

Abstract
The E2F transcription factors are critical regulators of cell cycle and cell fate control. Several classes of E2F target genes have been categorized based on their roles in DNA replication, G2/M and mitosis, apoptosis, DNA repair, etc. How E2Fs coordinate the appropriate and timely expression of these functionally disparate gene products is poorly understood at a molecular level. We previously showed that the E2F1 binding partner Jab1/CSN5 promotes E2F1-dependent induction of apoptosis but not proliferation. To better understand how Jab1 regulates E2F1 dependent transcription, we performed gene expression analysis to identify E2F target genes most and least affected by shRNA depletion of Jab1. We find that a significant number of apoptotic and mitotic E2F target genes are poorly expressed in cells lacking Jab1/CSN5, whereas DNA replication genes are generally still highly expressed. Chromatin immunoprecipitation analysis indicates that both Jab1 and E2F1 co-occupy apoptotic and mitotic, but not DNA replication target genes. We explored a potential connection between PI3K activity and Jab1/E2F1 target gene induction, and found that E2F1/Jab1 co-induction of apoptotic target genes can be inhibited by activated PI3K. Furthermore, PI3K activity interferes with formation of the E2F1/Jab1 complex by co-immunoprecipitation. Jab1/CSN5 is upregulated in a variety of human tumors, but it’s unclear how its pro-proliferatory and apoptotic functions are regulated in this context. We explored the link between increased Jab1 levels and PI3K function in tumors and detected a highly significant correlation between elevated Jab1/CSN5 levels and PI3K activity in breast, ovarian, lung and prostate cancers.
Key words: E2F, apoptosis, gene regulation, Jab1, CSN5, PI3K, G2/M
Introduction
The E2F transcription factors are critical regulators of cell cycle entry, proliferation and cell fate control. E2F1, the founding member of the E2F family, is able to induce transcription of gene products involved in DNA replication, G2/M, DNA repair and programmed cell death.1,2 How E2Fs regulate the appropriate and timely expression of these functionally distinct gene products is poorly understood. E2F1 is not expressed in quiescent cells, but is transcriptionally induced in G1 subsequent to mitogenic cell cycle entry. E2Fs generally contain DNA binding and transactivation domains, and repression domains that bind to the pocket proteins Rb, p107 and p130.3 Binding of pocket proteins to E2Fs represses transcriptional activity of the activator E2Fs and also allows co-recruitment of chromatin modifying enzymes to E2F promoters to further regulate target gene expression.4 This causes repression of E2F target genes in quiescence and expression during cell cycle entry following cyclin/Cdk activation. Functional inactivation of RB1, either through direct loss of the RB1 gene itself or through mutations in upstream regulators (cyclins and CDKs) occurs frequently in various human cancers. Loss of RB1 function can lead to activation of E2Fs, causing the excess expression of proliferative target genes frequently seen activated in tumors. In some settings, E2F activity also promotes p53 dependent and independent apoptosis as a tumor suppressive mechanism.5,6 Mechanisms distinguishing E2F1-dependent proliferation verses apoptosis target gene expression are poorly understood.
E2F1 normally accumulates as a part of growth stimulation and during cellular exit from quiescence. Accumulation of E2F1 has the potential to induce context dependent proliferation or apoptosis. However, many apoptotic genes are not expressed during normal cell cycle entry but are robustly induced by E2F activity subsequent to inactivation of the RB pathway.7 Our previous work has identified two potential regulators of the balance between E2F replication and apoptotic target gene expression. First, the phosphatidyl inositol-3-kinase (PI3K), which is activated during exit from quiescence at the same time E2F1 begin to accumulate and activate target gene expression, acts as a negative regulator of E2F1 apoptosis and apoptotic transcriptional output.8,9 PI3K phosphorylates plasma membrane localized phosphoinositides that serve to recruit and activate downstream signaling molecules like Akt. PI3K is also frequently activated by oncogenic mutations in various human cancers.10 This raises the possibility that co-occurrence of RB1 pathway mutations that unleash E2F activity, and PI3K mutations that suppress Rb/E2F apoptosis, may be common in human tumorigenesis.
We also previously shed light on the distinction of E2F1 induction of apoptosis vs. proliferation by demonstrating that Jab1, subunit #5 (CSN5) of the Cop9 signalosome, binds to the “marked box” region of E2F1, but not to the highly similar region of E2F2 or E2F3, and enhances E2F1 apoptosis and p53 induction.11 Jab1 (c-Jun activation domain-binding protein) was originally identified as a specificity co-factor for c-Jun and JunD but not the JunB or v-Jun members of the AP-1 transcription factor family.12 Jab1/CSN5 resides as subunit five in the eight subunit containing COP9 signalosome complex (CSN). The COP9 signalosome complex (CSN) was originally characterized in Arabidopsis as a transcriptional repressor of “light-activated” genes normally repressed in the dark.13,14 The CSN is conserved in mammals, D. melanogaster, C. elegans and yeast. Loss of most of the CSN components leads to complex destabilization; however CSN5 is stable outside the complex as a monomer and can form poorly defined sub-complexes that vary depending on cell cycle stage and in response to various stresses.15,16 CSN5/Jab1 binds numerous cellular regulators, many of which are oncogenes or tumor suppressors.17 Apoptosis and proliferation appear to be two of the most common cellular events regulated by CSN5/Jab1 and the full COP9 signalosome.18
In this study, we determined the range of E2F1 target genes that are affected by shJab1 and found that the most influenced targets are G2/M and apoptotic, whereas E2F1 induction of DNA replication targets is least affected overall by the loss of Jab1. We used chromatin immunoprecipitation to demonstrate that Jab1 and E2F1 are co-recruited to several mitotic and apoptotic promoters. By contrast, Jab1 is not co-recruited with E2F1 to DNA replication target genes. We noticed that many of the Jab1 regulated E2F1 apoptotic target genes strikingly overlapped with a set of previously identified E2F1 target genes whose expression is blocked by PI3K signaling.9 This suggests a potential antagonistic relationship between Jab1 and PI3K in the regulation of E2F1-dependent apoptosis and apoptotic target gene expression. We used RT-PCR to demonstrate that PI3K blocks E2F1/Jab1 co-induction of several of these targets. Furthermore, co-immunoprecipitation shows that PI3K signaling interferes with formation of the E2F1/Jab1 complex. Finally, analysis of breast, ovarian, lung and prostate tumor microarray data sets demonstrate that Jab1/CSN5 levels, which are elevated in a variety of human cancers, correlates strikingly with activation of PI3K in each of these tumor types. These findings suggest the novel hypothesis that the pro-apoptotic function of E2F1/Jab1 may be countered by co-activation of the oncogenic PI3K pathway in some human tumors.
