diagnostic yield based on our current study of 14 families is 35%, of which five pathogenic mutations are likely to be disease causing. The role of ZMYM3 as a XLID gene is still questionable and can further be substantiated by the identification of additional families and mutations.
In eight families, work is ongoing as a functionally relevant mutation has not yet been identified. This might be due to technical limitations or a non-X-chromosomal origin of the disorders. In five of the mutation “negative” families the affected are brother pairs, in which linkage is less strong than for the other families.
Conclusions
The first X-exome sequencing study of Finnish families with intellectual disability revealed the first two Finnish families with Allan-Herndon-Dudley syndrome, one family with a novel mutation in GRIA3, and two families with novel mutations in DLG3. In addition, a novel missense mutation in the candidate XLID gene ZMYM3 cosegregated with a novel syndrome. A seventh family was found to have a benign missense variant SYP and a syndromic phenotype. Further studies are needed to find the causative mutation in this extended family.
This study demonstrates the power of exome sequencing in finding rare mutations underlying intellectual disability. It has been estimated that roughly 10% of the protein coding genes causing ID are located on the X-chromosome. Based on our research results, the likelihood of finding a genetic cause for the disease is increased in families with several affected males. We hypothesize that in some of the families, rare autosomal recessive mutations may be identified due to the characteristics of the founder population of Finland [46].
Web resources
The URLs for data presented are as follows:
dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
ConSeq, http://conseq.tau.ac.il/
PolyPhen-2, genetics.bwh.harvard.edu/pph2/
SIFT, sift.jcvi.org
Phd-SNP, http://snps.biofold.org/phd-snp/phd-snp.html
Refseq, http://www.ncbi.nlm.nih.gov/refseq/
SNPs &GO, http://snps.biofold.org/snps-and-go/snps-and-go.html
PANTHER, http://www.pantherdb.org/tools/csnpScoreForm.jsp?
HOPE, http://www.cmbi.ru.nl/hope/input
MERAP, http://www.sourceforge.net/projects/merap/files/MERAP20131101/
OMIM, http://www.ncbi.nlm.nih.gov/omim/
NHLBI Exome Sequencing Project (ESP)
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Abbreviations
XLID: X linked intellectual disability; MCT8: Monocarboxylate transporter 8; ADHD: Attention deficit hyperactivity disorder; AHDS: Allan-Herndon-Dudley syndrome.
Competing interests
We declare that we have no competing interest.
Authors’ contributions
IJ conceived the idea of the study. AS, KA, MS, HK, MP and MA were responsible for the clinical diagnosis of the patients. AKP designed the experiments, performed mutation analysis for the families, X-inactivation study and wrote the manuscript. HVE and GF analyzed family L107. FD and MAh were involved in the mutation analysis study of the family D174 and family D172. SAH, HU and VMK performed X-exome sequencing and bioinformatic analysis. All authors contributed to the final manuscript version.
Supplementary Material
List of all the variants identified using X-exome sequencing.
X inactivation pattern of an affected carrier female III-6 in Family D301.
{kind=link}
Contributor Information
Anju K Philips, Email: anju.philips@helsinki.fi.
Auli Sirén, Email: auli.siren@pshp.fi.
Kristiina Avela, Email: kristiina.avela@hus.fi.
Mirja Somer, Email: mirja.somer@fimnet.fi.
Maarit Peippo, Email: maarit.peippo@fimnet.fi.
Minna Ahvenainen, Email: minna.ahvenainen@hotmail.com.
Fatma Doagu, Email: fatma.doagu@helsinki.fi.
Maria Arvio, Email: maria.arvio@phsotey.fi.
Helena Kääriäinen, Email: helena.kaariainen@thl.fi.
Hilde Van Esch, Email: hilde.vanesch@uzleuven.be.
Guy Froyen, Email: guy.froyen@med.kuleuven.be.
Stefan A Haas, Email: haas@molgen.mpg.de.
Hao Hu, Email: hu@molgen.mpe.de.
Vera M Kalscheuer, Email: kalscheu@molgen.mpg.de.
Irma Järvelä, Email: irma.jarvela@helsinki.fi.
Acknowledgements
We thank all the patients and their families for their active participation and cooperation. The work was funded by the Sigrid Jusélius Foundation, Finnish Medical Association and by the Project GENCODYS (241995), which is funded by the European Union Framework Program 7 (FP7). Nathalie Fieremans and Melanie Bienek are acknowledged for their excellent technical assistance.
References
- Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- Schalok RL, Borthwick-Duffy SA, Bradley VJ, Buntinx WHE, Coulter DL, Craig EM, Gomez SC, Reeve A, Shogren KA, Snell ME, Spreat S, Tasse MJ, Thompson JR, Verdugo-Alonso MA, Yeager MH. AAIDD’s 11th edition of Intellectual Disability: Definition, Classification, and Systems of Support. 2012.
