

Abstract
In rats over-expressing SOD1G93A, ventilation is preserved despite significant loss of respiratory motor neurons. Thus, unknown forms of compensatory respiratory plasticity may offset respiratory motor neuron cell death. Although mechanisms of such compensation are unknown, other models of respiratory motor plasticity may provide a conceptual guide. Multiple cellular mechanisms give rise to phrenic motor facilitation; one mechanism requires spinal serotonin receptor and NADPH oxidase activity whereas another requires spinal adenosine receptor activation. Here, we studied whether these mechanisms contribute to compensatory respiratory plasticity in SOD1G93A rats. Using plethysmography, we assessed ventilation in end-stage SOD1G93A rats after: 1) serotonin depletion with parachlorophenylalanine (PCPA), 2) serotonin (methysergide) and A2A (MSX-3) receptor inhibition, 3) NADPH oxidase inhibition (apocynin), and 4) combined treatments. The ability to increase ventilation was not decreased by individual or combined treatments; thus, these mechanisms do not maintain breathing capacity at end-stage motor neuron disease. Possible mechanisms giving rise to enhanced breathing capacity with combined treatment in end-stage SOD1G93A rats are discussed.
Keywords: plethysmography, compensatory plasticity, respiration, facilitation
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative disease that involves loss of both upper and lower motor neurons. Once diagnosed, expected survival is 3-5 years, and death most frequently results from respiratory failure (Lechtzin et al., 2002; Bourke et al., 2001; Lyall et al., 2001). Whereas more than 90% of ALS cases are sporadic, ~10% are linked to defined genetic mutations (Boillée et al., 2006). Frequently studied models of ALS include rats and mice over-expressing human mutated forms of the copper-zinc superoxide dismutase (SOD-1) gene (Rosen et al., 1993; Gurney et al., 1994; Wong et al., 1995; Bruijn et al., 1997; Nagai et al., 2001; Howland et al., 2002; Wong et al., 2002; Wang et al., 2003), including hSOD1G93A (Rosen et al., 1993; Gurney et al., 1994; Howland et al., 2002), hSOD1G37R (Wong et al., 1995), hSOD1G85R (Bruijn et al., 1997), hSOD1Quad (Wang et al., 2003), and hSOD1H46R (Nagai et al., 2001). These rodent models recapitulate important clinical and patho-physiological features of ALS, most notably delayed and progressive loss of spinal motor neurons (Boillée et al., 2006).
In hSOD1G93A mice, ventilation is maintained until late in disease progression; profound ventilatory failure then occurs over a two day period, and the mouse dies (Tankersly et al., 2007). hSOD1G93A rats maintain breathing capacity during maximal chemosensory stimulation, despite ~60-80% loss of phrenic, and ~65% loss of ventral thoracic (T5) motor neurons (Nichols et al., 2013a). We hypothesize that compensatory respiratory plasticity offsets the loss of respiratory motor neurons, thereby preserving breathing capacity until such compensation is no longer possible (i.e. motor neuron loss is too great to overcome). Compensatory plasticity may involve anatomical and/or functional plasticity in the CNS, neuromuscular junction and/or shifts between the inspiratory motor pools (eg. diaphragm versus accessory inspiratory muscles).
Mechanisms preserving breathing capacity despite impressive respiratory motor neuron loss are unknown. Here, we focus on the potential role of specific CNS neurochemicals known to be involved in other forms of phrenic motor plasticity.
Phrenic long-term facilitation (pLTF) following acute intermittent hypoxia (AIH) is a well studied model of phrenic motor facilitation (pMF; Dale-Nagle et al., 2010; Hayashi et al., 1993; Bach and Mitchell 1996). pLTF is serotonin and NADPH oxidase dependent (Kinkead and Mitchell 1999; Fuller et al., 2001; MacFarlane and Mitchell, 2007; MacFarlane and Mitchell, 2008a; MacFarlane and Mitchell, 2008b; MacFarlane and Mitchell, 2009; MacFarlane et al., 2009), and requires new synthesis of brain derived neurotrophic factor (Baker-Herman et al., 2004). pMF can be pharmacologically-induced via spinal, episodic application of serotonin, and 5-HT2A, 5-HT2B or 5-HT7 receptor agonists (MacFarlane and Mitchell, 2007; MacFarlane and Mitchell, 2008a; Hoffman and Mitchell, 2011; MacFarlane and Mitchell, 2009), as well as adenosine 2A receptor agonists (A2A; Golder et al., 2008). Distinct cellular cascades give rise to pMF following activation of Gq (5-HT2A/B) versus Gs (5-HT7 and A2A) protein coupled metabotropic receptors (Figure 1); these mechanisms are referred to as the Q (Gq) and S (Gs) pathways to pMF, respectively (Dale-Nagle et al., 2010).
Figure 1.

Working model of phrenic motor facilitation (pMF). pMF is initiated by multiple Gq (5-HT2A/B) or Gs (5-HT7 and A2A) protein coupled metabotropic receptors (the Q and S pathways, respectively). NADPH oxidase is necessary for the Q pathway to pMF. Here, we have given PCPA (tryptophan hydroxylase inhibitor), with and without methysergide (broad spectrum 5-HT antagonist), with and without apocynin (NADPH oxidase inhibitor), and/or MSX-3 (adenosine A2A receptor antagonist) in an effort to identify Q and S pathway contributions to the mechanism of compensatory respiratory plasticity in motor neuron disease. By blocking these mechanisms, we originally hypothesized that ventilatory capacity would decrease in end-stage SOD1G93A rats.
NADPH oxidase activity is necessary for AIH- and serotonin-induced pMF, most likely via inhibition of okadaic-acid sensitive protein phosphatases (MacFarlane and Mitchell, 2007; MacFarlane and Mitchell, 2008a; MacFarlane and Mitchell, 2008b; MacFarlane and Mitchell, 2009; MacFarlane et al., 2009; Figure 1).
The ability of serotonin and adenosine receptors to elicit pMF suggests that these neurochemicals have the potential to underlie spontaneous compensatory respiratory plasticity in motor neuron disease. We hypothesized that compensatory respiratory plasticity in end-stage hSOD1G93A mutant rats can be achieved through either or both of these cellular cascades. Thus, we predicted that ventilatory capacity would be undermined in end-stage SOD1G93A rats following inhibition of serotoninergic, adenosinergic and/or NADPH oxidase function. Ventilatory capacity was assessed during maximal chemoreceptor stimulation in a whole body plethysmograph after: 1) systemic serotonin depletion with the tryptophan hydroxylase inhibitor, parachlorophenylalanine (PCPA); 2) serotonin receptor inhibition with the broad spectrum 5-HT receptor antagonist, methysergide; 3) NADPH oxidase inhibition with apocynin; and 4) adenosine A2A receptor inhibition with MSX-3 (Figure 1). These treatments were administered alone, and in combination, to test the hypothesis that they would diminish ventilatory capacity in end-stage SOD1G93A rats.
