

Abstract
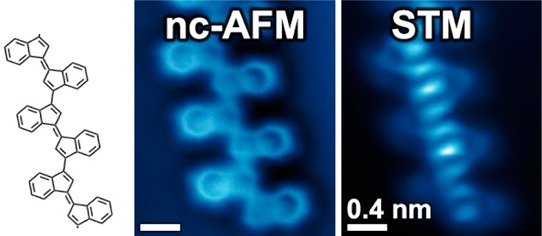
Semiconducting π-conjugated polymers have attracted significant interest for applications in light-emitting diodes, field-effect transistors, photovoltaics, and nonlinear optoelectronic devices. Central to the success of these functional organic materials is the facile tunability of their electrical, optical, and magnetic properties along with easy processability and the outstanding mechanical properties associated with polymeric structures. In this work we characterize the chemical and electronic structure of individual chains of oligo-(E)-1,1′-bi(indenylidene), a polyacetylene derivative that we have obtained through cooperative C1–C5 thermal enediyne cyclizations on Au(111) surfaces followed by a step-growth polymerization of the (E)-1,1′-bi(indenylidene) diradical intermediates. We have determined the combined structural and electronic properties of this class of oligomers by characterizing the atomically precise chemical structure of individual monomer building blocks and oligomer chains (via noncontact atomic force microscopy (nc-AFM)), as well as by imaging their localized and extended molecular orbitals (via scanning tunneling microscopy and spectroscopy (STM/STS)). Our combined structural and electronic measurements reveal that the energy associated with extended π-conjugated states in these oligomers is significantly lower than the energy of the corresponding localized monomer orbitals, consistent with theoretical predictions.
Keywords: Conducting polymers, C1−C5 thermal enediyne cyclization, radical step-growth polymerization, noncontact atomic force microscopy (nc-AFM), scanning tunneling microscopy (STM), density functional theory (DFT)
Conjugated polymers have attracted considerable interest into their fundamental properties as well as their potential for industrial applications.1−3 Their tunable electronic structure makes them a useful material for applications in the fields of molecular electronics and photonics.4−6 Numerous chemical reactions yielding conjugated polymers have been adapted for the synthesis of well-defined molecular wires on surfaces.7−17 These reactions include Ullmann-type cross-coupling,10−12 diyne polymerization,13,14 and alkyne homocoupling,17 most of which require catalytically active metal substrates to facilitate polymerization. Thermally induced enediyne cyclization on surfaces,18,19 a newer polymerization technique, has received increased attention due to its greater flexibility regarding noncatalytic growth substrates.20 Effective utilization of carbon-based nanostructures resulting from these reactions requires the development of synthetic tools to control the chemical and electronic structure of the polymer products. Of particular importance for advanced electronics applications21 is the formation of polymeric structures featuring extended π-conjugation during the growth process from small-molecule building blocks.22,23
Here we report the synthesis and characterization of individual chains of oligo-(E)-1,1′-bi(indenylidene)24 obtained through thermally induced cooperative C1–C5 radical cyclizations of enediyne precursors followed by step-growth polymerization on Au(111). We have gained new insight into the relationship between the chemical structure and electronic properties in these oligomers by utilizing nc-AFM to determine the precise atomic-scale structure of the enediyne starting material, cyclized monomers, and covalently linked oligomeric chains, while simultaneously utilizing STM/STS measurements to probe the localized and extended electronic states of these species (including orbital energies). These measurements help us to understand the oligomer formation reaction mechanism and show that the development of oligomer extended electronic states can be rationalized as the result of an efficient π-orbital overlap between monomer building blocks. Increased spatial delocalization is associated with a decrease in oligomer electronic energy, as confirmed by our theoretical simulations.
