

Abstract
Infection with invasive Shigella species results in intestinal inflammation in humans but no symptoms in adult mice. To investigate why adult mice are resistant to invasive shigellae, 6~8-week-old mice were infected orally with S. flexneri 5a. Shigellae successfully colonized the small and large intestines. Mild cell death was seen but no inflammation. The infected bacteria were cleared 24 hours later. Microarray analysis of infected intestinal tissue showed that several genes that are involved with the sphingosine-1-phosphate (S1P) signaling pathway, a lipid mediator which mediates immune responses, were altered significantly. Shigella infection of a human intestinal cell line modulated host S1P-related genes to reduce S1P levels. In addition, co-administration of S1P with shigellae could induce inflammatory responses in the gut. Here we propose that Shigella species have evasion mechanisms that dampen host inflammatory responses by lowering host S1P levels in the gut of adult mice.
Keywords: Shigella flexneri, Inflammation, Sphingosine-1-phosphate, Intestine
INTRODUCTION
Invasive Shigella species cause bacillary dysentery, or shigellosis, which is identified by stomach cramps, fever, diarrhea, and bloody feces (1,2). It is common in infants in developing countries (3). Infection with Shigella organisms occurs primarily by the oral route, entering through the M cells of the intestinal epithelium and spreading to the basolateral side. Infection promotes inflammatory responses in the human intestine (4). In the animal model, only newborn mice to age 5 days and guinea pigs show inflammatory responses in the gut (5,6). These phenomenon are partially ascribable to crypt development producing anti-bacterial cryptdin and commensal micro flora (7). However, adult mice infected by invasive shigellae do not show any inflammation in the intestine, and the reason why adult mice are resistant to invasive shigellae remains unknown.
Shigellae infect macrophages, followed by pyroptosis and initiation of host inflammatory responses (8,9), but they also have evasion mechanisms to host defense. The Shigella flexneri effector OspI deamidates UBC13 to inhibit the inflammatory response (10), and the Shigella effector IcsB competitively inhibits host autophagy induction by binding inhibition of VirG and host ATG5 (11). The Tecpr1-dependent pathway is important in selective autophagy to Shigella organisms (12). Recently we suggested that Shigella infection of adult mice induces acute host cell death accompanying breakdown of host barrier and that autophagy represses inflammation (13). Therefore, autophagy controls an intrinsic host defense to bacteria by promoting epithelial cell survival in the murine gut. Intra-peritoneal (but not oral) challenge with shigellae results in diarrhea, severe body weight loss, and inflammation in adult C57BL/6 (B6) mice that is similar to human shigellosis (14). Collectively, these results suggest that non-inflammatory host responses in the guts of adult mice after oral Shigella infection are complicated.
In this study, we investigated the host modulation mechanism of shigellae in adult mice by infecting adult B6 mice (6~8 weeks old) with a virulent Shigella strain and observing histological and gene expression changes in the gut.
MATERIALS AND METHODS
Mice and bacterial strains
C57BL/6 (B6) mice were purchased from Charles River Laboratories (Orient Bio Inc., Sungnam, Korea). All mice were maintained under specific pathogen-free conditions in the animal research center at the International Vaccine Institute (Seoul, Korea) where they received sterilized food and water ad libitum. All animal experiments described were approved by Institutional Animal Care and Use committees (IACUC PN2011-002). S. flexneri 5a (M90T), IpaB4 deletion mutant S. flexneri 5a (M90TΔIpaB4), wild-type Salmonella typhimurium (UK-1), and non-invasive Shigella strains (BS176) were used.
Bacterial infection
For oral infection, each mouse was orally administered 5×109 bacteria. For ileal loop infection, mice were anesthetized and abdomens were surgically opened for about 10 mm. The ileum was looped with a suture about 40-mm long and then 5×109 bacteria and/or 200µM sphingosine-1-phosphate (S1P; Avanti Polar Lipids, Inc., Alabaster, AL) were injected into lumen in the loop. The looped tissues were harvested and analyzed for histological changes.