Contrary to predictions, no treatment decreased ventilatory capacity. Ventilatory capacity was unaffected by PCPA, apocynin or MSX-3 alone or by combined PCPA + methysergide or PCPA + MSX-3 in end-stage SOD1G93A rats; surprisingly, ventilatory capacity actually appeared to increase with combined apocynin + MSX-3 + methysergide. These results provide no evidence that serotonergic, adenosinergic or NADPH oxidase dependent mechanisms are necessary to maintain ventilatory capacity at end-stage motor neuron disease, although they do not rule out a role in initiating mechanisms of compensatory respiratory plasticity. A preliminary report of these findings was previously published (Nichols et al., 2009).
2. Methods
2.1 Animals
Experiments were conducted on Taconic adult male rats with transgenic sires overexpressing the human SOD1G93A gene (Taconic Laboratories, Germantown, NY) bred to female wildtype Taconic Sprague Dawley rats. Heterozygous SOD1G93A progeny were identified with polymerase chain reaction (PCR) of tail DNA with primers specific for hSOD1. Only male rats showing disease onset ~120-140 days were used as breeders in order to minimize genetic drift in the colony that could increase variability in age at the onset of symptoms and the age at end-stage. Seven groups of rats (identified in next section) were used for this study; all groups included the heterozygous progeny age-matched to progeny with SOD-1 negative (wild-type or WT) rats. All rats were maintained on a 12:12 light:dark cycle and had free access to food and water. At approximately 120-140 days, mutant (MT) rats began to show signs of muscle weakness, weight loss, and gait changes. At approximately 150-180 days, MT rats show gradual fore- and hind-limb paresis, followed by paralysis. Rats were considered end-stage when they had lost more than 20% of their peak body weight (WT rats = 495±9g; MT rats = 360±6g). When body mass of MT rats decreased by 15%, MT and WT littermates were scheduled for experiments as described below. All procedures involving rats were approved by the Institutional Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin, and were in agreement with standards set forth in the National Institutes of Health Guide (NIH) for Care and Use of Laboratory Animals. The University of Wisconsin is accredited by AAALAC and is covered by NIH Assurance (A3368-01.).
2.2 Drugs and rat treatment groups
Seven MT groups were studied and compared to age-matched WT littermates. After MT rats had decreased by at least 15% in body mass, MT and WT groups were given either: 1) PCPA (300 mg/kg IP; MT: n = 3, WT: n = 3) for three days, 2) PCPA (for two days) + methysergide (4 mg/kg IP; given on the day of study, which was the third day after the 15% decrease in body mass; MT: n = 3, WT: n = 3), 3) MSX-3 (3 mg/kg IP; MT: n = 3, WT: n = 3; given the day of study), 4) PCPA (for two days) + MSX-3 (MT: n = 3, WT: n = 3; given the day of study), 5) apocynin (80 μg/mL in water supply for three days; MT: n = 2, WT: n = 2), 6) methysergide (given the day of study) + MSX-3 (given the day of study) + apocynin (MT: n = 3, WT: n = 3; in water supply for three days) or 7) vehicle injections (0.9% NaCl, 7.5 ml/kg IP; MT: n = 9, WT: n = 8; for three days). Whole body serotonin depletion with PCPA and serotonin receptor inhibition with methysergide block pLTF (Millhorn et al., 1980; Eldridge and Millhorn, 1986; Bach and Mitchell, 1996; McGuire et al., 2004; Ling, 2008; Figure 1), as does NADPH oxidase inhibition with apocynin (MacFarlane et al. 2009). Although MSX-3 enhances AIH-induced pLTF (Hoffman et al., 2010), it suppresses the S pathway to pMF (Golder et al., 2008; Figure 1). The doses of apocynin (given in water supply) and MSX-3 (IP) used here have been shown to be selective (Schindler et al., 2005; Hougee et al., 2006). All chemicals were purchased from Sigma (St. Louis, MO), except where indicated.
2.3 Plethysmography
Three days after MT rats lost 15% of their body mass, both WT and MT rats were placed in a whole-body flow through plethysmograph (BUXCO Electronics, Troy, NY). This technique allows for quantitative measurement of ventilation in freely-behaving animals while simultaneously altering inspired gas concentrations. The system was calibrated, the rats were weighed, and their body temperature was measured with a rectal thermometer (Type T Themocouple Thermometer, Model 600-1020, Barnant Company, Barrington, IL) before the rats were placed in the plethysmograph chamber (~2 L volume). Ventilation was assessed in WT (n=25) and MT (n=26) rats. The rats acclimated to the chamber while breathing room air (21% O2, balance N2; flushed at ~3 liters/min). Recording commenced once the rats were quiet but awake, and ventilatory measurements were then made during baseline conditions, before exposing them to hypoxic (10.5% O2, balance N2; 20 min), hypercapnic (21% O2, 7% CO2, balance N2; 20 min) and hypoxic+hypercapnic gas mixtures (10.5% O2/7% CO2; 20 min). A pressure calibration signal, plethysmograph temperature, rat body temperature, ambient and chamber pressures, and rat body mass were used to calculate breath-by-breath tidal volume (VT; Drorbaugh and Fenn, 1955; Jacky, 1978), respiratory frequency, mean inspiratory flow (VT/TI) and minute ventilation (). Data were rejected if there was evidence of pressure fluctuations caused by gross body movements or sniffing behavior. At the conclusion of the study, rats were removed from the chambers and their body temperatures recorded. Data were averaged over the last 10 min of each exposure period.
2.4 Histology and motor neuron counting
WT and MT rats were deeply anesthetized with Beuthanasia®-D (100μg/g IP; Schering-Plough Animal Health) and perfused transcardially with 4% paraformaldehyde (Fisher Scientific, Pittsburgh, PA) in 0.1M phosphate buffered saline (PBS, pH~7.4). The C4-C5 cervical spinal cord was isolated, post-fixed overnight in 4% paraformaldehyde, and cryoprotected in 30% sucrose at 4°C. Transverse sections (40 μm) were cut using a freezing microtome (Leica SM 200R, Germany) and processed for motor neuron counting. Sections were mounted on coated-glass slides (Fisherbrand Colorfrost® / Plus Microscope Slides, Fisher Scientific, Pittsburgh, PA) and air dried. They were then stained in 0.1% cresyl-violet for 15 min, rinsed in distilled water, dehydrated through graded alcohol (70% to 100%), cleared in xylene, and cover-slipped using Eukitt (Electron Microscope Science, PA).
For morphometric analysis, C4 phrenic motor neurons were manually counted. Every 8th section was selected to count neurons. Putative phrenic motor neurons were defined as a cluster of large, medio-lateral neurons in the cervical ventral horn (Boulenguez et al., 2007). Only intact motor neurons with an intact soma and nucleus were counted. Six sections from each animal were used in this study as described previously (Nichols et al. 2013a). Briefly, labeled neurons (0.1% cresyl-violet stained positive-neurons) were counted rostro-caudal in the phrenic area as the average from both sides. The number of 0.1% cresyl-violet-positive motor neurons in the C4 phrenic motor nucleus were counted from all rats in each group and compared between MT and WT rats.
2.5 Statistical analysis
For both plethysmography and histology, values are reported as means ± 1 S.E.M. Plethysmography data are not normalized to body mass. However, when normalized in this way (per 100g body mass), all fundamental conclusions remained the same. Our arguments for presenting non-normalized data in this experimental model are outlined in an earlier publication (Nichols et al., 2013a). Plethysmography data were analyzed via two-way repeated measures ANOVA, with group (WT vs. MT) and treatment level (baseline, hypoxia, etc.) as factors (SigmaStat v.2.03, Systat Software, Inc., Chicago, IL); Tukey’s post hoc test was used to identify individual differences. Motor neuron counts were compared between groups via one-way ANOVA (Systat Software, Inc., Chicago, IL) and Tukey’s post hoc tests. P<0.05 was considered statistically significant.