The enediyne precursor 1,2-bis(2-ethynylphenyl)ethyne (1) (Figure 1) used in this study was synthesized through iterative Sonogashira cross-coupling reactions. 1 was deposited in ultrahigh vacuum onto a Au(111) surface held at room temperature (T = 293 K). Cryogenic nc-AFM measurements (T = 4 K) (Figure 1a) reveal that 1 adopts two conformational isomers on the surface: a C2h symmetric trans-conformation (Figure 1b) and a C2v symmetric cis-conformation (Figure 1c). The atomic structure of the phenyl rings and the positions of the single and triple bonds are clearly resolved in the nc-AFM image. Contrast in nc-AFM measurements at the small oscillation amplitudes used here (60 pm) is dominated by short-range chemical forces that arise from repulsive interactions between the CO functionalized AFM tip and molecular adsorbates on the surface.25,26 This imaging technique allows precise spatial resolution of surface-bound atoms and bonds at a level that is not attainable by other surface probes,18,26,27 as indicated by comparison of the nc-AFM image in Figure 1a to the wire-frame structures in Figure 1b,c.
Figure 1.
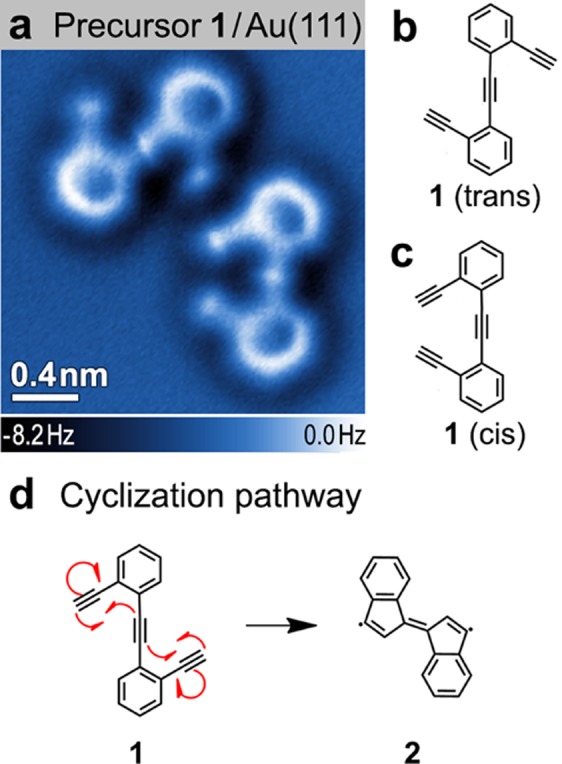
Precursor molecule 1. (a) nc-AFM image of the two conformational isomers of 1 on Au(111) (T = 4 K; tip height corresponds to tunnel current set point Vs = 50 mV and I = 10 pA above Au(111)); (b) Schematic representation of trans conformation and (c) cis conformation. (d) Cyclization reaction: The precursor 1 can transform to the monomer 2 via two C1–C5 thermal enediyne cyclizations.
Thermal annealing of the Au(111) surface decorated with a submonolayer coverage of 1 at 160 °C induces two intramolecular C1–C5 radical cyclization reactions (Figure 1d) as well as intermolecular carbon–carbon coupling reactions between monomer units. Following this annealing step more than 70% of the material on the surface becomes part of covalently linked molecular assemblies with lengths n ≥ 3 (where n denotes the number of monomer subunits). As depicted in the nc-AFM image of Figure 2a, the most common structures observed are covalently linked oligo-(E)-1,1′-bi(indenylidene) chains (an oligo-acetylene derivative) containing the common monomer subunit 2 (Figure 2b, dashed box). Most chains are composed of 5–10 monomer units, but sometimes exceed 20. Defect-free translational symmetry along the oligomer backbones is typically retained over segments of three to five monomers. While 2 (Figure 2b, dashed box) is representative of the dominant monomer incorporated into extended oligomer chains, other monomer subunits were also observed within the chains (see Supporting Information).
Figure 2.
