

Background: The POLB gene is mutated in 40% of human colorectal tumors.
Results: The S229L variant is a slow DNA polymerase that leads to the accumulation of BER intermediates and induces cellular transformation.
Conclusion: The S229L variant transforms cells by inducing genomic instability.
Significance: Tumor-associated variants of DNA polymerase β can affect DNA repair efficiency and drive cancer.
Keywords: Base Excision Repair, Cancer, Genetics, Genomic Instability, Mutagenesis
Abstract
DNA polymerase β (Pol β) plays a key role in base excision repair (BER) by filling in small gaps that are generated after base adducts are excised from the DNA. Pol β is mutated in a large number of colorectal tumors, and these mutations may drive carcinogenesis. In the present study, we wished to determine whether the S229L somatic Pol β variant identified in a stage 3 colorectal tumor is a driver of carcinogenesis. We show that S229L does not possess any defects in binding to either DNA or nucleotides compared with the WT enzyme, but exhibits a significant loss of polymerization efficiency, largely due to an 8-fold decrease in the polymerization rate. S229L participates in BER, but due to its lower catalytic rate, does so more slowly than WT. Expression of S229L in mammalian cells induces the accumulation of BER intermediate substrates, chromosomal aberrations, and cellular transformation. Our results are consistent with the interpretation that S229L is a driver of carcinogenesis, likely as a consequence of its slow polymerization activity during BER in vivo.
Introduction
Faithful DNA repair is critical to maintaining genomic integrity. If DNA repair does not function properly, cells acquire a mutator phenotype that contributes to tumorigenesis (1, 2). Endogenous DNA damage occurs >20,000 times per cell per day (3, 4) with base excision repair (BER)2 being the primary pathway responsible for repairing oxidative and alkylating lesions. After excision of the damaged base by a specific DNA glycosylase, apurinic/apyrimidinic endonuclease 1 cuts the DNA backbone 5′ to the abasic site, leaving a 3′-OH and a deoxyribose phosphate (5′-dRP) moiety. Some DNA glycosylases are bifunctional, removing the damaged base and incising the DNA backbone, abrogating the requirement for apurinic/apyrimidinic endonuclease 1. Pol β is the major polymerase that fills the resulting single-nucleotide gap and removes the dRP group (5). Once gap filling and dRP removal have taken place, DNA ligase IIIα seals the nick, and repair is complete. Pol β is a small 39-kDa protein that contains four subdomains. The 8-kDa subdomain is responsible for the dRP lyase activity (5). The thumb subdomain binds the DNA, the palm subdomain contains the active site required for catalysis, and the fingers subdomain is responsible for selecting and binding nucleotides.
Pol β is mutated in 40% of human colorectal tumors (6). Our recent study identified 51 different nonsynonymous mutations in the POLB gene in a collection of 134 human colon tumors. The mutations were not clustered in any specific region of the protein and were located in all four subdomains. Some of these nonsynonymous mutations are likely a significant contributor to cancer progression because the mutations are selected for with many of the mutations being identified in late stage tumors, and there were no corresponding mutations identified in the normal matched tissues (6). In fact, two variants identified in this screen were found to induce cellular transformation (7, 8).
In this study, we sought to determine whether the S229L variant identified in a stage 3 colorectal tumor could drive carcinogenesis. S229L is located in the palm domain and lies in close proximity to the DNA, but is not near the active site of the protein. We found that the expression of S229L in mammalian cells induces genomic instability and cellular transformation. Biochemically, S229L has a reduced catalytic rate compared with WT Pol β and impaired BER capacity. In combination, our results suggest that the S229L variant results in aberrant BER and that S229L can act as a driver of tumorigenesis.
EXPERIMENTAL PROCEDURES
Plasmids and Cloning
The S229L variant of Pol β was generated using the Stratagene QuikChange® Site-directed Mutagenesis kit according to manufacturer's instructions. pET28a rat WT Pol β containing a N-terminal His6 tag was used as a template for protein expression and biochemical studies. For cell culture experiments, the C-terminal hemagglutinin (HA)-tagged rat WT Pol β in the pRVYTet retroviral vector was used as a template. The primers used for these reactions were purchased from Invitrogen, and the sequences are available upon request. Positive clones were confirmed by sequencing at the W. M. Keck facility at Yale University School of Medicine.
Protein Expression and Purification
The N-terminal His-tagged rat WT and S229L Pol β pET28a plasmids were transformed into the Escherichia coli strain Rosetta2 (DE3) competent cells and purified by FPLC as described previously (9). All proteins were purified to >90% homogeneity as confirmed by Coomassie Blue staining of 10% SDS-polyacrylamide gels. The final protein was aliquoted, flash frozen in liquid nitrogen, and stored at −80 °C. The final concentration was determined using the absorbance at 280 nm and the extinction coefficient for Pol β (ϵ = 21,200 m−1 cm−1).