3. Results
3.1 Phrenic motor neurons are decreased in MT rats vs. WT littermates
Regardless of treatment, MT rats had a fewer phrenic motor neurons in the C4 spinal segment versus WT littermates in every group, and there were no apparent differences among groups (WT rats were: 197±6 sham, 196±11 PCPA, 185±17 PCPA + methy, 198±11 MSX-3, 183±3 MSX-3 + PCPA, and 191±6 Apo + MSX-3 + methy, demonstrating good consistency among WT rat groups; in contrast, MT rats were: 24±2 sham, 30±9 PCPA, 37±4 PCPA + methy, 41±9 MSX-3, 27±4 MSX-3 + PCPA, and 43±10 Apo + MSX-3 + methy, demonstrating consistency among groups with no significant differences between them; p<0.05 comparing WT vs. MT in all groups, and p>0.05 when comparing treatment groups within WT or MT). Thus, motor neuron counts for all WT and all MT rats were grouped together in the final analysis, revealing 17±1% phrenic motor neuron survival in MT vs. WT rats at disease end-stage (P < 0.05; Figure 2). Consistent with our previous report, phrenic motor neurons degenerate severely at this defined disease end-stage in MT rats (Nichols et al., 2013a). Further, the extent of degeneration was equivalent among rat treatment groups.
Figure 2.
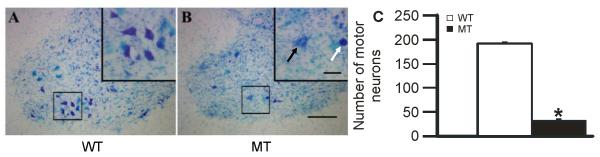
A and B: Photomicrographs (10X) depicting representative cresyl violet stained sections from the C4 spinal ventral horn; these sections contain the highest density of phrenic motor neurons (framed regions on which morphometric analyses were carried out). Images are from an age-matched, wild-type littermate (WT; A), and an SOD1G93A mutant (MT) rat at disease end-stage (B). The region surrounding phrenic motor neurons is magnified in the upper right hand panels (40X). Notice the number and shape of healthy pyramidal-shaped putative phrenic motor neurons in the WT littermate (A) versus the paucity of surviving phrenic motor neurons in the end-stage MT rat (B). The phrenic motor neuron in the MT rat looks much larger (swollen) and did not appear to be healthy (depicted by black arrow). A dying/dead phrenic motor neuron is depicted by the white arrow. C: The white and black bars represent the number of surviving phrenic motor neurons in WT littermates (n = 25) and MT rats (n = 26), respectively. MT rats had a significantly lower number of phrenic motor neurons versus WT littermates in all experimental groups. Values are means ± 1 SEM and * = p < 0.05. Scale bar in C4 spinal ventral horn = 200 μm, small panel= 50 μm.
3.2 Ventilatory response in sham treated rats
We determined the impact of motor neuron disease on ventilation during both normoxia and chemoreflex activation. Ventilation was monitored (VT, frequency, and VT/TI) in sham-treated WT and MT rats at baseline, and during hypoxia (HX), hypercapnia (HC), and hypoxia + hypercapnia (HX + HC). There were significant differences between treatment levels and treatment groups; there were no significant interactions between treatment level and group in any variable. VT was not affected in MT rats during baseline or with chemoreflex activation versus sham WT rats (Figure 3A; p>0.05). However, frequency was decreased in MT rats in all conditions (Figure 3B; p<0.05). Frequency may have decreased in sham MT rats because the mutation had direct effects on brainstem mechanisms of rhythm generation or indirect effects via afferent feedback to these centers. was significantly impaired during HX + HC in MT rats (619 ± 72 ml/min in sham MT rats vs. 776 ± 180 in sham WT rats; p<0.05; Figure 3C). In contrast, VT/TI was not impaired in sham MT rats in any condition studied (Figure 3D; p>0.05).
Figure 3.

Tidal volume (VT), frequency, minute ventilation () and mean inspiratory flow (VT/TI) in sham wild-type (WT; triangles) and mutant (MT; circles) rats during baseline, hypoxia (HX), hypercapnia (HC) and hypoxia plus hypercapnia (HX + HC). Notice that frequency is impaired in sham MT rats compared to sham WT rats; was impaired in sham MT rats versus WT rats only with HX + HC. In contrast, neither VT nor VT/TI were impaired in MT rats in any condition. Values are means ± SEM and * = from sham MT, # = from baseline, + = from HX, and $ = from HC where all p < 0.05.
For all other treatment groups, ventilatory variables did not differ significantly between HX, HC, and HX + HC. Only baseline and HX + HC were compared in Figures 4-6; variables in all conditions are reported in Tables 1-4.
Figure 4.

Tidal volume (VT), frequency, minute ventilation () and mean inspiratory flow (VT/TI) during baseline and HX + HC in wild-type (WT; black bars) and mutant (MT; white bars) rats treated with PCPA alone, or PCPA + methysergide (methy) versus sham treated rats. In all groups, HX + HC values were significantly larger than baseline values. PCPA alone had no effect on VT, frequency, or VT/TI in WT or MT rats during baseline or HX + HC versus sham treated rats. PCPA + methy had no effect on VT, or VT/TI during baseline or HX + HC in WT or MT rats versus sham treated rats. PCPA + methy decreased frequency significantly in WT rats during baseline and HX + HC versus sham treated WT rats. Values are means ± 1 SEM and = + = from sham WT where all p < 0.05.
Figure 6.

Tidal volume (VT), frequency, minute ventilation () and mean inspiratory flow (VT/TI) during baseline and HX + HC in wild-type (WT; black bars) and mutant (MT; white bars) rats treated with apocynin (Apo) + MSX-3 + methy versus sham treated rats. In all groups, HX + HC values were significantly larger than baseline values. Apo + MSX-3 + methy significantly increased VT, and VT/TI during HX + HC in MT rats versus sham treated MT rats. Values are means ± 1 SEM and * = from sham MT, + = from sham WT, where all p < 0.05.
Table 1.