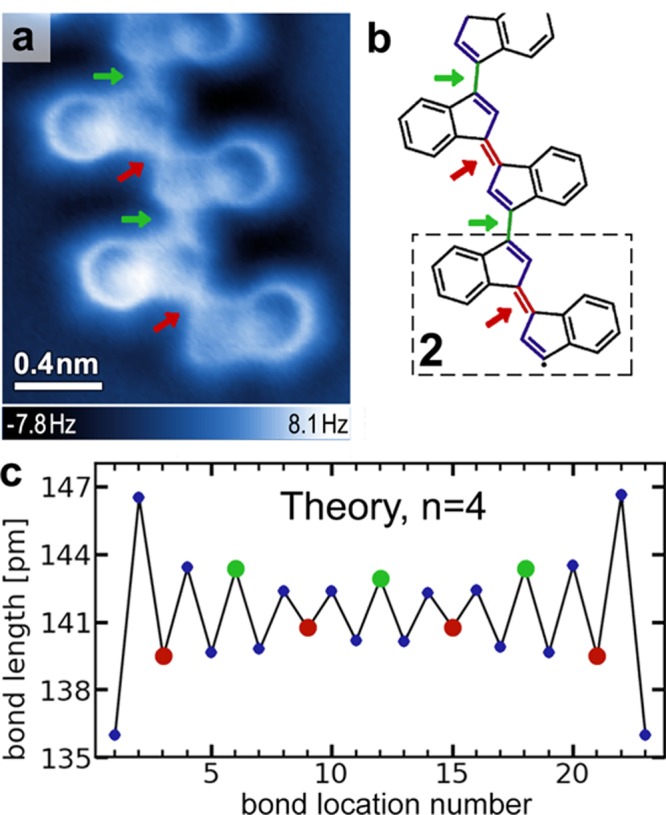
Oligomer containing monomer 2 subunits. (a) nc-AFM image of an oligomer chain on Au(111) (T = 4 K; tip height corresponds to tunnel current set point Vs = 50 mV and I = 30 pA above Au(111)). (b) Schematic representation of chemical structure of the oligomer in a. Arrows indicate short (red) and long (green) bonds between indenyl groups. The dashed box shows monomer structure 2. (c) DFT calculated bond lengths for a four-unit oligomer chain (composed of units of 2 as seen in b) as a function of the location of the bond along the chain. Red and green dots indicate short and long bonds between indenyl groups, while blue dots indicate bonds within the five-membered rings (calculation shown for an oligomer chain having unsaturated radical valences at the chain ends; results are similar for hydrogen-terminated chains).
nc-AFM imaging of the atomic structure of the oligomer chains (Figure 2a) reveals that the π-conjugated carbon–carbon double bonds along the backbone exhibit alternating lengths. Even though the symmetry of their local chemical structure is equivalent, bonds between the five-membered rings of adjacent indenyl groups alternate in length and are marked by red arrows (short bonds) and green arrows (long bonds) in the image. A schematic representation of the alternating pattern of short (C=C) and long (C–C) carbon–carbon bonds along the conjugated backbone is depicted in Figure 2b. The shorter (double) bonds between indenyl groups appear to be roughly 50% of the length of the longer (single) bonds for the tip–sample distance used to obtain the image in Figure 2a (a discussion of bond length measurement and associated bond length variation with tip height can be found in the Supporting Information (SI), see Figure S3). It is important to note that while our nc-AFM measurement correctly identifies alternating trends in bond lengths, the magnitude of this effect is greatly exaggerated by this imaging technique27 (see SI). Bonds within the five-membered rings are also seen to exhibit a bond length modulation. In particular, the double bonds in indenyl end-groups show a distinctive deviation compared to indenyl groups along the extended oligomer chain (Figure 2a).
Our STM spectroscopy of oligo-(E)-1,1′-bi(indenylidene) reveals that the electronic structure of the covalently linked oligomer chains exhibits extended-state behavior (Figure 3). An AFM image of a representative oligomer chain on the Au(111) surface is depicted in Figure 3a along with the STM dI/dV spectrum (Figure 3d) measured at one point along the backbone of the oligomer chain (dI/dV measurement reflects the electronic local density of states (LDOS) at the energy selected by the tip–sample bias; spectra taken at different points on the oligomer differ only in the intensity of the observed resonance). A well-defined electronic resonance is observed at an energy approximately 0.125 V above EF (blue arrow) compared to the dI/dV spectrum on bare Au(111). A dI/dV spatial map of the oligomer chain at a tip bias of 0.125 V (Figure 3b) reveals that the intensity of this state is localized along the oligomer backbone and extends continuously along the full length of the π-conjugated chain (excluding the ends, which are often composed of different types of monomer subunits). This extended-state spatial distribution was observed for oligomer chains of different lengths composed of monomer units 2 (see Figure S4).