Results
Jab1/CSN5 mediates E2F1-dependent expression of mitotic and apoptotic but not DNA replication targets
We previously described the generation of shJAB1 REF52 cell lines showing significantly reduced Jab1 protein levels.11 Our previous results indicated that the loss of Jab1 does not affect E2F1 induction of the DNA replication PCNA gene. In contrast, E2F1 failed to induce p53 in Jab1 depleted cells. It was unclear if this was due to a failure of E2F1 to transcriptionally induce a p53 regulator like p19ARF, or if Jab1 loss affected p53 stability post-translationally. We hypothesized that E2F1 and Jab1 complexes may regulate expression of a subset of E2F1 target genes, owing to the fact that this complex promotes apoptosis but not excess proliferation. We used microarray analysis to test the validity of this model and identify the putative members of this category of E2F target genes. Control and shJAB1 targeted quiescent REF52 cells were infected with control or E2F1 expressing adenovirus. The wild-type E2F1 gene is present but not expressed in quiescent REF52 cells. RNA was isolated and applied to RAE230A rat Affymetrix arrays in duplicate to assess target gene expression in these genetic backgrounds. This analysis confirmed that Jab1 mRNA levels were reduced in shJAB1 cells by approximately 90% (Table S1).
Gene expression data was sorted based on fold-induction (FI) by E2F1 compared with CMV infection in wild-type cells (E2F WT/CMV WT). We focused only on genes induced at least 2-fold by E2F1 for further analysis of the effects of Jab1 loss. shJab1 pertains to E2F1 induced gene levels in shJab1 cells compared with E2F1 induced gene levels in wild-type cells (E2F-shJ/E2F WT). E2F1-induced genes were then sorted by comparing their percent induction in shJAB1 cells comparing with induction levels in control cells (shJab1). At one extreme of this list, shown in Figure 1, are genes that fail to be induced by E2F1 in the absence of Jab1/CSN5 (“least induced”). At the other end are E2F1-induced genes that display full, or near full, expression in shJab1 cells (“most induced”). We observed very few E2F1 induced genes that were further induced by Jab1 loss. E2F1 induced genes were split into two categories based on the effects of Jab1 loss to facilitate classification of genes. Genes induced only 4%–50% by E2F1 following Jab1 loss were placed in one category (least induced), and genes whose induction only 50% to 100% by E2F1 were placed in the other category (most induced). The entire list of genes is included in supplementary data (Table S1). Selected genes from each category are displayed in Figure 1.

Figure 1. Identification of E2F1 target genes requiring Jab1/CSN5 for expression. Control and shJab1 REF52 cells were infected with equal multiplicities of infection (25 min.o.i.) of control or E2F1-expressing adenovirus. RNA was harvested from infected cells and analyzed by rat Affymetrix arrays (RAE230A). E2F1 target genes induced at least 2-fold were sorted based on the effect that loss of Jab1/CSN5 had on E2F1-dependent expression. FI refers to “fold-induction” by E2F1 compared with CMV infection in control REF52 cells. shJAB1 is the percent expression of the gene in shJAB1 REF52 cells compared with E2F1-induction of the gene in control cells.
GO annotation and TRANSFAC analysis of all genes poorly induced by E2F1 in shJab1 indicated a surprising over-representation of genes involved in the mitotic cell cycle (Fig. S1A). Examples of E2F induced mitotic genes in this category include Bub1b, cyclin B1 and B2, Cdc2a, Cdc25b, Cenp3, Espl1, several mitotic kinesins, and Plk1 (p < 0.0001 for many M-phase related annotations). Promoter analysis (TRANSFAC) of this set of genes suggested a significant overrepresentation of nuclear factor Y (NF-Y) and E2F promoter elements. Similar analysis was performed for the set of E2F1 induced genes that remain highly expressed in shJab1 cells. GO annotation indicates a high probability (p < 0.0001) overrepresentation of DNA replication, chromosome cycle and DNA metabolism genes (Fig. S1B). Examples of genes in this category are Dut, Fen1, several MCMs, Orc6, several polymerases, replication factors (RFC3, RFC4, RPA1, RPA3) and ribonucleotide reductases M1 and M2. These genes were also highly enriched for E2F transcription factor binding sites (p < 0.0001). These results suggest that E2F gene induction can be affected by the presence of the E2F1 binding partner Jab1/CSN5. Furthermore, it would appear that genes involved in mitosis are generally more dependent on E2F1/Jab1 induction than DNA replication and other G1/S genes.
We directly tested the expression of several of the Jab1 affected and unaffected E2F1 target genes using quantitative real-time PCR (qRT-PCR) (Fig. 2). Control and shJab1 REF52 cells were brought to quiescence by serum deprivation and infected with control or E2F1 expressing adenovirus. mRNA was isolated for qRT-PCR at 24 h post-infection. We compared fold-induction by E2F1 in control and shJAB1 cells and are reporting results as fraction of the level of E2F induction in shJAB1 divided by fold-induction in control cells. Several of the genes from our microarray analysis failed to be induced by E2F1 in shJAB1 cells, compared with induction in control cells. For example, the mitotic genes Cdc2 and cyclin B1 were induced only ~30% by E2F1 in shJAB1 cells comparing with induction in control cells. Other genes, not relating to mitosis, displayed a similar pattern. For example, p19ARF was induced only 38% in shJab1 cells compared with wild type. Furthermore, Cyp26b1 and Prkaa2 (AMPK>2), two pro-apoptotic E2F1 target genes we previously identified in reference 9, were also poorly expressed in shJab1 cells. In contrast, the DNA replication genes RFC4, Dut and PCNA retain at least 80% E2F1-induction in shJab1 cells compared with induction in control cells. These findings suggest that E2F1 and Jab1 coordinate the expression of some (mitotic and apoptotic), but not all (DNA replication) target genes. RT-PCR consequently agreed with the findings from our microarray analysis.