Summary of tidal volume (VT; mL ± SEM) data in baseline, hypoxia (HX), hypercapnia (HC) and hypoxia + hypercapnia (HX + HC) in all treatment groups. Groups were: Sham (WT: n=8; MT: n=9), PCPA (WT: n=3; MT: n=3), PCPA + Methy (WT: n=3; MT: n=3), MSX-3 (WT: n=3; MT: n=3), PCPA + MSX-3 (WT: n=3; MT: n=3), and Apo + MSX-3 + Methy (WT: n=3; MT: n=3). Significance was accepted at p < 0.05; 3 = Sham MT vs. treatment group MT..
| Tidal Volume | Baseline | HX | HC | HX + HC |
|---|---|---|---|---|
| Sham WT | 3.0 ±0.3 | 3.5 ±0.4 | 4.8 ±0.6 | 5.8 ±0.6 |
| Sham MT | 3.4 ±0.2 | 3.9 ±0.3 | 4.9 ±0.3 | 5.3 ±0.4 |
| PCPA WT | 3.8 ± 0.8 | 3.8 ± 0.5 | 5.1 ± 0.6 | 5.8 ± 1.1 |
| PCPA MT | 3.32 ± 0.04 | 3.7 ± 0.2 | 5.2 ± 0.4 | 5.8 ± 0.7 |
| PCPA + Methy WT | 4.3 ± 0.3 | 4.1 ± 0.3 | 7.0 ± 0.4 | 7.1 ± 0.4 |
| PCPA + Methy MT | 3.9 ± 0.2 | 3.8 ± 0.3 | 6.6 ± 0.1 | 7.0 ± 0.1 |
| MSX-3 WT | 3.4 ± 0.1 | 3.5 ± 0.2 | 4.9 ± 0.2 | 5.4 ± 0.1 |
| MSX-3 MT | 3.8 ± 0.2 | 3.9 ± 0.1 | 4.9 ± 0.2 | 5.51 ± 0.04 |
| MSX-3 + PCPA WT | 4.2 ± 0.1 | 4.3 ± 0.2 | 6.3 ± 0.2 | 7.0 ± 0.3 |
| MSX-3 + PCPA MT | 4.5 ± 0.2 | 4.1 ± 0.2 | 6.1 ± 0.1 | 6.9 ± 0.2 |
| Apocynin + MSX-3 + Methy WT | 4.1 ± 0.2 | 4.4 ± 0.1 | 6.3 ± 0.3 | 7.2 ± 0.7 |
| Apocynin + MSX-3 + Methy MT | 4.1 ± 0.3 | 4.4 ± 0.3 | 6.7 ± 0.4 | 7.6 ± 0.53 |
Table 4.
Summary of mean inspiratory flow (VT/TI; mL/sec ± SEM) during baseline, hypoxia (HX), hypercapnia (HC) and hypoxia + hypercapnia (HX + HC) in all treatment groups. Groups were: Sham (WT: n=8; MT: n=9), PCPA (WT: n=3; MT: n=3), PCPA + Methy (WT: n=3; MT: n=3), MSX-3 (WT: n=3; MT: n=3), PCPA + MSX-3 (WT: n=3; MT: n=3), and Apo + MSX-3 + Methy (WT: n=3; MT: n=3). Significance was accepted at p < 0.05; 3 = Sham MT vs. treatment group MT.
| Mean Inspiratory Flow | Baseline | HX | HC | HX + HC |
|---|---|---|---|---|
| Sham WT | 13 ± 4 | 18 ± 4 | 26 ± 6 | 29 ± 7 |
| Sham MT | 9 ± 0.6 | 16 ± 2 | 23 ± 3 | 24 ± 3 |
| PCPA WT | 16 ± 4 | 17 ± 4 | 26 ± 5 | 28 ± 6 |
| PCPA MT | 12 ± 0.5 | 14 ± 0.4 | 27 ± 2 | 27 ± 3 |
| PCPA + Methy WT | 14 ± 1 | 16 ± 1 | 32 ± 2 | 31 ± 2 |
| PCPA + Methy MT | 13 ± 1 | 15 ± 2 | 31 ± 2 | 32 ± 1 |
| MSX-3 WT | 17 ± 1 | 18 ± 0.6 | 27 ± 0.2 | 28 ± 0.6 |
| MSX-3 MT | 14 ± 2 | 15 ± 0.8 | 26 ± 1 | 28 ± 2 |
| MSX-3 + PCPA WT | 19 ± 2 | 18 ± 1 | 33 ± 1 | 33 ± 1 |
| MSX-3 + PCPA MT | 18 ± 0.8 | 17 ± 0.2 | 33 ± 1 | 34 ± 0.8 |
| Apocynin + MSX-3 + Methy WT | 18 ± 0.4 | 22 ± 1 | 37 ± 2 | 37 ± 3 |
| Apocynin + MSX-3 + Methy MT | 16 ± 2 | 18 ± 0.8 | 36 ± 43 | 39 ± 43 |
3.3 Ventilatory capacity is not decreased by PCPA with or without methysergide
To determine if diminished serotoninergic function impairs ventilatory capacity in MT rats, serotonin was depleted with PCPA, with or without the serotonin receptor antagonist methysergide (methy). There were significant differences due to treatment level and group; there were also significant interactions between treatment level and group in VT (0.001) and VT/TI (0.01) (although neither showed significant individual comparisons). In all groups, VT, frequency, and VT/TI were significantly increased during HX + HC versus baseline (all p<0.05; Figure 4). PCPA alone had no effect on VT, frequency, or VT/TI (Figure 4) in either WT or MT rats during baseline or HX + HC when compared to sham rats (all p>0.05).
The combination of PCPA + methy had no effect on VT, frequency, or VT/TI (Figure 4) in MT rats during baseline or HX + HC when compared to sham MT rats (all p>0.05). PCPA + methy significantly decreased frequency in WT rats during baseline (57 ± 5 breaths/min; Figure 4B) and HX + HC (116 ± 5 breaths/min; Figure 4B) versus sham WT rats (80 ± 6 breaths/min at baseline and 146 ± 4 breaths/min at HX + HC; all p < 0.05; Figure 4B). Thus, serotonin depletion, with or without concurrent serotonin receptor inhibition, did not impair breathing capacity during HX + HC in MT rats.
3.4 Ventilatory capacity is not decreased by serotonin depletion plus adenosine receptor inhibition
To examine the role of impaired adenosinergic function, with or without concurrent impairment of serotonergic function, A2A receptors were inhibited with MSX-3 with and without serotonin depletion. There were significant differences due to the effect of treatment level only; in addition, there were significant interactions between treatment level and group in VT (0.017) and VT/TI (0.007). As expected, VT, frequency, and VT/TI were significantly increased during HX + HC versus baseline in all groups (all p<0.05; Figure 5). MSX-3 alone or in combination with PCPA (MSX-3 + PCPA) had no effect on VT, frequency, or VT/TI in WT or MT rats during baseline or HX + HC versus sham rats (all p>0.05; Figure 5).Thus, adenosine receptor inhibition with or without serotonin depletion did not impair breathing capacity during HX + HC in MT rats.
Figure 5.

Tidal volume (VT), frequency, minute ventilation () and mean inspiratory flow (VT/TI) during baseline and HX + HC in wild-type (WT; black bars) and mutant (MT; white bars) rats treated with MSX-3 alone, or MSX-3 + PCPA. In all groups, HX + HC values were significantly larger than baseline values. MSX-3 alone or in combination with PCPA had no effect on VT, frequency, or VT/TI in either WT or MT rats during baseline or HX + HC. Values are means ± 1 SEM and + = from sham WT, where all p < 0.05.