- de Brouwer AP, Yntema HG, Kleefstra T, Lugtenberg D, Oudakker AR, de Vries BB, van Bokhoven H, Van Esch H, Frints SG, Froyen G, Fryns JP, Raynaud M, Moizard MP, Ronce N, Bensalem A, Moraine C, Poirier K, Castelnau L, Saillour Y, Bienvenu T, Beldjord C, des Portes V, Chelly J, Turner G, Fullston T, Gecz J, Kuss AW, Tzschach A, Jensen LR, Lenzner S. et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat. 2007;28:207–208. doi: 10.1002/humu.9482. [DOI] [PubMed] [Google Scholar]
- Bassani S, Zapata J, Gerosa L, Moretto E, Murru L, Passafaro M. The neurobiology of X-linked intellectual disability. Neuroscientist. 2013;19:541–552. doi: 10.1177/1073858413493972. [DOI] [PubMed] [Google Scholar]
- Willemsen M, Kleefstra T. Making headaway with genetic diagnostics of intellectual disabilities. Clin Genet. 2013;85:101–110. doi: 10.1111/cge.12244. [DOI] [PubMed] [Google Scholar]
- Ellison JW, Rosenfeld JA, Shaffer LG. Genetics basis of intellectual disability. Annu Rev Med. 2013;64:441.450. doi: 10.1146/annurev-med-042711-140053. [DOI] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Schuurs-Hoeijmakers JH, Vulto-van Silfhout AT, Vissers LE, van de Vondervoort II, van Bon BW, de Ligt J, Gilissen C, Hehir-Kwa JY, Neveling K, Del Rosario M, Hira G, Reitano S, Vitello A, Failla P, Greco D, Fichera M, Galesi O, Kleefstra T, Greally MT, Ockeloen CW, Willemsen MH, Bongers EM, Janssen IM, Pfundt R, Veltman JA, Romano C, Willemsen MA, van Bokhoven H, Brunner HG. et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. 2013;50:802–811. doi: 10.1136/jmedgenet-2013-101644. [DOI] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weissmann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi SG, Cohen M, Fattahi Z. et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H. et al. Range of genetic mutations associated with severe non syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O’Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S. et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Hu H, Wrogemann K, Kalscheuer V, Tzschach A, Richard H, Haas SA, Menzel C, Bienek M, Froyen G, Raynaud M, Van Bokhoven H, Chelly J, Ropers H, Chen W. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 2009;3:41–49. doi: 10.1007/s11568-010-9137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2002;22:2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large scale gene function analysis with the PANTHER classification system. Nat Protocols. 2013;8:1551–1556. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 2007;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N. ConSeq: The Identification of Functionally and Structurally Important Residues in Protein Sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- van der Maarel SM, Scholten IH, Huber I, Philippe C, Suijkerbuijk RF, Gilgenkrantz S, Kere J, Cremers FP, Ropers HH. Cloning and characterization of DXS6673E, a candidate gene for X-linked mental retardation in Xq13.1. Hum Mol Genet. 1996;5:887–897. doi: 10.1093/hmg/5.7.887. [DOI] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005;77:41–53. doi: 10.1086/431313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frints SG, Lenzner S, Bauters M, Jensen LR, Frints SG, Lenzner S, Bauters M, Jensen LR, van Esch H, Des Portes V, Moog U, Macville MV, van Roozendaal K, Schrander-Stumpel CT, Tzschach A, Marynen P, Fryns JP, Hamel B, van Bokhoven H, Chelly J, Beldjord C, Turner G, Gecz J, Moraine C, Raynaud M, Ropers HH, Froyen G, Kuss AW. MCT8 mutation analysis and identification of the first female with Allan-Herdon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet. 2008;16:1029–1037. doi: 10.1038/ejhg.2008.66. [DOI] [PubMed] [Google Scholar]
- Namba N, Etani Y, Kitaoka T, Nakamoto Y, Nakacho M, Bessho K, Miyoshi Y, Mushiake S, Mohri I, Arai H, Taniike M, Ozono K. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. 2008;167:785–791. doi: 10.1007/s00431-007-0589-6. [DOI] [PubMed] [Google Scholar]
- Capri Y, Friesema EC, Kersseboom S, Touraine R, Monnier A, Eymard-Pierre E, Des Portes V, De Michele G, Brady AF, Boespflug-Tanguy O, Visser TJ, Vaurs-Barriere C. Relevance of different cellular models in determining the effects of mutations on SLC16A2/MCT8 thyroid hormone transporter function and genotype-phenotype correlation. Hum Mut. 2013;34:1018–25. doi: 10.1002/humu.22331. [DOI] [PubMed] [Google Scholar]
- Karapanou O, Papadimitriou A. Thyroid hormone transporters in the human. Hormones. 2011;10:270–279. doi: 10.14310/horm.2002.1318. [DOI] [PubMed] [Google Scholar]