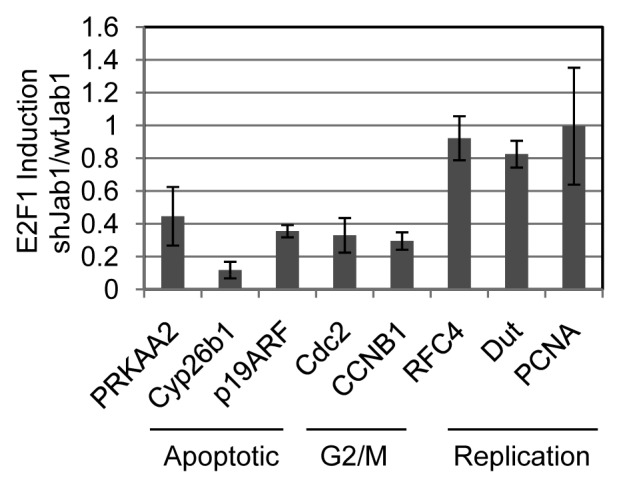
Figure 2. Depletion of Jab1/CSN5 impairs E2F1 dependent expression of mitotic and apoptotic but not replication targets. mRNA expression levels of PRKAA2 (AMPK>2), p19ARF, Cdc2, Cyclin B2 (CCNB2), Cyp26b1, RFC4, Dut and PCNA were measured using quantitative real-time PCR in control vs. shJAB1 REF52 cells. Expression levels were normalized to GAPDH levels and displayed as % expression in control REF52 cells compared with E2F1-dependent expression in shJAB1 cells.
E2F1 and Jab1 chromatin immunoprecipitation
We used chromatin immunoprecipitation (ChIP) to test the model that both E2F1 and Jab1 both bind to the promoters of co-regulated genes, but that Jab1 does not bind to E2F DNA replication promoters whose expression is unaffected by Jab1 loss. REF52 cells were cross-linked with formaldehyde, collected, lysed and sonicated to DNA fragment sizes around 500–800 nucleotides. DNA fragments were precipitated with control, anti-E2F1 or anti-Jab1 antisera. Recovered DNA was used in SYBR green real-time PCR to detect enrichment of E2F1 target gene promoters by anti-E2F1 or -Jab1 immunoprecipitation. Results were reported as fold-increased binding with antisera vs. control. We observed an increase in binding of E2F1 to all promoters tested, except the control HBB (hemoglobin ®) (Fig. 3A). E2F1 binding to the apoptotic and mitotic target gene promoters varied from around 20-fold increase for Cyp26b1 and Cdc2a, to 100-fold increase for Prkaa2 (AMPK>2). E2F1 binding was generally detected as stronger on the replication target genes, ranging from 300- to 500-fold increased promoter precipitation compared with control.
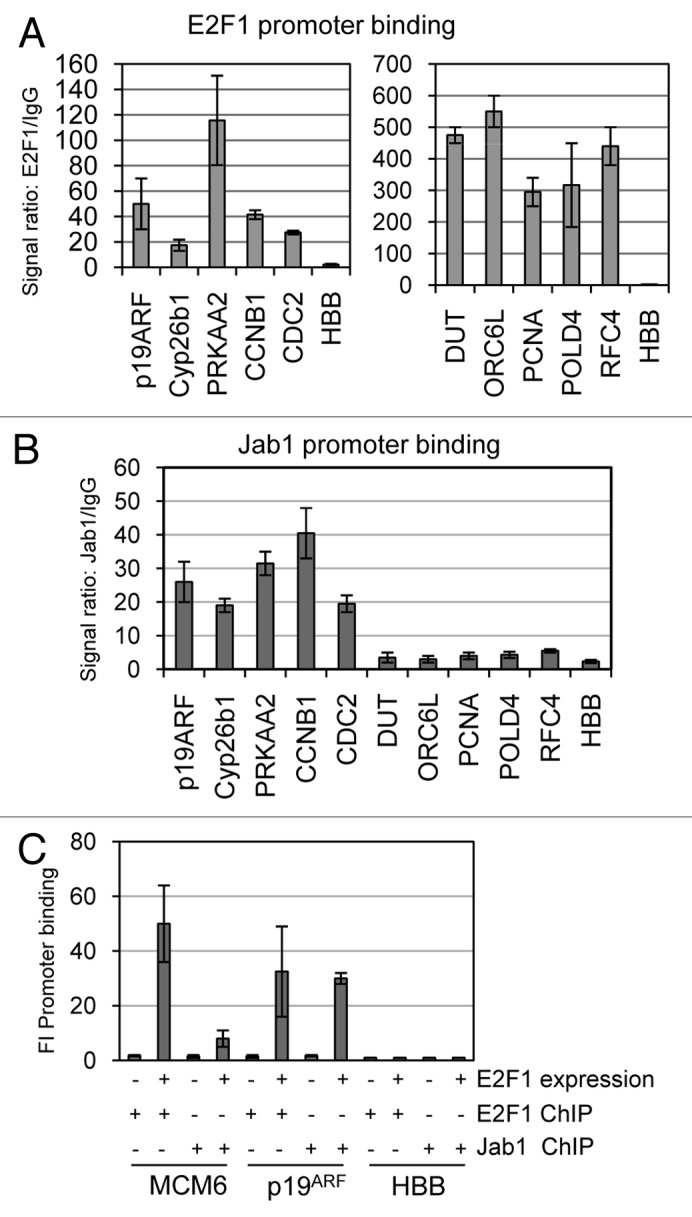
Figure 3. E2F1 and Jab1/CSN5 co-occupy mitotic and apoptotic target but not DNA replication gene promoters. E2F1 and Jab1 binding to E2F1 target gene promoters was assessed using chromatin IP (ChIP). REF52 cells were cross-linked with formaldehyde, chromatin was isolated and sonicated and immunoprecipitated with anti-E2F1 (A) or anti-Jab1/CSN5 (B) antisera. Precipitated DNA was analyzed by SYBR green real-time PCR using primers and results are displayed as a comparative fold-induced binding from precipitate using no antibody vs. experimental antibody. Specific promoters analyzed are listed below each graph. ChIP results are separated into two parts in section A because of the very different scales for the two classes of genes. (C) Serum deprived quiescent REF52 cells were transfected with E2F1 or vector control. Chromatin was isolated and immunoprecipitated with anti-E2F1 or anti-Jab1 antisera.