3.5 Ventilatory capacity is not decreased by combined PCPA, MSX-3 and apocynin
Lastly, we studied simultaneous serotonin receptor, adenosine receptor and NADPH oxidase inhibition (Apo + MSX-3 + methy) in WT and MT rats. There were significant differences in treatment level and group. Further, there were significant interactions between treatment level and group in VT (0.015), (0.001), and VT/TI (0.035). All variables were significantly increased during HX + HC versus baseline (all p<0.05; Figure 6). Apo + MSX-3 + methy significantly increased VT (8.0 ± 0.5 mL in treated MT rats vs. 5.0 ± 0.4 mL in sham MT rats; p < 0.05; Figure 6A), (1023 ± 80 mL/min in treated MT rats vs. 619 ± 72 mL/min in sham MT rats; p < 0.05; Figure 6C) and VT/TI (39 ± 4 mL/s in treated MT rats vs. 24 ± 3 mL/s in sham MT rats; p < 0.05; Figure 6D) during HX + HC in MT rats. Frequency was unaffected by Apo + MSX-3 + methy in any group (p>0.05, Figure 6B). VT, frequency, and VT/TI were compared in only two WT and MT rats treated with Apo alone (n=2 each); all variables studied were increased during HX + HC versus baseline (data not shown), but Apo alone had no apparent effects on any variable studied in either WT or MT rats (data not shown). Thus, breathing capacity was not impaired during HX + HC in MT rats when serotonin levels and serotonin receptor activation were inhibited (PCPA + methy), or when serotonin receptors, NADPH oxidase, and adenosine receptors were all inhibited simultaneously (Apo + MSX-3 + methy).
4. Discussion
The main findings of this study are: 1) PCPA, MSX-3 and apocynin (drugs all shown to block respiratory plasticity) alone or in combination did not decrease ventilatory capacity in MT rats; and 2) the extent of motor neuron survival was equivalent among drug treatment groups in MT rats. Thus, although more than 80% of phrenic motor neurons had died at disease end-stage, ventilatory capacity was not significantly impaired in MT rats. We provide no evidence that the neurochemicals investigated are necessary to maintain ventilatory capacity at disease end stage, although we cannot rule out some role in initiating processes that subsequently maintain compensatory respiratory plasticity in these rats.
4.1 Serotonergic function and compensatory plasticity
Many forms of respiratory plasticity are serotonin-dependent, including long-term facilitation of phrenic motor output (Millhorn et al., 1980b; Kinkead and Mitchell 1999; Fuller et al., 2001; MacFarlane and Mitchell 2007; MacFarlane and Mitchell, 2008a; MacFarlane and Mitchell 2009) and sensory long-term facilitation in rats pre-conditioned with chronic intermittent hypoxia (Prabhakar et al., 2006; Peng et al., 2009; Prabhakar 2011). Here, PCPA was used to deplete whole-body serotonin levels through inhibition of its rate-limiting biosynthetic enzyme, tryptophan hydroxylase. Systemic PCPA transiently depletes serotonin levels over a period of days (Kuhar et al. 1971; Aghajanian et al., 1972; Deguchi et al., 1973; Steinman et al., 1987), differentially affects serotonin levels within the CNS (Carlton et al., 1987; Steinman et al., 1987), and may affect norepinephrine levels at high concentrations (Sloviter et al., 1978; Olson et al., 1979; Alexander et al., 1980). Although we did not measure serotonin levels in our study, PCPA attenuates serotonin-dependent respiratory plasticity (Millhorn et al., 1980; Eldridge and Millhorn, 1986), suggesting that it is sufficient to impair other forms of serotonin-dependent respiratory plasticity. Although we hypothesized that PCPA would at least partially block the Q (via 5-HT2 receptors) and S pathways (via 5-HT7 receptors; Figure 1), and reduce compensatory respiratory plasticity necessary to maintain breathing capacity in end-stage MT rats, ventilatory capacity was unaffected by this treatment (Figure 4). Thus, serotonin is not necessary for compensatory respiratory plasticity in MT rats. An important caveat is that it remains possible that serotonin was not sufficiently depleted, that serotonin was not depleted at the relevant site or time-frame, or that concurrent inhibition of 5-HT2 and 5-HT7 receptor function (due to decreased serotonin levels which affect both) has offsetting effects due to cross-talk inhibition between the Q and S pathways to pMF (Dale-Nagle et al., 2010).
Since PCPA doesn’t completely deplete serotonin (Kuhar et al. 1971; Aghajanian et al., 1972; Deguchi et al., 1973; Steinman et al., 1987), methysergide was combined with PCPA to further inhibit serotonergic function. Methysergide is a non-selective serotonin receptor antagonist that is well tolerated by rats and is known to block AIH-induced pLTF (Millhorn et al., 1980; Eldridge and Millhorn, 1986; Bach and Mitchell, 1996; McGuire et al., 2004; Ling, 2008). To some extent, both the Q and S pathways to pMF are expected to be affected by PCPA plus methysergide, since serotonin depletion and antagonism of multiple serotonin receptors affect receptors known to initiate each of these pathways (i.e. 5-HT2 and 5-HT7; Dale-Nagle et al., 2010; Figure 1). However, with this dual treatment, ventilatory capacity was not decreased in MT rats versus sham MT rats (Figure 4). Although methysergide is a broad-spectrum serotonin receptor antagonist, it is also a 5-HT1A agonist (Silberstein, 1998). Auto-inhibitory 5-HT1A receptors on raphe, serotonergic neurons restrain their activity (Glennon et al., 2000). Thus, by activating 5-HT1A receptors with methysergide, serotonin release is expected to decrease (Jacobs and Azmitia, 1992; Zifa and Fillion, 1992; McCrimmon et al., 1995; Wu et al., 1991; Hoyer et al., 1994). This effect is also expected to reduce serotonergic function, strengthening the argument that serotonergic function is not necessary to maintain compensatory respiratory plasticity in MT rats.
In these studies, many rat groups were studied, each with a relatively small n value (often n = 3). Although the small sample size of each individual group may compromise our statistical ability to discriminate differences if they had occurred, our main goal was to test the hypothesis that drugs known to block distinct forms of respiratory plasticity would decrease ventilatory capacity in end-stage SOD1G93A rats. Thus, we used multiple drug treatments (each in an independent rat group) targeting the same mechanism of respiratory plasticity. Since each treatment or treatment combination led to the same conclusion, the collective evidence that distinct treatments/combinations (each targeting the same neural mechanism) yield similar results increases confidence in our conclusions. Indeed, if we were to lump all rats receiving PCPA, for example, the functional n would be 9 rather than the value of 3 reported for the PCPA, PCPA + methy and PCPA + MSX3 groups. It is very unlikely that additional rats would reverse our conclusion that PCPA is not sufficient to reverse the compensatory respiratory plasticity observed in SOD1G93A rats at disease end-stage.
4.2 Adenosinergic function and compensatory plasticity
Since activation of spinal adenosinergic (A2A) receptors induces pMF (Golder et al., 2008; Hoffman et al. 2010), we hypothesized that A2A activation underlies compensatory respiratory plasticity with motor neuron degeneration. A2A agonist-induced pMF requires new synthesis of an immature TrkB isoform, believed to auto-dimerize, auto-phosphorylate and signal from within the cell (Lee and Chao 2001). We inhibited A2A receptors with systemic MSX-3 to at least transiently and partially block the S pathway (Figure 1). With MSX-3, baseline ventilation and the ability to increase ventilation during chemoreceptor activation was unaffected (Figure 5), suggesting that ongoing adenosine-dependent mechanisms are not critical for compensatory plasticity in MT rats.