Histology
Intestinal tissue was washed with PBS containing gentamicin and fixed in 4% formaldehyde for 1 hour at 4℃. The tissues were dehydrated by gradually soaking them in alcohol and xylene and then embedded in paraffin. The paraffin-embedded specimens were cut into 5-µm sections, stained with H&E or Alcian blue periodic acid-Schiff (PAS; Merck, Nottingham, UK), and viewed with a digital light microscope (Olympus, Tokyo, Japan). To detect green fluorescent protein (GFP)-expressing bacteria in the intestinal tissue, the frozen sections of ileal or colon tissues were prepared and viewed under a confocal scanning laser microscope (Carl Zeiss, Göttingen, Germany).
Disease score
Histological changes of small intestine were assessed using H&E-stained samples of ileum after bacterial infection as previously reported (13). The category and parameters include host cell death (crypt, epithelium, lamina propria, and muscles), epithelium shedding, barrier integrity, inflammation, and goblet cell hyperplasia.
Bacteria count (CFU)
To assess the numbers of bacteria from intestinal tissues of non-infected and Shigella-infected mice, tissues were extensively washed in PBS with gentamicin (50µg/ml) to remove luminal and simply attached bacteria. Tissues were homogenized and plated onto TSB agar plates containing streptomycin since the M90T strain is streptomycin resistant. After overnight culture at 37℃, colonies were counted. For in vitro colony-forming unit (CFU) counts, a Caco-2 cell line originated from human intestinal epithelium was seeded at 5×105/well in a 6-well plate. The next day Shigella strains were infected at 100 MOI (multiplicity of infection) in antibiotic-free culture medium for 1 hour. After extensively washing with culture media containing antibiotics to remove non-infected bacteria, Caco-2 cells were harvested at the indicated time point to evaluate CFU.
Real time PCR and gene chip analysis
For real time PCR analysis, total RNA was extracted using an RNeasy kit (Qiagen, Hilden, Germany) and cDNA was synthesized by Superscript II reverse transcriptase with oligo(dT) primer (Invitrogen). Then, gene expression quantification was performed using an ABI PRISM sequence detection system (Applied Biosystems, Foster, CA). The levels of mRNA expression were displayed as the expression units of each target gene relative to the expression units of GAPDH. Murine Ppap2a (208 bp, forward: CTTACGACCCATGCTCCAGT, reverse: CGTGTGTGGATCCTCTTCCT), murine Sgpl1 (169 bp, forward: CTGATGACCGTACCATGTGC, reverse: GAATCCCTGAGAAGGGGAAG), human S1P lyase (HS1PL, 244 bp, forward: CTGGAGCACCCATTTGATTT, reverse: TCTCACCGAAGTGCATCAAG), human S1P phosphatase 2 (hS1PP2, 164 bp, TCCTCTTGGTTCGTCAGCTT, reverse: CACAAAGGTTGTAGCGCAGA), human S1P phosphatase 1 (hS1PP1, 188 bp, forward: TATTGCGGATCCTCATAGGG, reverse: TGTGATGGAGAAACCAACCA), human sphingosine kinase 2 (hSK2, 200 bp, forward: TGGCAGTGGTGTAAGAACCA, reverse: CAGTCAGGGCGATCTAGGAG), human sphingosine kinase 1 (hSK1, 227 bp, forward: ACCCATGAACCTGCTGTCTC, reverse: CAGGTGTCTTGGAACCCACT). For gene chip analysis, total RNA was amplified and purified using the Ambion Illumina RNA amplification kit (Ambion, Austin, TX) to yield biotinylated cRNA according to the manufacturer's instructions. In total, 750 ng of labeled cRNA samples were hybridized to MouseRef-8 V2.0 expression bead array for 16~18 hours at 58℃, according to the manufacturer's instructions (Illumina, San Diego, CA). Array signal detection was carried out using Amersham fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Little Chalfont, UK) as described in the bead array manual. Arrays were scanned with an Illumina bead array reader confocal scanner according to the manufacturer's instructions.