Immunoprecipitation with anti-Jab1 antisera failed to enrich any of the E2F1 replication target gene promoters like Dut, Orc6l, PCNA, POLD4, RFC4 or the control HBB (Fig. 3B). This result is consistent with our finding that Jab1 is also not required for E2F1 dependent expression of this class of E2F targets. By contrast, anti-Jab1 was able to immunoprecipitate the pro-apoptotic promoters p19ARF, Prkaa2 and Cyp26b1. Similarly, the mitotic promoters Cdc2 and CCNB1 were also enriched by ChIP by 20- and 40-fold. Jab1/CSN5 did not bind to any of these promoters in serum deprived REF52 cells that express no E2F1, suggesting that E2F1 expression and binding to the promoters is a prerequisite for Jab1 binding (Fig. 3C). However, transfection of E2F1 in quiescent cells restores binding of Jab1 to the p19ARF, but not the MCM6 promoter. These results indicate that Jab1/CSN5 and E2F1 both reside on the promoters of the genes requiring Jab1 and E2F1 for their expression (mitotic and apoptotic). DNA replication E2F1 targets, however, which generally do not appear to require Jab1/CSN5 for induction by E2F, have E2F1 but not Jab1 bound on their promoters in asynchronously growing cells.
Jab1/E2F1 induced genes counter-regulated by PI3K
We previously demonstrated that expression of a significant number of E2F1 target genes are transcriptionally repressed by serum activated PI3K signaling.9 Some of these genes function as mediators of E2F1-induced apoptosis. By contrast, PI3K did not repress expression of E2F1 DNA replication target genes. Inspection of the list of genes requiring Jab1/CSN5 for induction by E2F1 revealed a striking number of genes that were also identified in the microarray for PI3K repressed E2F1 target genes. For example, both Cyp26b1 and Prkaa2/AMPK>2 require Jab1 for E2F1 induced expression and are also repressed by PI3K signaling. To determine which other E2F1-induced genes are counter- regulated by Jab1/CSN5 and PI3K, we combined and normalized the microarray data sets from both the shJab1 (this study) and PI3K modulation of E2F1 target gene inductions. We performed unsupervised clustering on the genes and included only the lanes with E2F1 gene induction to simplify the visualization of the effects of Jab1 loss (shJab1) or Serum activation on gene induction. Expression of most of the E2F target genes were not reduced either by shJAB1 or by serum treatment. However, two unique subsets of genes emerged from this analysis (Fig. 4A). The first cluster was comprised of E2F1 induced target genes showing significantly reduced expression in shJAB1 cells, but maintaining high E2F1 induction in the presence of serum. Analysis of the genes in this category revealed a significant presence of E2F mitotic target genes. The second cluster of E2F-induced genes showed impaired expression in shJab1 cells and repression by serum activated PI3K signaling. Examples of genes in this cluster are CDKN2a (the locus containing p19ARF), Cyp26b1 and Prkaa2 (AMPK>2). Our results indicate that E2F induced genes requiring Jab1 can be divided into two categories, those that are repressed by Serum/PI3K signaling, and those that are not.
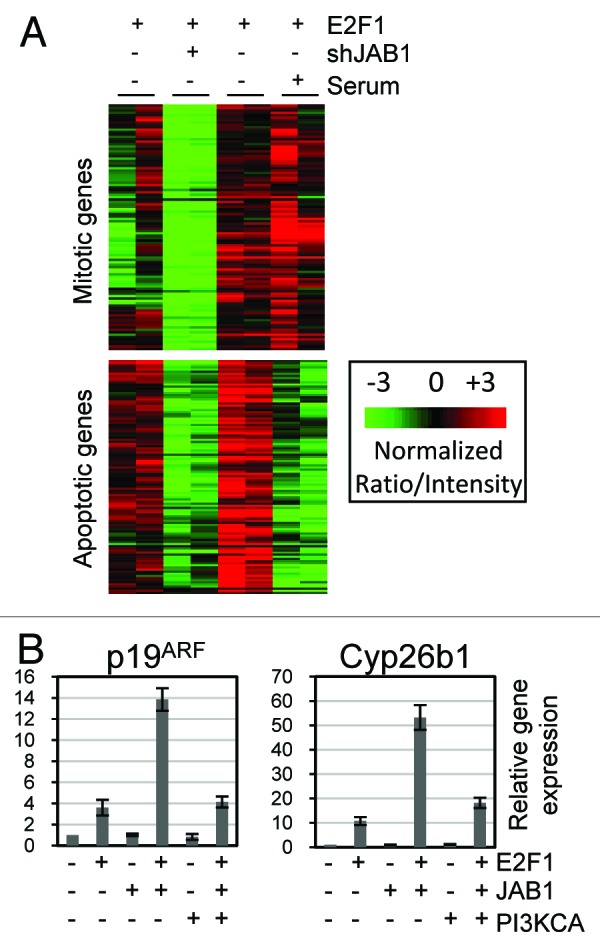
Figure 4. PI3K counter-regulates Jab1 induction of E2F1 apoptotic target genes. (A) Microarray data sets from this study (shJab1) and a previous study looking at Serum/PI3K repression of E2F1 target genes were combined, normalized using distance weighted discrimination (DWD), and subjected to unsupervised hierarchical clustering. Clusters were visualized using Java TreeView. Red shading represents relatively higher gene expression, and green shading corresponds to relatively lower gene expression. Only expression arrays with E2F1 added are included, to better display reductions in gene expression caused by the shJAB1 or Serum/PI3K lanes. (B) PI3K activity blocks Jab1/E2F1 target gene induction. Quiescent REF52 cells were infected with control, E2F1, Jab1 and PI3K expressing adenovirus, mRNA was isolated 24 h post-infection and used for real-time PCR analysis looking at p19ARF and Cyp26b1 gene induction. Results are reported as fold-inductions compared with gene expression in control (CMV) infected cells.