4.3 Combined inhibition of S and Q pathways
Compensatory respiratory plasticity in MT rats does not appear to require the Q or S pathways for its maintenance. Thus, we used combined drug treatments to determine if either pathway alone can assume this function. Serotonin depletion via PCPA combined with A2A receptor inhibition should effectively impair both the Q and S pathways (Figure 1). However, since ventilation was not decreased in MT rats treated with PCPA + MSX-3 (Figure 5), other mechanisms are necessary to maintain compensatory plasticity during motor neuron disease. Nevertheless these data do not rule out a role for the S and/or Q pathways in initiating compensatory respiratory plasticity.
In another attempt to more completely block the Q and S pathways, we simultaneously applied apocynin, MSX-3 and methysergide. We predicted that NADPH oxidase inhibition with apocynin would decrease ROS formation, enabling phosphatases to inhibit the Q pathway (Figure 1), and thereby reduce compensatory respiratory plasticity at disease end-stage. NADPH oxidase activity is necessary for AIH-induced pLTF (MacFarlane and Mitchell, 2007; MacFarlane and Mitchell, 2008a; MacFarlane and Mitchell, 2009; MacFarlane et al. 2009). Since pLTF is constrained by okadaic acid sensitive serine/threonine protein phosphatases (Wilkerson et al. 2008), we suggested that NADPH derived ROS inhibit these phosphatases, thereby enabling full pLTF expression. When spinal phosphatases are inhibited with okadaic acid, AIH-induced pLTF is restored in rats pretreated with ROS scavengers (MacFarlane et al. 2008). The complex of NADPH oxidase, ROS and protein phosphatases has been hypothesized to form a “regulatory cassette” for pLTF expression (Wilkerson et al. 2007; MacFarlane et al. 2008; Dale-Nagle et al., 2010). With the combination of apocynin, MSX-3 and methysergide, both the Q and S pathways should be affected because: 1) serotonin receptor activation is sufficient to induce both pathways via actions on 5-HT2 and 5-HT7 receptors, respectively (i.e. methysergide sensitive), 2) NADPH oxidase-derived ROS are necessary to inhibit phosphatases that constrain the Q pathway (i.e. apocynin sensitive), and 3) A2A receptors are sufficient to induce the S pathway (i.e. MSX-3 sensitive; Figure 1). However, ventilation was not decreased and, surprisingly, was actually enhanced in MT rats treated with combined apocynin + MSX-3 + methysergide (Figure 6); this observation reinforces our conclusion that neither the Q nor S pathways are necessary to maintain compensatory plasticity in end-stage MT rats. Regardless, we cannot rule out roles for either or both mechanisms in initiating compensatory plasticity. Enhanced ventilatory capacity with this combined treatment suggests that the Q and/or S pathways constrain the mechanism giving rise to spontaneous compensatory plasticity with respiratory motor neuron degeneration.
4.4 Initiation versus maintenance of compensatory plasticity
We targeted cellular pathways using drugs that would exert only transient effects, well after compensatory plasticity had been established. Thus, it is possible that the Q and/or S pathways are necessary to initiate (versus maintain) spontaneous compensatory respiratory plasticity in MT rats. Short-term (versus chronic) receptor inhibition may not be sufficient to reverse established compensatory respiratory plasticity in MT rats. Alternately, the timing of drug delivery may not have been appropriate. If either of these pathways is necessary or sufficient to induce compensatory plasticity, earlier drug delivery may be required (i.e. pre-symptomatic or early in disease progression). Mechanisms underlying compensatory plasticity may be resistant to agents administered after the plasticity has been induced. Another possibility is that compensatory plasticity may take longer to reverse, and that we need to administer drugs for longer periods in MT rats. PCPA (which causes serotonin depletion) has a time course of days (Kuhar et al. 1971; Aghajanian et al., 1972; Deguchi et al., 1973; Steinman et al., 1987), making this possibility less likely.
4.5 Alternative mechanisms for compensatory plasticity in MT rats
Other mechanisms beyond the Q and S pathways are capable of eliciting pMF, including VEGF, erythropoietin and PKCζ dependent pMF (Dale-Nagle et al., 2010). Q-dependent pMF can also be induced by adrenergic receptors; for example, spinal administration of α-1 receptor agonists (Gq coupled metabotropic receptors; Dale-Nagle et al., 2010; Huxtable et al., 2010) elicits pMF, although these receptors are not necessary for AIH-induced pLTF (Huxtable et al., unpublished). Although we thoroughly tested the roles of serotonin and adenosine-dependent mechanisms in maintaining compensatory plasticity, other neuromodulatory systems (such as norepinephrine) could be involved. Since α-1 receptors most likely elicit pMF via the Q pathway, it is logical to suspect that this mechanism requires NADPH oxidase dependent ROS formation. Although the failure of apocynin to reverse compensatory plasticity argues against this possibility, there appear to be differential roles for NADPH oxidase in the Q pathway elicited by different ligands (MacFarlane et al., 2011).
Other mechanisms of pMF have not been investigated here; intrathecal VEGF (Dale-Nagle et al., 2011) and erythropoietin (Dale et al., 2012) both elicit pMF via ERK MAP kinase and AKT dependent pathways. Lastly, phrenic inactivity due to hypocapnia, vagal stimulation and/or anesthetic depression (Zhang et al., 2004; Mahamed et al., 2011) induces pMF by a mechanism that requires spinal PKCζ activity (Strey et al., 2012). One or all of these pathways to pMF may contribute to compensatory plasticity.
Finally, we cannot rule out that compensatory plasticity in MT rats arises at other sites, including: 1) plasticity at the neuromuscular junction (Johnson and Mitchell, 2013); 2) a shift in the relative contributions to breathing between the diaphragm versus accessory inspiratory muscles; 3) increased lung compliance, making breathing easier for a given level of respiratory muscle force generation (assuming not enough to induce dynamic compression of the airways on expiration); and/or 4) the functional types of phrenic motor neuron axons are differentially susceptible as the disease progresses in MT rats, which may explain the differential impact of the disease on motor neuron survival versus ventilation (recently reviewed in Nichols et al., 2013b). Each of these possibilities remains to be explored.
Highlights.
Ventilation is preserved despite respiratory motor neuron loss in ALS rats.
Few studies have been done to understand how breathing is preserved in ALS rats.
Treatments used in this study in combination did not impair breathing in ALS rats.
Table 2.