Figure 3.
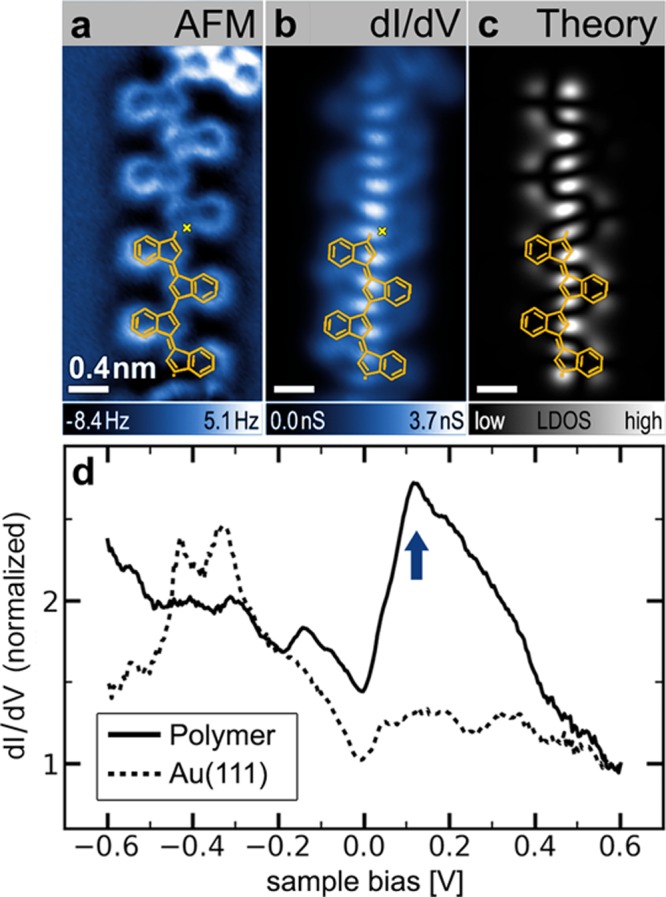
Electronic structure of an individual oligomer. (a) nc-AFM image of oligomer chain (tip height corresponds to tunnel current set point Vs = 50 mV and I = 20 pA above Au(111)). (b) Experimental STM dI/dV map (constant height) at Vs = 0.125 V reveals an extended electronic state along the conjugated backbone of oligomer shown in a. (c) GW calculation of electronic local density of states of the LUMO for a free-standing oligomer chain containing four monomer 2 subunits (n = 4). Orange overlays in a–c show the chemical structure of two units of the n = 4 oligomer chain used in the calculation. (d) STM dI/dV point spectroscopy of oligomer chain shown in a at the position indicated by the yellow cross reveals an electronic resonance at Vs ≈ 0.13 V (blue arrow) compared to a reference spectrum on bare Au(111) (spectra are normalized by their respective values at Vs = 0.6 V, open feedback spectroscopy starting parameters Vs = 0.6 V, I = 0.8 nA, T = 4 K).