We further explored the relationship between Jab1 and PI3K counter-regulation of E2F1 target gene expression using RT-PCR analysis looking at the p19ARF and Cyp26b1 genes (Fig. 4B). Quiescent REF52 cells were infected with adenovirus expressing control, E2F1, Jab1, and/or PIK3CA. A reduced multiplicity of infection was used for E2F1 to have minimal effect on target gene induction on its own. Jab1 expression had no effect on either p19ARF or Cyp26b1 gene expression. Both p19ARF and Cyp26b1 were induced strongly, however, by co-expression of E2F1 and Jab1. Induction of p19ARF increased from 4-fold with E2F1 alone to 14-fold with E2F1 plus Jab1. Similarly, Cyp26b1 expression increased from 10-fold with E2F1 alone to 50-fold with both E2F1 and Jab1. Since our combined microarray experiments discussed in 4a suggested that both p19ARF and Cyp26b1 are PI3K repressible E2F1 target genes, we asked if addition of PI3K was capable of blocking the synergistic co-activation of these genes by E2F1 and Jab1. PI3K expression alone failed to induce or repress either gene. However, co-expression of PIK3CA significantly blocked the induction of both genes by co-expression of E2F1 and Jab1. In the case of p19ARF, expression was reduced from 14-fold (E2F1 + Jab1) to 4-fold (E2F1 + Jab1 + PIK3CA). Similarly, Cyp26b1 was reduced from ~50-fold to 20-fold by co-expression of PIK3CA. These experiments are consistent with the idea that PI3K impairs gene expression by modulating the function of the E2F1/Jab1 complex but do not rule out the possibility that PI3K affects E2F1 target gene expression by another mechanism.
PI3K inhibits Jab1/CSN5 binding to E2F1
We demonstrated that Jab1 and E2F1 contribute to gene expression, and that this effect on gene expression can be inhibited by PI3K expression. We tested the notion that PI3K activity may negatively control E2F1 and Jab1/CSN5 protein interaction using co-immunoprecipitation. REF52 cells were infected with Ad-Jab1 and treated with serum, either with or without the PI3K inhibitor LY294002. Protein lysates were immunoprecipitated with an anti-E2F1 antibody and immunoblotted with anti-Jab1 or anti-E2F1 antisera. Immunoblots of cell extracts indicate that Jab1 levels do not change following treatment with LY294002. Immunoprecipitated E2F1 protein levels were reduced in LY294002 treated cells and in whole cell extracts, suggesting that inhibiting PI3K activity reduces E2F1 protein levels. In spite of this reduction in E2F1 levels, we observe a large increase in co-immunoprecipitated Jab1/CSN5 following treatment with LY294002 (see Fig. 5). These findings suggest that PI3K activation may block expression of E2F1 apoptotic target genes and apoptosis induction in part by interfering with formation of the pro-apoptotic E2F1/Jab1 complex.
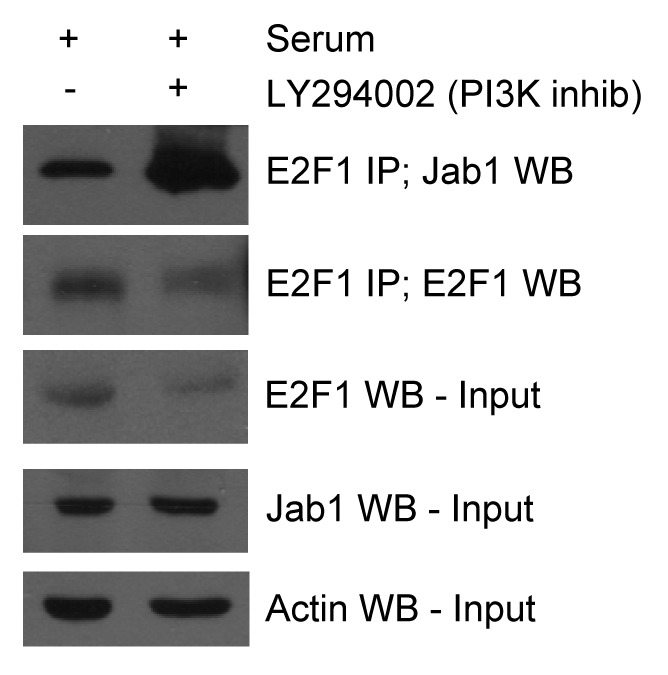
Figure 5. PI3K inhibition promotes Jab1 binding to E2F1. PI3K inhibition promotes Jab1/E2F1 complex formation. REF52 cells were infected with Ad-Jab1 (100 min.o.i.) and treated with vehicle or the PI3K inhibitor LY294002. Protein lysates were isolated and endogenous E2F1 was immunoprecipitated with anti-E2F1 antisera and immunoblotted with anti-Jab1 antisera. Anti-Jab1 and -Actin were used to confirm equal protein input levels.
PI3K activity significantly elevated in tumors with increased Jab1/CSN5
Jab1/CSN5 appears to be an important positive regulator of cellular proliferation. Jab1/CSN5 and other CSN subunits have been recovered from several genome-wide proliferation screens looking at human cancer cells or D. melanogaster cells.19,20 CSN5 can promote proliferation by interacting directly with the cell cycle inhibitor p27KIP1, inducing its relocalization to the cytoplasm and degradation.21 CSN5 is amplified in diverse human tumor types, for example breast, lung, lymphoma, ovarian, pancreatic and prostate.18 Furthermore, elevated CSN5 levels significantly correlates with reduced p27 levels in some tumor types.22,23 Jab1 appears to function like an oncogene in some tumors and is associated with enhanced proliferation, but Jab1 can also induce E2F1 mediated apoptosis. It is unclear what mechanism control Jab1/CSN5 directed proliferation in some conditions and apoptosis in others. Because we have demonstrated a role for PI3K/Akt activity in suppressing E2F1 induced apoptosis, and E2F1/Jab1 mediated apoptotic target gene expression, we assessed if tumors with high levels of Jab1/CSN5 also displayed elevated PI3K activity. We downloaded tumor data sets from NCBI, including two breast (GSE1456, GSE4922), an ovarian (GSE3149), two lung (GSE12667 and GSE3141) and a prostate (GSE6919). We applied a PI3K gene expression signature across each NCBI data set to obtain a putative PI3K activation score for each tumor, as we and others have done in previous studies using gene expression signatures of oncogenic pathways to predict the activation status of the pathway.9,24 The PI3K score is displayed as a color bar below the Jab1 graph, where blue shades represent poor predicted PI3K activation and red colors represent high activity. Each data set was sorted based on Jab1/CSN5 MAS5 levels, from lowest to highest. Jab1 levels vary widely across each tumor data set. We noticed a striking correlation between increased Jab1/CSN5 levels and elevated PI3K activity across these tumors. Tumors of each origin (breast, ovarian, lung and prostate) showed a highly significant increase in PI3K activity when Jab1/CSN5 levels were elevated (Fig. 6). We used linear regression to determine a correlation coefficient between Jab1/CSN5 MAS5 levels and PI3K activity and determined that the p values for this correlation were <0.0001 for each tumor, except for the ovarian (GSE3149) which had a p value of p = 0.0003. Analysis of the genes included in the “PI3K signature” indicates that Jab1/CSN5 was not included. We tested whether increased Jab1/CSN5 levels in the breast cancer data sets (GSE1456 and GSE4922) correlated with increased E2F1 or other members of the COP9 signalosome. Jab1/CSN5 showed a positive correlation with E2F1 in both data sets (p = 0.0071 for GSE1456 and p = 0.0007 for GSE4922). However, of the other CSN components, only CSN6 showed a positively correlated relationship with Jab1/CSN5 in both data sets (p = 0.202 in GSE1456 and p = 0.0008 in GSE4922). This suggests that at least in these two breast cancer data sets, increased levels of Jab1/CSN5 occurs independently of increases in the full CSN.