Summary of frequency (breaths/min ± SEM) data in baseline, hypoxia (HX), hypercapnia (HC) and hypoxia + hypercapnia (HX + HC) in all treatment groups. Groups were: Sham (WT: n=8; MT: n=9), PCPA (WT: n=3; MT: n=3), PCPA + Methy (WT: n=3; MT: n=3), MSX-3 (WT: n=3; MT: n=3), PCPA + MSX-3 (WT: n=3; MT: n=3), and Apo + MSX-3 + Methy (WT: n=3; MT: n=3). Significance was accepted at p < 0.05; 1 = treatment group WT vs. treatment group MT, and 2 = Sham WT vs. treatment group WT.
| Frequency | Baseline | HX | HC | HX + HC |
|---|---|---|---|---|
| Sham WT | 80 ± 6 | 112 ± 3 | 156 ± 7 | 146 ± 4 |
| Sham MT | 57 ± 21 | 89 ± 41 | 131 ± 41 | 127 ± 31 |
| PCPA WT | 74 ± 4 | 91 ± 2 | 136 ± 4 | 125 ± 3 |
| PCPA MT | 72 ± 3 | 108 ± 8 | 141 ± 6 | 128 ± 3 |
| PCPA + Methy WT | 57 ± 52 | 85 ± 42 | 123 ± 72 | 116 ± 52 |
| PCPA + Methy MT | 65 ± 1 | 94 ± 2 | 124 ± 5 | 116 ± 6 |
| MSX-3WT | 88 ± 13 | 102 ± 9 | 154 ± 10 | 146 ± 6 |
| MSX-3 MT | 60 ± 4 | 90 ± 8 | 144 ± 9 | 138 ± 7 |
| MSX-3 + PCPA WT | 82 ± 4 | 89 ± 6 | 141 ± 3 | 123 ± 1 |
| MSX-3 + PCPA MT | 74 ± 6 | 100 ± 8 | 141 ± 9 | 129 ± 5 |
| Apocynin + MSX-3 + Methy WT | 71 ± 3 | 100 ± 7 | 164 ± 7 | 140 ± 5 |
| Apocynin + MSX-3 + Methy MT | 64 ± 4 | 89 ± 2 | 141 ± 7 | 135 ± 4 |
Table 3.
Summary of minute ventilation (; mL/min ± SEM) during baseline, hypoxia (HX), hypercapnia (HC) and hypoxia + hypercapnia (HX + HC) in all treatment groups. Groups were: Sham (WT: n=8; MT: n=9), PCPA (WT: n=3; MT: n=3), PCPA + Methy (WT: n=3; MT: n=3), MSX-3 (WT: n=3; MT: n=3), PCPA + MSX-3 (WT: n=3; MT: n=3), and Apo + MSX-3 + Methy (WT: n=3; MT: n=3). Significance was accepted at p < 0.05; 1 = treatment group WT vs. treatment group MT, and 3 = Sham MT vs. treatment group MT.
| Minute Ventilation | Baseline | HX | HC | HX + HC |
|---|---|---|---|---|
| Sham WT | 223 ± 49 | 355 ± 88 | 696 ± 173 | 776 ± 180 |
| Sham MT | 173 ± 18 | 311 ± 43 | 586 ± 75 | 619 ± 721 |
| PCPA WT | 281 ± 63 | 337 ± 79 | 690 ± 138 | 718 ± 149 |
| PCPA MT | 240 ± 13 | 387 ± 33 | 723 ± 66 | 739 ± 75 |
| PCPA + Methy WT | 238 ± 9 | 336 ± 38 | 851 ± 33 | 819 ± 58 |
| PCPA + Methy MT | 254 ± 14 | 384 ± 23 | 793 ± 49 | 818 ± 41 |
| MSX-3 WT | 290 ± 39 | 344 ± 16 | 738 ± 15 | 785 ± 22 |
| MSX-3 MT | 223 ± 12 | 336 ± 19 | 698 ± 18 | 761 ± 42 |
| MSX-3 + PCPA WT | 340 ± 18 | 375 ± 29 | 868 ± 38 | 856 ± 37 |
| MSX-3 + PCPA MT | 329 ± 24 | 396 ± 12 | 853 ± 32 | 885 ± 16 |
| Apocynin + MSX-3 + Methy WT | 272 ± 14 | 413 ± 42 | 1004 ± 61 | 1002 ± 109 |
| Apocynin + MSX-3 + Methy MT | 250 ± 12 | 372 ± 27 | 946 ± 933 | 1023 ± 803 |
Acknowledgements
This work was supported by NIH R01 NS057778 and T32 HL007654 (NLN), the Francis Families Foundation (NLN), and the Clinical and Translational Science Award (CTSA) program through the NIH National Center for Advancing Translational Sciences (NCATS) grant UL1TR000427 (RAJ). We thank Jacalyn McHugh for maintenance and genotyping of the ALS rodent colony.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aghajanian GK. Influence of drugs on the firing of serotonin-containing neurons in brain. Fed. Proc. 1972;31:91–96. [PubMed] [Google Scholar]
- Alexander GJ, Kopeloff LM, Alexander RB. Serotonin and norepinephrine. Long-term decrease in rate of synthesis in brain of rats primed with p-chlorophenylalanine. Neurochem. Res. 1980;5:879–883. doi: 10.1007/BF00965787. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir. Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat. Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Gestreau C, Vinit S, Stamegna JC, Kastner A, Gauthier P. Specific and artifactual labeling in the rat spinal cord and medulla after injection of monosynaptic retrograde tracers into the diaphragm. Neurosci. Lett. 2007;417(2):206–211. doi: 10.1016/j.neulet.2007.02.047. [DOI] [PubMed] [Google Scholar]
- Bourke SC, Shaw PJ, Gibson GJ. Respiratory function vs. sleep-disordered breathing as predictors of QOL in ALS. Neurology. 2001;57:2040–2044. doi: 10.1212/wnl.57.11.2040. [DOI] [PubMed] [Google Scholar]
- Bruijn L,I, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Steinman JL, Hillman GR, Willis WD. Differential effects of p-chlorophenylalanine on indoleamines in brainstem nuclei and spinal cord of rats. II. Identification of immunohistochemically stained structures using computer-assisted image enhancement techniques. Brain Res. 1987;426:310–322. doi: 10.1016/0006-8993(87)90884-5. [DOI] [PubMed] [Google Scholar]
- Dale EA, Satriotomo I, Mitchell GS. Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and Akt signaling. J. Neurosci. 2012;32(17):5973–5983. doi: 10.1523/JNEUROSCI.3873-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv. Exp. Med. Bio. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J. Neurosci. 2011;31(21):7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi T, Sinha AK, Barchas JD. Biosynthesis of serotonin in raphe nuclei of rat brain: effect of p-chlorophenylalanine. J. Neurochem. 1973;20:1329–1336. doi: 10.1111/j.1471-4159.1973.tb00244.x. [DOI] [PubMed] [Google Scholar]
- Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics. 1955;16:81–87. [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE. Handbook of Physiology, Section 3: The Respiratory System, Vol. II: Control of Breathing, part 1. American Physiological Society; Bethesda, MD: 1986. Oscillation, gating and memory in the respiratory control system; pp. 93–114. [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity chemosensitivity. Annu. Rev. Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Zabka A, Baker TL, Mitchell GS. Physiological and genomic consequences of intermittent hypoxia: Selected contribution: Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J. Appl. Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Glennon RA, Dukat M, Westkaemper RB. Serotonin Receptor Subtypes and Ligands. American College of Neurophyscopharmacology. 2000 http://www.acnp.org/g4/GN401000039/Ch039.html. [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J. Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Hinrichsen CF, McCrimmon DR. Short-term plasticity of descending synaptic input to phrenic motoneurons in rats. J Physiol. 2003;94(4):1421–1430. doi: 10.1152/japplphysiol.00599.2002. [DOI] [PubMed] [Google Scholar]
- Hoffman MS, Golder FJ, Mahamed S, Mitchell GS. Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J Physiol. 2010;588(Pt 1):255–266. doi: 10.1113/jphysiol.2009.180075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J. Physiol. 2011;589(6):1397–1407. doi: 10.1113/jphysiol.2010.201657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougee S, Hartog A, Sanders A, Graus YM, Hoijer MA, Garssen J, van den Berg WB, van Beuningen HM, Smit HF. Oral administration of the NADPH-oxidase inhibitor apocynin partially restores diminished cartilage proteoglycan synthesis and reduces inflammation in mice. Eur. J. Pharmacol. 2006;531(1-3):264–269. doi: 10.1016/j.ejphar.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PPA. International union of pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol.Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc. Natl. Acad. Sci. U S A. 2002;99(3):1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, MacFarlane PM, Mitchell GS. Episodic spinal alpha 1 receptor activation induces phrenic motor facilitation in adult rats. Soc. Neurosci. 2010 Abst. Program No. 298.20. [Google Scholar]