RESULTS
Shigellae invade inside tissue and evoke host responses in the gut without inflammation
To investigate host responses in adult mice, 6~8-week-old B6 mice were challenged orally with M90T (5×109 cfu), a wild type invasive strain of S. flexneri 5a. Consistent with results of a previous study (13), acute epithelial cell death followed by epithelial barrier breakage could be observed 1 hour after M90T infection in the terminal ileum of the small intestine. The damaged epithelium had mostly recovered 24 hours later (Fig. 1A). There was no inflammatory sign despite loss of barrier functions and entry of shigellae and commensal bacteria in the lumen. Infection with T3SS mutant strain M90TΔipaB4 produced no histological changes. Degranulation of secretory granules in the Paneth cells of crypt was seen from 1 to 3 hours after infection (Fig. 1B). To ensure that M90T infected by the route colonized, and not simply adhered to the small intestine, we tracked down GFP-expressing M90T in the small intestine. Of note, M90T-GFP bacteria were found on the epithelium and even deep within the crypts (Fig. 1C). We observed similar findings in the colon. There was only mild epithelial tissue damage 3 hours after oral M90T infection in the ascending and descending colon but no inflammation (Fig. 2A). Bacterial colonization was successful as indicated by detection of M90T-GFP in the epithelium and lamina propria of the colon (Fig. 2B). shigellae were detected in the colon and cecum 3 hours after infection and were completely cleared 24 hours later (Fig. 2C). These results indicate that shigellae successfully invade and induce host responses in the murine gut only during the acute phase and do not cause inflammation.
Figure 1.
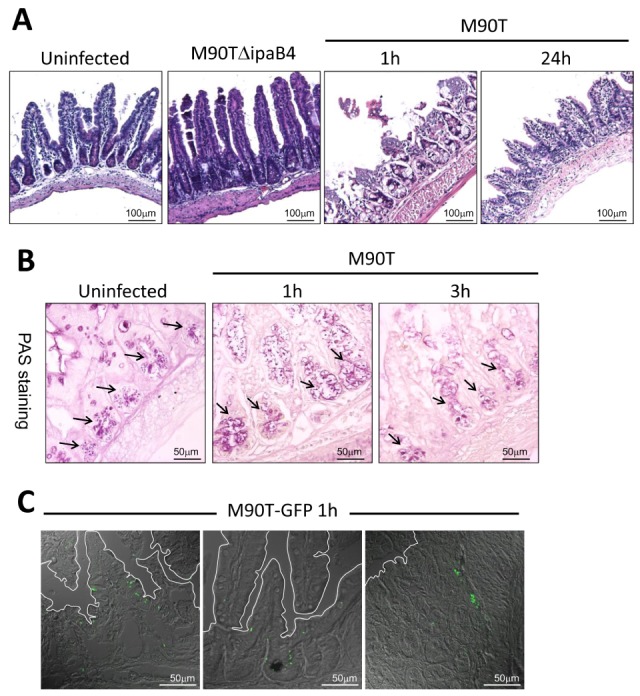
S. flexneri evade and evoke host cell death in the small intestine without inflammation. (A) Hematoxylin and eosin (H&E) staining of terminal ileum 1 and 24 h after oral infection with 5×109 CFU wild-type M90T or T3SS mutant M90TΔipaB4 (n=5 per group). (B) Periodic acid-Schiff (PAS) staining of terminal ileum 1 and 3 hours after oral infection with 5×109 CFU M90T. Arrows indicate secretory granules of crypt Paneth cells and mucin of goblet cells (n=3 per group). (C) Localization of GFP expressing M90T in the villi and crypts of the terminal ileum 1 h after oral infection (n=3 per group).
Figure 2.
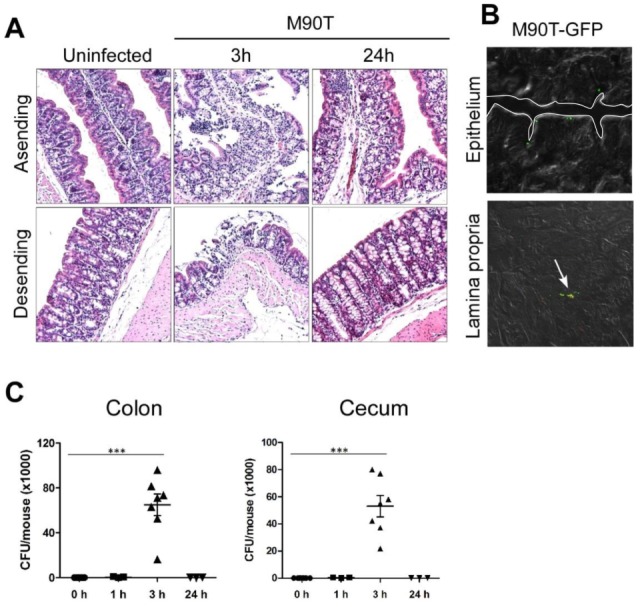
S. flexneri evade colon epithelium without inflammation. (A) H&E staining of ascending and descending colon orally infected with M90T after 3 and 24 h (n=5 per group). (B) M90T-GFP in the epithelium and lamina propria of the colon 3 h after oral infection (n=3 per group). (C) CFU of M90T in the colon and cecum 1, 3, and 24 h after infection. Graphs show mean±SEM. (one-way ANOVA with Tukey post test, ***p<0.0001).