Isolated monomer building blocks 2 not incorporated into oligomeric structures were observed to coexist with the chains on the Au(111) surface. These monomers account for a few percent of the material on the surface (other monomer structures have also been observed). Figure 4a depicts a nc-AFM image of an isolated (E)-1,1′-bi(indenylidene) monomer. Isolated monomer building blocks 2 exhibit a geometry that resembles the indenyl end-groups in oligomer chains (Figure 2a, bottom), including the presence of highly distorted five-membered rings (such distortion might arise due to the interaction of a radical with the surface28). The electronic structure of bi(indenylidene) monomers 2 was characterized by STM spectroscopy. A representative dI/dV spectrum measured on an isolated (E)-1,1′-bi(indenylidene) monomer and the corresponding spectrum of the bare Au(111) surface are depicted in Figure 4d (spectra taken at different points on the monomer differ only in the intensity of the observed resonance). A prominent resonance at Vs = 1.2 V (blue arrow) marks the lowest unoccupied molecular orbital (LUMO) of the molecule adsorbed on the Au(111) surface. The spatial distribution of this molecular electronic state was imaged using dI/dV mapping (Figure 4b).
Figure 4.
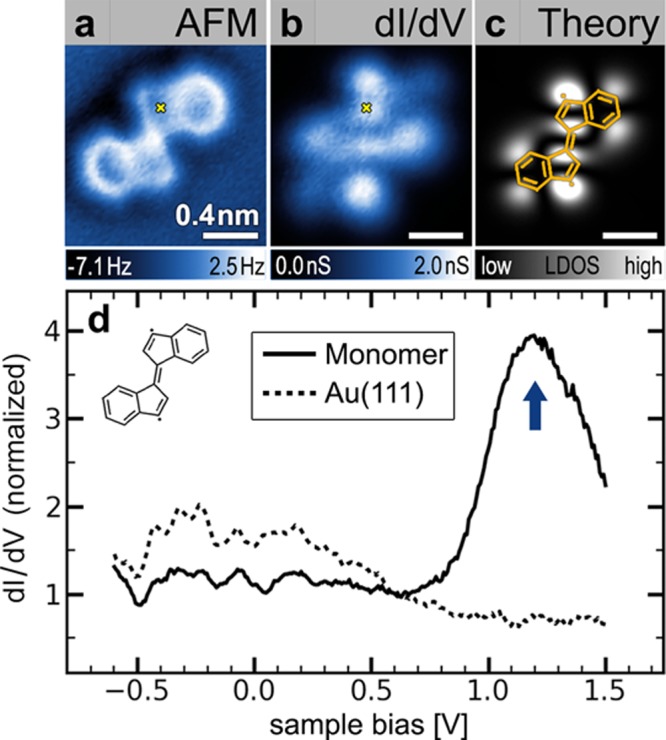
Isolated monomer building block 2: (a) nc-AFM image of an isolated monomer 2 on Au(111) (T = 4 K, tip height corresponds to tunnel current set point Vs = 50 mV and I = 35 pA above Au(111)). (b) Experimental constant-height dI/dV map of monomer 2 shown in a at Vs = 1.2 V depicts the spatial distribution of the monomer LUMO. (c) GW calculation of the local density of states of the LUMO for a free-standing monomer 2. (d) STM dI/dV point spectroscopy performed on the monomer shown in a at the position indicated by the yellow cross reveals the monomer LUMO at Vs ≈ 1.2 V (blue arrow) compared to a reference spectrum on bare Au(111) (spectra are normalized by their respective values at Vs = 0.6 V, open feedback spectroscopy starting parameters Vs = 1.5 V, I = 0.5 nA, T = 4 K).
The combination of nc-AFM and STM spectroscopy measurements on individual oligomer chains and small molecule precursors reveals both the underlying reaction mechanism that leads to the formation of oligo-(E)-1,1′-bi(indenylidene), as well as the origin of the oligomer electronic structure. Two thermally induced intramolecular C1–C5 enediyne cyclizations of 1 yield the monomer building-block 2 that has been observed on the surface (Figure 4). This highly reactive 3,3′-diradical intermediate diffuses along the Au(111) surface and recombines with other monomers in a step-growth process. While theoretical models in the gas phase have reported an activation barrier of ∼40 kcal mol–1 for the C1–C5 cyclization of benzannulated enediynes,29 we have previously shown that metal surface-supported C1–C5 cyclizations can proceed at temperatures below 100 °C.18 In some oligomer chains we have observed uncyclized precursor molecules 1 that are covalently linked via their terminal alkyne carbon atoms (see Supporting Information, Figure S1). We attribute this structural defect to the reaction of a radical at the end of the growing oligo-(E)-1,1′-bi(indenylidene) chain with a terminal alkyne carbon atom of an uncyclized building block 1.