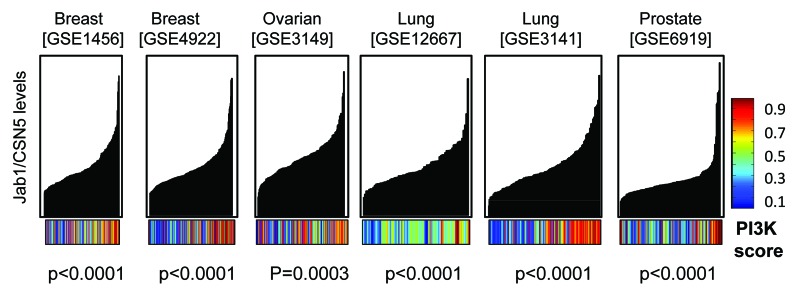
Figure 6. Increased Jab1/CSN5 levels in various tumor types correlates with elevated PI3K activity. Publically available data sets for different tumor types were downloaded from the NCBI website. Tumor data sets were sorted based on increasing Jab1/CSN5 mas5 levels. The x-axis are individual tumors, and the y-axis is relative Jab1/CSN5 mas5 levels in each tumor. A “score” for putative PI3K activity based on gene expression patterns was derived from binary regression analysis in MATLAB. PI3K scores are displayed as a color bar with blue representing low activity and red, high activity. Correlations between PI3K score and Jab1/CSN5 levels were determined by linear regression analysis using GraphPad software.
Discussion
The E2F transcription factors control a myriad of genes involved in all stages of cell cycle progression. This control extends from repression of genes in quiescent cells, and activation of the same genes during G1/S or G2/M. The mechanisms controlling differential E2F gene expression during G0 and G1/S are becoming clearly understood. For example, repressive E2Fs like E2F4 can inhibit expression of an E2F target gene during G0, and binding to the E2F promoter element is shifted toward activator E2F occupation during G1/S. Also, both positive and negative acting E2F elements have been identified in a single promoter.25 However, it remains largely unclear what mechanisms allow distinguished expression patterns of subsets of E2F target genes, including DNA replication, G2/M and apoptosis genes. It has been demonstrated that at least some of E2F promoter specificity can be coordinated through interactions with other sequence-specific transcription factors. For example, E2F3 interacts with TFE3 to activate expression of the DNA pol> p68 promoter, which contains adjacent E2F and E-Box binding elements.26 Likewise, E2F2 and E2F3 bind to RYBP/YY1 to activate expression of Cdc6, which contains E2F and YY1 promoter elements.27 Finally, B-Myb has been implicated in G1/S vs. G2/M E2F target gene expression.28 B-Myb is induced by E2Fs during G1/S and then coordinates expression of some G2/M genes like Cdc2. In this case, expression of the G2/M genes is delayed since B-Myb is not expressed until G1/S. E2Fs and B-Myb thus define a feed-forward loop between E2Fs and E2F/Myb temporal control of G2/M target genes. Our results indicate that Jab1 binding to E2F1 facilitates preferential expression of apoptotic and G2/M but not DNA replication target genes. Our results differ from previous efforts toward understanding E2F target gene specificity in that Jab1 is not thought to be a sequence specific DNA binding transcription factor. In fact, we do not detect Jab1/CSN5 binding to E2F target gene promoters by ChIP when E2F1 is not expressed. Jab1/CSN5 binding to these promoters is restored when E2F1 expression is restored, suggesting an indirect association with DNA through binding to promoter associated E2F1.
Several other reports have linked Jab1 and the CSN to target gene expression. CSN5 interacts with a myriad of cancer associated transcription factors like p53, HIF-1>, c-jun, JunD, SMAD4, SMAD7, Estrogen receptor >, Progesterone receptor and others.12,29-34 Despite the ability to interact with a wide variety of different factors, Jab1 also exhibits specificity in transcription factor family binding. Jab1 was originally described as c-jun binding protein, and was then shown to also interact with JunD but not the related family members JunB or v-jun.12 Furthermore, we reported Jab1 binding to E2F1 but not to the structurally and functionally related E2F2 or E2F3.
The CSN was first discovered in Arabidopsis as a repressor of light-dependent transcription and growth.13,14 Interestingly, conditional deletion of Csn8 in T-lymphocytes and MEFs indicate that the CSN is crucial for cell cycle entry from quiescence but not essential for G1/S transition in already cycling cells.35 This phenotype was linked to altered gene regulation in these cells. Loss of Csn8 led to increased background expression of cell cycle genes like E2F1, Cyclin D2 and Cyclin E1, genes that are normally induced by antigen stimulation, suggesting a repressive role for the CSN in the expression of these E2F target genes. Further, these genes fail to be further induced following induction. In these instances CSN1 and CSN8 bound directly to the promoters of Ccnd2, CDK4 and Cdkn1a by ChIP assay. These data indicate that the CSN can negatively regulate expression of some E2F target genes prior to cell cycle induction and act as a positive regulator after stimulation. It is possible that the exact nature of the complex and subunits controlling expression of these targets varies with the stage of the cell cycle.