Shigellae modulate host molecular expression in the gut
To investigate the reason why Shigella infection results in loss of gut epithelial barrier but does not induce inflammation, we compared gene expression profiles of non-infected and M90T-infected ileum of the small intestine. In contrast with the uninfected mice and those infected with the T3SS mutant strain M90TΔipaB4, many genes were dramatically changed (Fig. 3A). S1P-related genes such as sphingosine phosphate lyase 1 (Sgpl1) and phosphatidic acid phosphatase 2a (Ppap2a) were up-regulated from the M90T-infected group (Fig. 3B). These findings were confirmed by real time RT-PCR of M90T-infected ileal homogenates. To interpret Shigella-specific modulation of host gene expression, wild-type S. typhimurium (UK-1) was used. Spgl1 expression was up-regulated in both Shigella- and Salmonella-infected tissues; however, Ppap2a was up-regulated only in M90T-infected tissues (Fig. 3C). These results suggested that oral Shigella infection could modulate host gene expression, especially the S1P-related genes.
Figure 3.
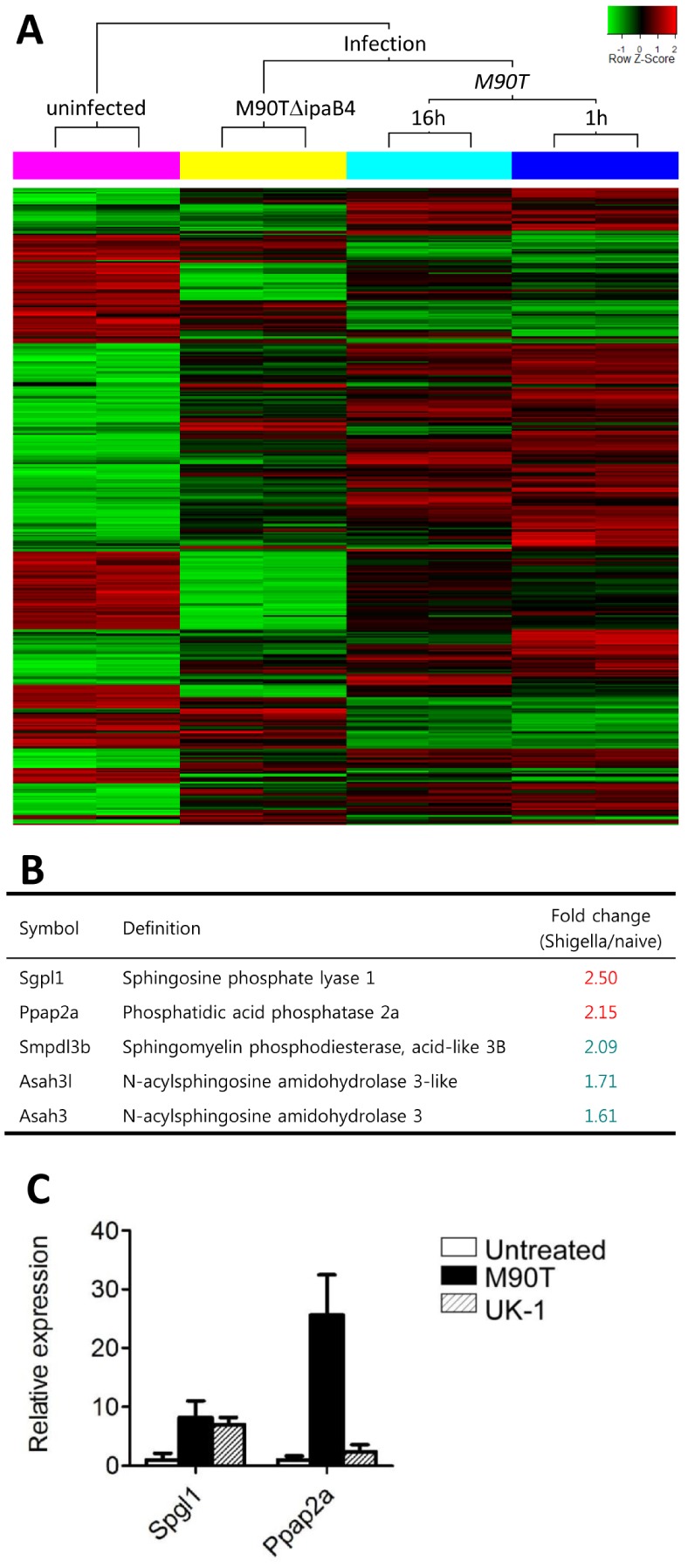
S. flexneri modulate host gene expression. (A) Microarray analysis of ileal tissue uninfected or infected with M90T (1 or 16 h after infection) and T3SS mutant M90TΔipaB4 (1 h). (B) Representative gene expression of ileal tissue infected with M90T (1 h after infection) regarding lipid metabolism. (C) mRNA expression of Spgl1 and Ppap2a of ileal tissue 1 h after oral infection with M90T or wild-type Salmonella typhimurium (UK-1).
Shigellae down-regulate S1P in host cells
S1P is well known to induce immune responses by inducing lymphocyte egress from lymphoid organs (15). We hypothesized that shigellae may have the ability to modulate host S1P levels to suppress inflammation. To investigate this hypothesis, we used the Caco-2 cell line, a human colorectal epithelial cell line, and infected the cells with non-invasive