Preparation of DNA Substrates
The DNA substrates used are shown in Table 1. Oligonucleotides were synthesized by the W. M. Keck facility and purified by polyacrylamide gel electrophoresis. 3C2M45 was used for kinetic studies. 45AG was used for gel mobility shift assays. LPSD was used for the BER assay. All primers were radiolabeled at the 5′-end using T4 polynucleotide kinase and [γ-32P]ATP. The downstream oligonucleotides were 5′-phosphorylated with nonradiolabeled ATP. The reactions were purified using Micro Bio-Spin® 30 Chromatography columns (Bio-Rad) to remove unincorporated ATP. The oligonucleotides were annealed by combining the radiolabeled primer, the nonradiolabeled downstream oligonucleotide, and the template in 50 mm Tris-HCl, pH 8.0, and 0.25 m NaCl. The mixture was incubated at 95 °C for 5 min, slowly cooled to 50 °C over 30 min, and incubated at 50 °C for an additional 20 min. Reactions were placed on ice and resolved on a 12% native polyacrylamide gel to verify complete annealing of the oligonucleotides.
TABLE 1.
DNA Substrates
The template base is in boldface.
| Substrate | Sequence | Assay |
|---|---|---|
| 45AG | 5′-GCCTCGCAGCCGTCCAACCAAC CAACCTCGATCCAATGCCGTCC-3′ | Gel mobility shift assay |
| 3′-CGGAGCGTCGGCAGGTTGGTTGAGTTGGAGCTAGGTTACGGCAGG-5′ | ||
| 3C2M45 | 5′-GCCTCGCAGCCGGCTGATGCGC GTCGGTCGATCCAATGCCGTCC-3′ | Pre-steady-state kinetics; single-turnover kinetics |
| 3′-CGGAGCGTCGGCCGACTACGCGGCAGCCAGCTAGGTTACGGCAGG-5′ | ||
| LPSD | 5′-CTGCAGCTGATGCGCUGTACGGATCCCCGGGTAC-3′ | BER assay |
| 3′-GACGTCGACTACGCGGCATGCCTAGGGGCCCATG-5′ |
Cell Lines and Cell Culture
The Pol β+/+ (92 TAg) and Pol β−/− (88 TAg) mouse embryonic fibroblasts (MEFs) were a kind gift from Leona Samson (Massachusetts Institute of Technology). These cells were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products), 1% penicillin-streptomycin, 1% l-glutamine, and 220 μg/ml hygromycin B (Invitrogen) (used for selection) and grown at 37 °C in 5% CO2. GP2-293 cells (Clontech) were maintained in high glucose DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, 1% l-glutamine, and 1 mm Hepes and grown at 37 °C in 5% CO2. C127 cells were obtained from ATCC. C127 cells are a nontransformed clonal line derived from a mammary tumor of a RIII mouse (10). C127 cells were grown in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin (Invitrogen), 1600 μg/ml G418, and 180 μg/ml hygromycin B (for selection of exogenous Pol β) at 37 °C in a 5% CO2 humidified incubator as described previously (8).
Transfection, Infection, and Expression Analysis
To obtain high titer retrovirus expressing WT and S229L Pol β, the GP2-293 packaging cell line was transfected with pRVY Pol β and pVSVG plasmids (11) using calcium phosphate as described previously (8). Western blots of Pol β confirmed exogenous expression in these cell lines. For the C127 cells, single clones were picked and screened for approximately equal ratios of endogenous to exogenous expression.
Western Blotting
Cells were washed once with PBS, lysed in boiling 1× SDS buffer (50 mm Tris-HCl, pH 6.8, 100 mm DTT, 2% SDS, 10% glycerol), and boiled for 5 min. Lysates were resolved on by 10% SDS-PAGE and transferred to a nitrocellulose membrane. Pol β was detected by incubating the membrane with monoclonal mouse anti-Pol β antibody (Abcam 1831). Blots were imaged using Bio-Rad ChemiDocTM XRS+ and quantified using Image Lab software.
Pre-steady-state Burst Assay
Radiolabeled 1-bp gapped DNA (300 nm 3C2M45) and Pol β (100 nm) were combined with the correct dNTP and 10 mm MgCl2 in a rapid quench apparatus at 37 °C (KinTek Corporation) with the reactions quenched by the addition of 0.5 m EDTA. The reaction products were separated on a 20% denaturing polyacrylamide gels, visualized, and quantified using a Storm 860 PhosphorImager with ImageQuant software. WT data were fit to the burst equation,
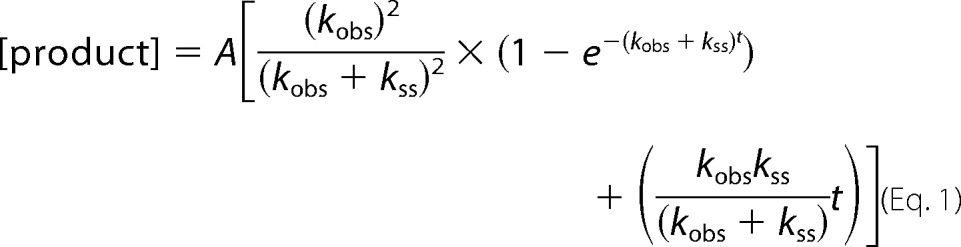 |
where A is the amplitude, kobs is the observed rate constant of the exponential phase, and kss is the steady-state rate constant of the linear phase (13).