The electronic structure of the oligo-acetylene derivative oligo-(E)-1,1′-bi(indenylidene) can be understood as a consequence of spatial delocalization caused by extended conjugation of π-systems between monomer building blocks. While an individual monomer exhibits localized electronic states separated by a large energy gap, the efficient π-overlap between monomer orbitals in the oligomer chains results in the formation of lower energy extended states. This behavior is evident in the spectroscopic data in Figures 3 and 4, where the LUMO of an isolated monomer lies ∼1.1 eV higher in energy than the extended state (relative to EF).
These conclusions were confirmed by GW calculations30,31 performed to model both an isolated monomer 2 and an n = 4 oligomer chain having the same structure indicated by the partial wireframe image (orange overlay) in Figure 3a–c. The calculated LDOS of the isolated monomer LUMO is depicted in Figure 4c. It closely resembles the experimental dI/dV map (Figure 4b), with strong intensity arising from the 3- and 3′-positions in the (E)-1,1′-bi(indenylidene). When monomer subunits are joined in the n = 4 oligomer chain, GW calculations indicate the formation of an extended electronic state that is lower in energy by 1.0 ± 0.1 eV compared to the monomer LUMO, assuming typical physisorption distances of 0.33 ± 0.03 nm32 (these values were obtained using an image-charge model of the surface combined with the GW calculation,33 see the Supporting Information). This state extends continuously along the oligomer backbone as depicted in the theoretical LDOS of Figure 3c. The energetic lowering and spatial extent of the calculated oligomer LDOS closely resembles the experimental data. The nodal pattern of the LUMO can also be qualitatively explained via a simple one-dimensional particle-in-a-box model whereby for an oligomer of length n (with 6n carbon atoms along the backbone) the 6n π-electrons fill the first 3n levels. The LUMO level, being the next highest level, thus exhibits 3n+1 antinodes.
The agreement between theory and experiment extends to the appearance of alternating bond lengths in the oligomer. Figure 2c shows that the calculated bond lengths along the conjugated backbone of an n = 4 oligomer chain alternate in the same pattern as observed experimentally for the C–C/C=C bond lengths between indenyl groups (green circles represent long bonds between indenyl groups, red circles represent short bonds between indenyl groups). In the calculation this behavior arises from a combination of Peierls distortion and boundary effects (since the boundary fixes the bond phase). As expected, there is a discrepancy between the nc-AFM experimental results and the theoretical calculations regarding the absolute magnitude of the bond-alternation effect. Whereas the AFM images (Figure 2a) largely exaggerate bond length variations (∼50%), the calculation predicts bond-to-bond variations of only ∼3%. This discrepancy does not arise from oligomer interaction with the substrate lattice, since similar bond variations are experimentally observed for oligomers lying along different substrate crystallographic directions. Also, artifacts caused by long-range forces27 can be excluded because the respective bonds within the chains are in a symmetrically equivalent chemical environment, which leads to an equivalent van der Waals background. The anomalously large bond alternation observed experimentally is likely an image distortion caused by tilting of the CO molecule adsorbed to the AFM tip.27,34
In conclusion, we have grown 1D chains of the oligo-acetylene derivative oligo-(E)-1,1′-bi(indenylidene) on a surface through a thermally induced radical cyclization/step-growth polymerization process. Individual oligomer chains exhibit extended 1D electronic states and alternating bond lengths. This radical polymerization process on surfaces provides a new route toward fully conjugated low-bandgap derivatives of all-trans polyacetylene.