- Gécz J, Barnett S, Liu J, Hollway G, Donnelly A, Eyre H, Eshkevari HS, Baltazar R, Grunn A, Nagaraja R, Gilliam C, Peltonen L, Sutherland GR, Baron M, Mulley JC. Characterization of the human glutamate receptor subunit 3 gene (GRIA3), a candidate for bipolar disorder and nonspecific X-linked mental retardation. Genomics. 1999;62:356–368. doi: 10.1006/geno.1999.6032. [DOI] [PubMed] [Google Scholar]
- Kuusinen A, Arvola M, Keinänen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327–32. doi: 10.1002/j.1460-2075.1995.tb00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wo ZG, Oswald RE. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995;18:161–168. doi: 10.1016/0166-2236(95)93895-5. [DOI] [PubMed] [Google Scholar]
- Wu Y, Arai AC, Rumbaugh G, Srivastava AK, Turner G, Hayashi T, Suzuki E, Jiang Y, Zhang L, Rodriguez J, Boyle J, Tarpey P, Raymond FL, Nevelsteen J, Froyen G, Stratton M, Futreal A, Gecz J, Stevenson R, Schwartz CE, Valle D, Huganir RL, Wang T. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci U S A. 2007;104:18163–18168. doi: 10.1073/pnas.0708699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiyonobu T, Hayashi S, Kobayashi K, Morimoto M, Miyanomae Y, Nishimura A, Nishimoto A, Ito C, Imoto I, Sugimoto T, Jia Z, Inazawa J, Toda T. Partial tandem duplication of GRIA3 in a male with mental retardation. Am J Med Genet. 2007;143A:1448–1455. doi: 10.1002/ajmg.a.31798. [DOI] [PubMed] [Google Scholar]
- Bonnet C, Leheup B, Béri M, Philippe C, Gregoire MJ, Jonveaux P. Aberrant GRIA3 Transcripts With Multi-Exon Duplications in a Family With X-Linked Mental Retardation. Am J Med Genet. 2009;149A:1280–1289. doi: 10.1002/ajmg.a.32858. [DOI] [PubMed] [Google Scholar]
- Bonnet C, Masurel-Paulet A, Khan AA, Beri-Dexheimer M, Callier P, Mugneret F, Philippe C, Thauvin-Robinet C, Faivre L, Jonveaux P. Exploring the potential role of disease-causing mutation in a gene desert: duplication of noncoding elements 5′ of GRIA3 is associated with GRIA3 silencing and X-linked intellectual disability. Hum Mutat. 2012;33:355–358. doi: 10.1002/humu.21649. [DOI] [PubMed] [Google Scholar]
- Philippe A, Malan V, Jacquemont ML, Boddaert N, Bonnefont JP, Odent S, Munnich A, Colleaux L, Cormier-Daire V. Xq25 duplications encompassing GRIA3 and STAG2 genes in two families convey recognizable X-linked intellectual disability with distinctive facial appearance. Am J Med Genet A. 2013;161:1370–1375. doi: 10.1002/ajmg.a.35307. [DOI] [PubMed] [Google Scholar]
- Smith SA, Holik P, Stevens J, Mazoyer S, Melis R, Williams B, White R, Albertsen H. Isolation of a gene (DLG3) encoding a second member of the discs-large family on chromosome 17q12-q21. Genomics. 1996;31:145–150. doi: 10.1006/geno.1996.0025. [DOI] [PubMed] [Google Scholar]
- Müller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron. 1996;17:255–265. doi: 10.1016/S0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- Tarpey P, Parnau J, Blow M, Woffendin H, Bignell G, Cox C, Cox J, Davies H, Edkins S. Holden. Mutations in the DLG3 gene cause nonsyndromic X-linked mental retardation. Am J Hum Genet. 2004;75:318–324. doi: 10.1086/422703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanni G, van Esch H, Bensalem A, Saillour Y, Poirier K, Castelnau L, Ropers HH, de Brouwer AP, Laumonnier F, Fryns JP, Chelly P. A novel mutation in the DLG3 gene encoding the synapse-associated protein 102 (SAP102) causes non-syndromic mental retardation. Neurogenetics. 2010;11:251–255. doi: 10.1007/s10048-009-0224-y. [DOI] [PubMed] [Google Scholar]
- Huang L, Jolly LA, Willis-Owen S, Gardner A, Kumar R, Douglas E, Shoubridge C, Wieczorek D, Tzschach A, Cohen M. A Noncoding, Regulatory Mutation Implicated HCF1 in Nonsyndromic Intellectual Disability. Am J Hum Genet. 2012;91:694–702. doi: 10.1016/j.ajhg.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Hamoudi R, Lu YJ, Cooper C, Shipley J. Cloning and mapping of members of the MYM family. Genomics. 1999;60:244–247. doi: 10.1006/geno.1999.5918. [DOI] [PubMed] [Google Scholar]
- Piton P, Redin C, Mandel JL. XLID-Causing Mutations and Associated Genes Challenged in Light of Data From Large-Scale Human Exome Sequencing. Am J Hum Genet. 2013;93:368–383. doi: 10.1016/j.ajhg.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8:1913–1923. doi: 10.1093/hmg/8.10.1913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of all the variants identified using X-exome sequencing.
X inactivation pattern of an affected carrier female III-6 in Family D301.