or wild type Shigella strains. To validate bacterial infection, bacterial CFU were measured. At 2 hours after infection, Shigella organisms were amplified within the host cells (Fig. 4A). S1P lyase (S1PL) and S1P phosphatase (S1PP) break down the phosphate of S1P, while sphingosine kinase (SK) produces S1P from sphingosine (Fig. 4B). The mRNA expression of S1PL, S1PP1, and S1PP2 increased in Shigella-infected Caco-2 cells 3 hours after infection while SK2 levels decreased. There was no significant change of SK1 (Fig. 4C). This suggests that Shigella organisms down-regulate host S1P levels, leading to suppression of inflammation.
Figure 4.

S. flexneri modulate host enzymes to regulate S1P level. (A) CFUs of avirulent BS176 and M90T in Caco-2 cells 2 h following infection. ***p<0.0001. (B) S1P is formed by phosphorylation of sphingosine that is catalyzed by sphingosine kinase (SK) and breaks down by S1P lyase (S1PL) and S1P phosphatase (S1PP). (C) mRNA expression of S1PL, S1PP1, S1PP2, SK1, and SK2 of Caco-2 cells infected with avirulent BS176 (3 h) or M90T (1 and 3 h).
Shigellae evade the host immune system by S1P depravation in local tissues
To confirm that down-regulation of S1P by shigellae suppressed inflammation, we examined murine ileum following M90T infection with co-administration of exogenous S1P. To maintain local high concentrations of S1P, murine ileum were looped and then injected with M90T and S1P. When injected with M90T only, there was massive cell death but no inflammation. However, when M90T were injected together with S1P, severe inflammation resulted with shortened villi, loss of crypt, thickened serosa, leucocyte infiltration, and loss of tissue integrity observed 1 hour after injection (Fig. 5A and 5B). Similar results were seen 24 hours after infection (Fig. 5A and 5C), suggesting that Shigella organisms actively regulate host S1P levels to inhibit inflammatory responses.
Figure 5.