- Jacky JP. A plethysmograph for long-term measurements of ventilation in unrestrained animals. J. Appl. Physiol. 1978;45(4):644–647. doi: 10.1152/jappl.1978.45.4.644. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol. Rev. 1992;72:165–229. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Mitchell GS. Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respir. Physiol. Neurobiol. 2013;189(2):419–428. doi: 10.1016/j.resp.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: Effects of ketanserin and 5 carboxamidotryptamine. Am. J. Physiol. 1999;277(2 Pt2):R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Forth RH, Aghajanian GK. Selective reduction of tryptophan hydroxylase activity in rat forebrain after midbrain raphe lesions. Brain Res. 1971;35:167–176. doi: 10.1016/0006-8993(71)90602-0. [DOI] [PubMed] [Google Scholar]
- Lechtzin N, Rothstein J, Clawson L, Diette G, Wiener CM. Amyotrophic lateral sclerosis: evaluation and treatment of respiratory impairment. Amyotrophic Lateral Sclerosis. 2002;3:5–13. doi: 10.1080/146608202317576480. [DOI] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. U S A. 2001;98(6):3555–60. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L. Serotonin and NMDA receptors in respiratory long-term facilitation. Respir. Physiol. Neurobiol. 2008;164(1-2):233–241. doi: 10.1016/j.resp.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall RA, Donaldson N, Polkey MI, Leigh PN, Moxham J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain. 2001;124(10):110–118. doi: 10.1093/brain/124.10.2000. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Serotonin-induced phrenic long-term facilitation requires reactive oxygen species signaling via the NADPH oxidase complex. Soc. Neurosci. 2007 Abst. Program No. 520.15. [Google Scholar]
- MacFarlane PM, Mitchell GS. NADPH oxidase activity is necessary for phrenic motor facilitation induced by 5HT2B receptor activation. FASEB J. 2008a;22:1232–7. [Google Scholar]
- MacFarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neuroscience. 2008b;152:189–197. doi: 10.1016/j.neuroscience.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir. Physiol. Neurobiol. 2008;164:263–271. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587(Pt22):5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;587(Pt9):1931–1942. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Strey KA, Mitchell GS, Baker-Herman TL. Reduced respiratory neural activity elicits phrenic motor facilitation. Respir. Physiol. Neurobiol. 2011;175(3):303–309. doi: 10.1016/j.resp.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrimmon DR, Mitchell GS, Dekin M. Glutamate, GABA and Serotonin in Ventilatory Control. M. Dekker; New York: 1995. pp. 151–218. [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;286:334–341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by endogenous central serotonin. Respir. Physiol. 1980;42:171–188. doi: 10.1016/0034-5687(80)90113-9. [DOI] [PubMed] [Google Scholar]
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr., Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase trangenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J. Neurosci. 2001;21(23):9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am. J. Respir. Crit. Care Med. 2013a;187(5):535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Johnson RA, Satriotomo I, Nashold LJ, Mitchell GS. Serotonin depletion does not impair ventilation in the SOD1G93A rat, a model of amyotrophic lateral sclerosis (ALS) FASEB J. 2009;23:784–8. [Google Scholar]
- Nichols NL, Van Dyke J, Nashold L, Satriotomo I, Suzuki M, Mitchell GS. Ventilatory control in ALS. Respir. Physiol. Neurobiol. 2013b;189(2):429–437. doi: 10.1016/j.resp.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EB, Dempsey JA, McCrimmon DR. Serotonin and the control of ventilation in awake rats. J. Clin. Invest. 1979;64:689–693. doi: 10.1172/JCI109510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J. Neurosci. 2009;29(15):4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR. Sensory plasticity of the carotid body: role of reactive oxygen species and physiological significance. Respir. Physiol. Neurobiol. 2011;178(3):375–380. doi: 10.1016/j.resp.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Yuan G, Kumar GK. Reactive oxygen species facilitate oxygen sensing. Novartis Found. Symp. 2006;272:95–99. [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Schindler CV, Karcz-Kubicha M, Thorndike EB, Müller CE, Tella SR, Ferré S, Goldberg SR. Role of central and peripheral adenosine receptors in the cardiovascular responses to intraperitoneal injections of adenosine A1 and A2A subtype receptor agonists. Br. J. Pharmacol. 2005;144(5):642–650. doi: 10.1038/sj.bjp.0706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein SD. Methysergide. Cephalalgia. 1998;18(7):421–435. doi: 10.1046/j.1468-2982.1998.1807421.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Drust EG, Connor JD. Serotonin agonist actions of p-chlorophenylalanine. Neuropharmacology. 1978;17:1029–1033. doi: 10.1016/0028-3908(78)90029-1. [DOI] [PubMed] [Google Scholar]
- Steinman JL, Carlton SM, Haber B, Willis WD. Differential effects of p-chlorophenylalanine on indoleamines in brainstem nuclei and spinal cord of rats. I. Biochemical and behavior analysis. Brain Res. 1987;426:297–309. doi: 10.1016/0006-8993(87)90883-3. [DOI] [PubMed] [Google Scholar]
- Strey KA, Nichols NL, Baertsch NA, Broytman O, Baker-Herman TL. Spinal atypical protein kinase C activity is necessary to stabilize inactivity-induced phrenic motor facilitation. J. Neurosci. 2012;32(46):16510–16520. doi: 10.1523/JNEUROSCI.2631-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankersly CG, Haenggeli C, Rothstein JD. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J. Appl. Physiol. 2007;102:926–932. doi: 10.1152/japplphysiol.00193.2006. [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum. Mol. Genet. 2003;12(21):2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Wilkerson JE, MacFarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: Roles of protein phosphatases and reactive oxygen species. Biochem. Soc. Trans. 2007;35(Pt5):1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]
- Wilkerson JE, Satriotomo I, Baker-Herman TL, Watters JJ, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J. Neurosci. 2008;28:2949–2458. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat. Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wu SY, Wang MY, Dun NJ. Serotonin via presynaptic 5-HT1 receptors attenuates synaptic transmission to immature rat motoneurones in vitro. Brain Res. 1991;554:111–121. doi: 10.1016/0006-8993(91)90178-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McGuire M, White DP, Ling L. Serotonin receptor subtypes involved in vagus nerve stimulation-induced phrenic long-term facilitation in rats. Neurosci. Lett. 2004;363(2):108–111. doi: 10.1016/j.neulet.2004.03.067. [DOI] [PubMed] [Google Scholar]
- Zifa E, Fillion G. 5-Hydroxytryptamine receptors. Pharmacol. Rev. 1992;44:401–458. [PubMed] [Google Scholar]