We demonstrated that PI3K is able to block expression of Jab1/E2F1 apoptotic target genes but does not appear to block G2/M target gene expression. It is possible that this apparent difference is due to the unique timing of PI3K activation during G1/S transition, when repression of apoptotic targets would be most beneficial, but not during the G2/M transition of the cell cycle. If PI3K activation is able to block E2F1/Jab1 expression of potentially tumor-suppressive apoptotic target genes, it is possible that this could contribute to tumorigenesis. Since Jab1 levels are elevated in a variety of human tumors, we explored a potential connection between excess Jab1 and increase PI3K activity, as a potential mechanism to counteract E2F1/Jab1 apoptosis. We determined that predicted PI3K activity is significantly heightened in breast, ovarian, lung and prostate tumors displaying elevated Jab1/CSN5 levels. These findings suggest the possibility that Jab1/CSN5, when elevated in certain tumors, may evade triggering E2F1 apoptosis through the frequent, simultaneous co-activation of PI3K activity.
Materials and Methods
Cell culture, DNA plasmids and adenovirus
REF52 (rat embryo fibroblast) cells were cultured in DMEM media containing 10% fetal calf serum. Cells to be infected were brought to quiescence by plating (3 x 105 cells/60 mm plate) in serum containing media for 24 h and then washing and replacing with 0.25% serum containing media. Cells were deprived of serum for 48 h before infecting with adenovirus. Cell numbers were determined prior to infection to infect with equal adenoviral multiplicities. Floating and adherent cells were harvested at 40 h post-infection, and assayed for apoptosis using the BD PharMingen PE-conjugated caspase-3 antibody kit. LY294002 was purchased from Sigma and used at a concentration of 50 uM. Adenovirus expressing hemagglutin (HA)-tagged E2F1, chimeric E2Fs, Jab1 and PI3K, and control adenovirus not expressing any gene have been described previously in references 8, 11 and 24. shJAB1 REF52 cells were previously generated and verified for Jab1 knockdown by immunoblotting.11 This cell line was also confirmed to have reduced Jab1 mRNA by our microarray analysis. This information is included at the bottom of Table S1.
RNA isolation, real-time PCR and microarray analysis
RNA was isolated from cells using QIAshredder and RNeasy Midi Kits from QIAGEN. We used the QuantiTect SYBR Green RT-PCR kit from QIAGEN according to manufacturer’s specifications for our quantitative real-time PCR. Each experimental condition used 100 ng of RNA for reverse transcription and RT-PCR and was performed in triplicate and normalized against expression of GAPDH expression levels. Analysis was done with a StepOnePlus real-time PCR system (Applied Biosystem) according to the manufacturer’s protocol.
Ten micrograms of mRNA was used for analysis with the RAE230a Affymetrix rat chip. We extracted MAS5 and RMA (robust multi-array average) data from the.CEL files using the “R” programming set (www.r-project.org). We used a distance weighted discrimination (DWD) algorithm (https://genome.unc.edu/dwd) to normalize microarray data sets from the independent shJab1 (this study) and the serum/PI3K effects on E2F1 gene expression.
CMV WT and E2F WT are the average gene expression levels in wild-type cells infected with control (CMV) or E2F1 adenovirus. CMV-shJ and E2F-shJ refer to the same, but in shJAB1 cells. FI refers to “fold-induction” by E2F1 compared with CMV infection in control REF52 cells (E2F WT/CMV WT). We focused only on genes induced at least 2-fold by E2F1 for further analysis of the effects of Jab1 loss. shJ pertains to fold induction by E2F1 in shJab1 cells compared with wild-type cells (E2F-shJ/E2F WT).
We used the GeneCluster 3.0 program (http://rana.lbl.gov/EisenSoftware.htm) to cluster our RMA data in an unsupervised, hierarchical fashion and visualized the heatmap using Java Treeview (available from the same location). Genes and tumors were clustered using average linkage with the uncentered correlation similarity metric. The GATHER bioinformatics tool has been described and can be found online at http://gather.genome.duke.edu.36
A signature representative of PI3-kinase activation was developed as previously described in reference 24. Control and PI3K gene expression training sets were used to create a gene expression signature by selecting the genes whose expression status across the training samples correlated most highly with the presence of PI3K. Then the signature was used to predict the phenotypic status in tumor expression data sets by fitting a Bayesian probit regression model to determine the probability that a tumor sample exhibits concordance with PI3K signature derived gene expression values. The analysis predicts a probability of relative pathway status.
Chromatin immunoprecipitation
Sonicated genomic extracts were prepared from REF52 cells following the protocol described by Bomsztyk.37 Two 15-cm plates of REF52 cells for each condition were fixed with formaldehyde for 15 min at room temperature and then quenched with 125 mM glycine for 5 min. Cells were collected, washed and resuspended with 300 μl lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.0), diluted 10x with dilution buffer (1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris, pH 8.1). The DNA pellet was resuspended in IP buffer and sonicated with a Branson model 450 Sonicator to an approximate length of 550 nucleotides. Immunoprecipitations were set up using mock IP, anti-E2F1 (C-20 Santa Cruz) or anti-Jab1 antibodies at 4 degrees overnight and then treated with protein A-agarose beads for 60 min. Complexes were centrifuged and washed three times with cold IP buffer (150 mM NaCl, 5 mM EDTA, 0.5% NP40, 1% Triton-X 100, 50 mM Tris pH 7.5). 100 microliters of 10% Chelex 100 (Bio-Rad) was added to precipitated DNA samples and then ethanol precipitated.