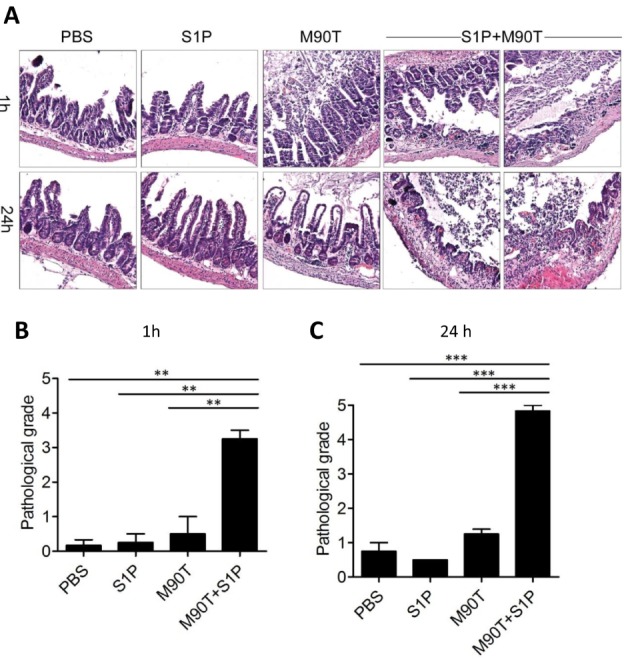
S. flexneri-provoked inflammation in the gut of adult mice in the presence of S1P. (A) Terminal ileum was looped and M90T injected directly into the looped tissue with or without S1P. After 1 h and 24 h, H&E staining was performed. Representative data were selected from three repetitive experiment sets. (B and C) Pathological grade of the looped ileum tissue injected with M90T and S1P after 1 h (B) and 24 h (C). **p<0.001, ***p<0.0001 (one-way ANOVA with Tukey post test).
DISCUSSION
Shigella species infect human colonic epithelium, resulting in severe inflammatory colitis and shigellosis (16,17). In our study in adult mice, a large number of Shigella organisms did not induce shigellosis. We found that shigellae could not enter the murine colonic epithelium via M cell translocation and then passively flow away without intrusion of the intestinal barrier. Recently, we reported finding clear evidence of aggressive intrinsic host defense in adult mice. After infected and bystander host cells were sacrificed, there was rapid renewal of epithelium (13). We found the host autophagy process was crucial for anti-bacterial defense and regulation of unnecessary inflammation. In the current study, we identified an active mechanism of enteric pathogens to evade host immune responses by modulation of host machinery.
S1P, which is made by phosphorylation catalyzed by sphingosine kinases, is activated by G protein-coupled S1P receptors (18). S1P has many crucial effects on regulation of the immune system, including inflammation (19). S1P causes lymphocytes to exit lymphoid organs to enter the circulation (20). With pro-inflammatory stimuli, sphingosine kinase is activated. Breakdown of S1P is done by dephosphorylation by S1P phosphatase or irreversible hydrolysis by S1P lyase (18). S1P also plays a role in inflammation by activating transcription factor NF-κB (21). Endothelial cells also express S1P receptor and its receptor agonist can inhibit S1P signaling (22). Blockade of S1P signals suppresses all features of influenza-inflicted pathological inflammation especially the cytokine storm.
Shigellae invade intestinal epithelial cells through M cells (23). Macrophages then engulf the shigellae but are killed by pyroptosis. Before death, the macrophages release pro-inflammatory cytokines that induce inflammation (24). However, S. flexneri have active mechanisms that regulate host immune defense. They inhibit acute inflammatory responses in the initial stage of infection by targeting the UBC13-TRAF6 complex (10). Our results indicate there is additional regulation mechanism for host immune defense that modulates the host S1P levels in order to evade inflammatory signals. In the case of humans, however, it seems that induction of inflammation by shigellae is stronger than evading it, which causes the difference of symptoms between human and mice. These findings help us in interpreting the pursuit-escape relationship between the host immune system and enteric pathogens.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP) (No. 2011-0006965, No. 2010-0029133 and No. 2012-0000805).
Abbreviations
- CFU
colony-forming unit
- GFP
green fluorescent protein
- H&E
hematoxylin-eosin
- MOI
multiplicity of infection
- PAS
periodic acid-Schiff
- PCR
polymerase chain reaction
- T3SS
type III secretion system
- S1P
sphingosine-1-phosphate
- S1PL
sphingosine-1-phosphate lyase
- S1PP
sphingosine-1-phosphate phosphatase
- SK
sphingosine kinase
Footnotes
The authors have no financial conflict of interest.