Acknowledgments
Research supported by the Office of Naval Research BRC Program (molecular synthesis, characterization, and STM imaging), by the U.S. Department of Energy Office of Basic Energy Sciences Nanomachine Program under contract no. DE-AC02-05CH11231 (STM and nc-AFM instrumentation development, AFM imaging, GW calculations, and surface renormalization analysis), and by the National Science Foundation awards DMR-1206512 (image analysis) and DMR10-1006184 (DFT calculations). A.R. acknowledges fellowship support by the Austrian Science Fund (FWF): J3026-N16. D.G.O. acknowledges fellowship support by the European Union under FP7-PEOPLE-2010-IOF. S.G.L. acknowledges support from a Simons Foundation Fellowship in Theoretical Physics. We acknowledge the assistance of the XSEDE computational cluster resource provided by NICS (Kraken), supported by the National Science Foundation.
Supporting Information Available
Additional information and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
A.R.: Institute of Applied Physics, Vienna University of Technology, Wiedner Hauptstrasse 8-10, 1040 Vienna, Austria.
Author Contributions
A.R. and S.W. contributed equally. A.R. and S.W. were responsible for the design of the experimental strategy and wrote the manuscript. A.R., S.W., and H.T. performed the nc-AFM and STM/STS measurements. P.G. and G.E. designed and synthesized the molecular building blocks used in this study. L.Z.T. conducted the DFT and GW calculations. D.G.O., Y.-C.C., A.B., and M.M.U. assisted in the experimental setup, the UHV deposition of the molecules, and the STM/STS measurements. S.G.L. supervised the DFT and GW calculations, helped with the interpretation, and edited the manuscript. F.F. supervised the molecular design and synthesis, helped with the interpretation of the experimental results, and edited the manuscript. M.F.C. supervised the nc-AFM and STM/STS measurements, helped with the interpretation of the experimental results, and edited the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Shirakawa H.; Louis E. J.; MacDiarmid A. G.; Chiang C. K.; Heeger A. J. J. Chem. Soc., Chem. Commun. 1977, 578–580. [Google Scholar]
- Friend R. H.; Gymer R. W.; Holmes A. B.; Burroughes J. H.; Marks R. N.; Taliani C.; Bradley D. D. C.; Dos Santos D. A.; Brédas J. L.; Lögdlund M.; Salaneck W. R. Nature 1999, 397, 121–128. [Google Scholar]
- Coropceanu V.; Cornil J.; Da Silva Filho D. A.; Olivier Y.; Silbey R.; Brédas J.-L. Chem. Rev. 2007, 107, 926–952. [DOI] [PubMed] [Google Scholar]
- Brabec C. J.; Sariciftci N. S.; Hummelen J. C. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar]
- Organic Light Emitting Devices: Synthesis, Properties and Applications; Müllen K., Scherf U., Eds.; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- Zaumseil J.; Sirringhaus H. Chem. Rev. 2007, 107, 1296–1323. [DOI] [PubMed] [Google Scholar]
- Barth J. V; Costantini G.; Kern K. Nature 2005, 437, 671–679. [DOI] [PubMed] [Google Scholar]
- Bartels L. Nat. Chem. 2010, 2, 87–95. [DOI] [PubMed] [Google Scholar]
- Elemans J. A. A. W.; Lei S.; De Feyter S. Angew. Chem., Int. Ed. 2009, 48, 7298–7332. [DOI] [PubMed] [Google Scholar]