Protein immunoblotting and immunoprecipitation
REF52 cells were harvested 40 h post-infection into microfuge tubes and re-suspended in boiling sample buffer. Equivalent amounts of protein were separated by SDS-PAGE, transferred to an Immobilon-P (Millipore) membrane and blocked in T-TBS containing 5% nonfat dry milk. Blots were then incubated with primary antiserum (1:1,000) at room temperature for four hours, washed three times with T-TBS buffer and then incubated with the appropriate secondary antiserum (1:2,000) for one hour at room temperature. Blots were processed using the ECL system (Amersham). Antiserum against E2F1 (c-20) was purchased from Santa-Cruz Biotechnology for immunoblot analysis.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by a University of Minnesota institutional American Cancer Society (A.C.S.) and Basil O’Connor March of Dimes Starter Scholar Research Award (5-FY09-134) research grants (T.C.H.).
Authors’ Contributions
T.C.H. designed research; X.L., H.L., O.A.I. and T.C.H. performed research; X.L., H.L., O.A.I. and T.C.H. analyzed data; and T.C.H. wrote the paper.
Glossary
Abbreviations:
- CSN
COP9 signalosome
- m.o.i.
multiplicity of adenoviral infection
- ChIP
chromatin immunoprecipitation
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/17618
References
- 1.Müller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–85. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21:4684–99. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol. 2008;9:713–24. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- 4.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci U S A. 1994;91:3602–6. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–11. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 7.Komori H, Enomoto M, Nakamura M, Iwanaga R, Ohtani K. Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J. 2005;24:3724–36. doi: 10.1038/sj.emboj.7600836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallstrom TC, Nevins JR. Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:10848–53. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 11.Hallstrom TC, Nevins JR. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–23. doi: 10.1101/gad.1345006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383:453–7. doi: 10.1038/383453a0. [DOI] [PubMed] [Google Scholar]
- 13.Wei N, Chamovitz DA, Deng XW. Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell. 1994;78:117–24. doi: 10.1016/0092-8674(94)90578-9. [DOI] [PubMed] [Google Scholar]
- 14.Chamovitz DA, Wei N, Osterlund MT, von Arnim AG, Staub JM, Matsui M, Deng XW. The COP9 complex, a novel multisubunit nuclear regulator involved in light control of a plant developmental switch. Cell. 1996;86:115–21. doi: 10.1016/S0092-8674(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 15.Kwok SF, Solano R, Tsuge T, Chamovitz DA, Ecker JR, Matsui M, Deng XW. Arabidopsis homologs of a c-Jun coactivator are present both in monomeric form and in the COP9 complex, and their abundance is differentially affected by the pleiotropic cop/det/fus mutations. Plant Cell. 1998;10:1779–90. doi: 10.1105/tpc.10.11.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukumoto A, Tomoda K, Kubota M, Kato JY, Yoneda-Kato N. Small Jab1-containing subcomplex is regulated in an anchorage- and cell cycle-dependent manner, which is abrogated by ras transformation. FEBS Lett. 2005;579:1047–54. doi: 10.1016/j.febslet.2004.12.076. [DOI] [PubMed] [Google Scholar]
- 17.Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Shackleford TJ, Claret FX. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Björklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenperä J, Taipale J. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature. 2006;439:1009–13. doi: 10.1038/nature04469. [DOI] [PubMed] [Google Scholar]
- 20.Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620–4. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–5. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 22.Kouvaraki MA, Rassidakis GZ, Tian L, Kumar R, Kittas C, Claret FX. Jun activation domain-binding protein 1 expression in breast cancer inversely correlates with the cell cycle inhibitor p27(Kip1) Cancer Res. 2003;63:2977–81. [PubMed] [Google Scholar]
- 23.Esteva FJ, Sahin AA, Rassidakis GZ, Yuan LX, Smith TL, Yang Y, Gilcrease MZ, Cristofanilli M, Nahta R, Pusztai L, et al. Jun activation domain binding protein 1 expression is associated with low p27(Kip1)levels in node-negative breast cancer. Clin Cancer Res. 2003;9:5652–9. [PubMed] [Google Scholar]
- 24.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 25.Araki K, Nakajima Y, Eto K, Ikeda MA. Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene. 2003;22:7632–41. doi: 10.1038/sj.onc.1206840. [DOI] [PubMed] [Google Scholar]
- 26.Giangrande PH, Hallstrom TC, Tunyaplin C, Calame K, Nevins JR. Identification of E-box factor TFE3 as a functional partner for the E2F3 transcription factor. Mol Cell Biol. 2003;23:3707–20. doi: 10.1128/MCB.23.11.3707-3720.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlisio S, Halperin T, Vidal M, Nevins JR. Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J. 2002;21:5775–86. doi: 10.1093/emboj/cdf577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004;23:4615–26. doi: 10.1038/sj.emboj.7600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, Dubiel W. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–9. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bae MK, Ahn MY, Jeong JW, Bae MH, Lee YM, Bae SK, Park JW, Kim KR, Kim KW. Jab1 interacts directly with HIF-1alpha and regulates its stability. J Biol Chem. 2002;277:9–12. doi: 10.1074/jbc.C100442200. [DOI] [PubMed] [Google Scholar]
- 31.Wan M, Cao X, Wu Y, Bai S, Wu L, Shi X, Wang N, Cao X. Jab1 antagonizes TGF-beta signaling by inducing Smad4 degradation. EMBO Rep. 2002;3:171–6. doi: 10.1093/embo-reports/kvf024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim BC, Lee HJ, Park SH, Lee SR, Karpova TS, McNally JG, Felici A, Lee DK, Kim SJ. Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol. 2004;24:2251–62. doi: 10.1128/MCB.24.6.2251-2262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calligé M, Kieffer I, Richard-Foy H. CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor alpha by the proteasome. Mol Cell Biol. 2005;25:4349–58. doi: 10.1128/MCB.25.11.4349-4358.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chauchereau A, Georgiakaki M, Perrin-Wolff M, Milgrom E, Loosfelt H. JAB1 interacts with both the progesterone receptor and SRC-1. J Biol Chem. 2000;275:8540–8. doi: 10.1074/jbc.275.12.8540. [DOI] [PubMed] [Google Scholar]
- 35.Menon S, Chi H, Zhang H, Deng XW, Flavell RA, Wei N. COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat Immunol. 2007;8:1236–45. doi: 10.1038/ni1514. [DOI] [PubMed] [Google Scholar]
- 36.Chang JT, Nevins JR., Jr. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006;22:2926–33. doi: 10.1093/bioinformatics/btl483. [DOI] [PubMed] [Google Scholar]
- 37.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–85. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.