References
- 1.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ. 1999;77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 2.Ogawa M, Handa Y, Ashida H, Suzuki M, Sasakawa C. The versatility of Shigella effectors. Nat Rev Microbiol. 2008;6:11–16. doi: 10.1038/nrmicro1814. [DOI] [PubMed] [Google Scholar]
- 3.Kweon MN. Shigellosis: the current status of vaccine development. Curr Opin Infect Dis. 2008;21:313–318. doi: 10.1097/QCO.0b013e3282f88b92. [DOI] [PubMed] [Google Scholar]
- 4.Ashida H, Ogawa M, Mimuro H, Kobayashi T, Sanada T, Sasakawa C. Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr Opin Immunol. 2011;23:448–455. doi: 10.1016/j.coi.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Shim DH, Suzuki T, Chang SY, Park SM, Sansonetti PJ, Sasakawa C, Kweon MN. New animal model of shigellosis in the Guinea pig: its usefulness for protective efficacy studies. J Immunol. 2007;178:2476–2482. doi: 10.4049/jimmunol.178.4.2476. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez MI, Thuizat A, Pedron T, Neutra M, Phalipon A, Sansonetti PJ. A newborn mouse model for the study of intestinal pathogenesis of shigellosis. Cell Microbiol. 2003;5:481–491. doi: 10.1046/j.1462-5822.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- 7.Shim DH, Ryu S, Kweon MN. Defensins play a crucial role in protecting mice against oral Shigella flexneri infection. Biochem Biophys Res Commun. 2010;401:554–560. doi: 10.1016/j.bbrc.2010.09.100. [DOI] [PubMed] [Google Scholar]
- 8.Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol. 2011;195:931–942. doi: 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, Ashida H, Kobayashi T, Koyama T, Nagai S, Shibata Y, Gohda J, Inoue J, Mizushima T, Sasakawa C. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature. 2012;483:623–626. doi: 10.1038/nature10894. [DOI] [PubMed] [Google Scholar]
- 11.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 12.Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, Koyama T, Goto Y, Nagatake T, Nagai S, Kiyono H, Kawalec M, Reichhart JM, Sasakawa C. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe. 2011;9:376–389. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Chang SY, Lee SN, Yang JY, Kim DW, Yoon JH, Ko HJ, Ogawa M, Sasakawa C, Kweon MN. Autophagy controls an intrinsic host defense to bacteria by promoting epithelial cell survival: a murine model. PLoS One. 2013;8:e81095. doi: 10.1371/journal.pone.0081095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang JY, Lee SN, Chang SY, Ko HJ, Ryu S, Kweon MN. A mouse model of shigellosis by intraperitoneal infection. J Infect Dis. 2014;209:203–215. doi: 10.1093/infdis/jit399. [DOI] [PubMed] [Google Scholar]
- 15.Le Stunff H, Mikami A, Giussani P, Hobson JP, Jolly PS, Milstien S, Spiegel S. Role of sphingosine-1-phosphate phosphatase 1 in epidermal growth factor-induced chemotaxis. J Biol Chem. 2004;279:34290–34297. doi: 10.1074/jbc.M404907200. [DOI] [PubMed] [Google Scholar]
- 16.Ashida H, Ogawa M, Kim M, Mimuro H, Sasakawa C. Bacteria and host interactions in the gut epithelial barrier. Nat Chem Biol. 2011;8:36–45. doi: 10.1038/nchembio.741. [DOI] [PubMed] [Google Scholar]
- 17.Sansonetti PJ. Shigellosis: an old disease in new clothes? PLoS Med. 2006;3:e354. doi: 10.1371/journal.pmed.0030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Stunff H, Milstien S, Spiegel S. Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem. 2004;92:882–899. doi: 10.1002/jcb.20097. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chi H. Sphingosine-1-phosphate and immune regulation: trafficking and beyond. Trends Pharmacol Sci. 2011;32:16–24. doi: 10.1016/j.tips.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashida H, Ogawa M, Mimuro H, Sasakawa C. Shigella infection of intestinal epithelium and circumvention of the host innate defense system. Curr Top Microbiol Immunol. 2009;337:231–255. doi: 10.1007/978-3-642-01846-6_8. [DOI] [PubMed] [Google Scholar]
- 24.Tran Van Nhieu G, Bourdet-Sicard R, Dumenil G, Blocker A, Sansonetti PJ. Bacterial signals and cell responses during Shigella entry into epithelial cells. Cell Microbiol. 2000;2:187–193. doi: 10.1046/j.1462-5822.2000.00046.x. [DOI] [PubMed] [Google Scholar]