- Cai J.; Ruffieux P.; Jaafar R.; Bieri M.; Braun T.; Blankenburg S.; Muoth M.; Seitsonen A. P.; Saleh M.; Feng X.; Müllen K.; Fasel R. Nature 2010, 466, 470–473. [DOI] [PubMed] [Google Scholar]
- Grill L.; Dyer M.; Lafferentz L.; Persson M.; Peters M. V.; Hecht S. Nat. Nanotechnol. 2007, 2, 687–691. [DOI] [PubMed] [Google Scholar]
- Hla S. W.; Bartels L.; Meyer G.; Rieder K. Phys. Rev. Lett. 2000, 85, 2777–2780. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Mandal S. K.; Hu C.; Tateyama Y.; Goedecker S.; Tsukamoto S.; Hasegawa T.; Gimzewski J. K.; Aono M. J. Am. Chem. Soc. 2011, 133, 8227–8233. [DOI] [PubMed] [Google Scholar]
- Okawa Y.; Aono M. Nature 2001, 409, 683–684. [DOI] [PubMed] [Google Scholar]
- Zwaneveld N. A. A.; Pawlak R.; Abel M.; Catalin D.; Gigmes D.; Bertin D.; Porte L. J. Am. Chem. Soc. 2008, 130, 6678–6679. [DOI] [PubMed] [Google Scholar]
- Treier M.; Richardson N. V.; Fasel R. J. Am. Chem. Soc. 2008, 130, 14054–14055. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Q.; Kepčija N.; Kleinschrodt M.; Diller K.; Fischer S.; Papageorgiou A. C.; Allegretti F.; Björk J.; Klyatskaya S.; Klappenberger F.; Ruben M.; Barth J. V. Nat. Commun. 2012, 3, 1286. [DOI] [PubMed] [Google Scholar]
- De Oteyza D. G.; Gorman P.; Chen Y.-C.; Wickenburg S.; Riss A.; Mowbray D. J.; Etkin G.; Pedramrazi Z.; Tsai H.-Z.; Rubio A.; Crommie M. F.; Fischer F. R. Science (New York, N.Y.) 2013, 340, 1434–1437. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Zhang C.; Li Z.; Kong H.; Tan Q.; Hu A.; Xu W. J. Am. Chem. Soc. 2013, 135, 8448–8451. [DOI] [PubMed] [Google Scholar]
- Mohamed R. K.; Peterson P. W.; Alabugin I. V. Chem. Rev. 2013, 113, 7089–7129. [DOI] [PubMed] [Google Scholar]
- Facchetti A. Chem. Mater. 2011, 23, 733–758. [Google Scholar]
- Zade S. S.; Zamoshchik N.; Bendikov M. Acc. Chem. Res. 2011, 44, 14–24. [DOI] [PubMed] [Google Scholar]
- Repp J.; Liljeroth P.; Meyer G. Nat. Phys. 2010, 6, 975–979. [Google Scholar]
- Wennerström O. Macromolecules 1985, 18, 1977–1980. [Google Scholar]
- Giessibl F. J. Rev. Mod. Phys. 2003, 75, 949–983. [Google Scholar]
- Gross L.; Mohn F.; Moll N.; Liljeroth P.; Meyer G. Science (New York, N.Y.) 2009, 325, 1110–1114. [DOI] [PubMed] [Google Scholar]
- Gross L.; Mohn F.; Moll N.; Schuler B.; Criado A.; Guitián E.; Peña D.; Gourdon A.; Meyer G. Science (New York, N.Y.) 2012, 337, 1326–1329. [DOI] [PubMed] [Google Scholar]
- Van der Lit J.; Boneschanscher M. P.; Vanmaekelbergh D.; Ijäs M.; Uppstu A.; Ervasti M.; Harju A.; Liljeroth P.; Swart I. Nat. Commun. 2013, 4, 2023. [DOI] [PubMed] [Google Scholar]
- Prall M.; Wittkopp A.; Schreiner P. R. J. Phys. Chem. A 2001, 105, 9265–9274. [Google Scholar]
- Hybertsen M. S.; Louie S. G. Phys. Rev. Lett. 1985, 55, 1418–1421. [DOI] [PubMed] [Google Scholar]
- Deslippe J.; Samsonidze G.; Strubbe D. A.; Jain M.; Cohen M. L.; Louie S. G. Comput. Phys. Commun. 2012, 183, 1269–1289. [Google Scholar]
- Henze S. K. M.; Bauer O.; Lee T.-L.; Sokolowski M.; Tautz F. S. Surf. Sci. 2007, 601, 1566–1573. [Google Scholar]
- Neaton J.; Hybertsen M. S.; Louie S. G. Phys. Rev. Lett. 2006, 97, 216405. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Boneschanscher M. P.; Swart I.; Vanmaekelbergh D.; Liljeroth P. Phys. Rev. Lett. 2011, 106, 046104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.