Circular Dichroism
Circular dichroism wavelength scans were performed with 1 μm Pol β protein in 10 mm K2HPO4 at 23 °C using the Chirascan circular dichroism spectrometer (Applied Photosystems). Ellipticity was measured in 0.5-nm steps from 185 to 290 nm. Thermal scans were also performed at a set wavelength of 222 nm with a 5 °C/min step from 4.5 to 62 °C. Three measurements were taken for each enzyme and averaged.
Preparation of MEF Extract
Extracts from Pol β−/− MEFs were collected as described previously (8). The extracts were aliquoted and flash frozen in liquid nitrogen. Protein concentrations were determined by measuring the absorbance (562 nm) after the addition of BCA Protein Assay Reagent (Thermo-Fisher Scientific) using BSA as a reference standard.
Reconstituted Base Excision Repair Assay
The base excision repair assay was performed with radiolabeled LPSD DNA and incubated with Pol β−/− extract containing purified WT or S229L protein as described previously (8). Equal amounts of active WT and S229L protein were used as determined by electrophoretic mobility shift assay (EMSA). Briefly, 5 μg of Pol β-deficient cell extract containing 10 nm purified Pol β enzyme was incubated with 5 nm UDG-treated DNA with 10 μm dCTP for 10–60 s at 37 °C. Samples were resolved on 20% denaturing polyacrylamide gels, visualized using a Storm 860 PhosphorImager, and quantified using ImageQuant software.
Clonogenic Survival Assays
Cell densities of 2.2 × 105 were plated in 60-mm dishes. After 48 h, cells were treated with varying concentrations of methylmethane sulfonate (MMS; Sigma-Aldrich). Following treatment, cells were trypsinized and plated at several dilutions. The cells were allowed to grow for 9–11 days and stained with crystal violet, and colonies were counted. Colonies were defined as having at least 50 cells. All experiments were performed at least three times.
Alkaline Comet Assay
Equal number of cells (4 × 105) were plated in 60-mm dishes. The following day, the cells were left untreated or treated with 2 mm MMS for 30 min with or without a 30-min recovery. After treatment, the cells were prepared and analyzed immediately using Cometslides (Trevigen 4250-200-03) as described previously (14). Image analysis of 50–100 cells was performed using CometScore software (TriTek, Sumerduck, VA). Data are represented as mean ± S.E.
Flow Cytometry
Pol β+/+ cells expressing WT or S229L Pol β were untreated or treated with 2 mm MMS for 2 h. Cells were rinsed with PBS and replaced with fresh medium. Cells were allowed to recover for 0 h and 2 h after treatment. Cells were harvested by trypsinization, washed once with PBS, and pelleted. The pellet was resuspended by adding 70% ice-cold ethanol dropwise while vortexing. Cells were fixed overnight at −20 °C. The cells were incubated with primary phospho-γH2AX antibody (Millipore 05-636) 1:500 overnight at 4 °C. Following the incubation, cells were washed twice with PBS and incubated with anti-mouse secondary antibody conjugated to FITC 1:500 for 1 h at room temperature. Cells were washed twice with PBS and resuspended in 400 μl of propidium iodide/RNase staining buffer (BD Biosciences). Fluorescence was analyzed by flow cytometry using the BD FACSCalibur and analyzed using FlowJo 8.8.6 software. Data are represented as mean ± S.E.
Gel Mobility Shift Assay
The DNA-binding constant was determined by gel EMSA as described previously (15). The dissociation constant for DNA (KD) was determined by fitting the fraction bound protein (Y) as a function of protein concentration with the equation
 |
where Y is the amount of bound protein, m is a scaling factor, and b is the apparent minimum Y value (16).
Single-turnover Kinetic Assay
Radiolabeled gapped DNA (50 nm 3C2M45) was incubated with WT Pol β (500 nm active sites) as was determined by active site titration experiments. For S229L Pol β, 500 nm protein was sufficient to bind 95% of DNA as was determined by EMSA. The DNA/Pol β mixture was incubated with varying concentrations of dNTP (5–2000 μm) as described previously (8). The concentration of extended product was plotted as a function of time, and the data were fit to a single-exponential curve to obtain the kobs for each dNTP concentration. A secondary plot of kobs for each dNTP concentration as a function of [dNTP] was fitted to a hyperbolic equation
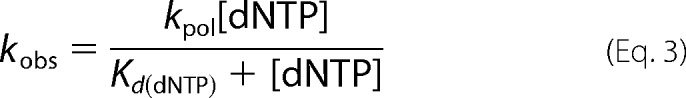 |
where kpol is the rate of polymerization and Kd(dNTP) is the equilibrium dissociation constant for dNTP.
Cellular Transformation
The focus formation assay was conducted as in (7). Typically, cells grow in a monolayer. Once cells become transformed, they grow on top of each other forming foci. The presence of foci was also monitored by microscopic examination as described previously (7, 12). Anchorage-independent growth was assessed as described previously (12).
Proliferation
High passage C127 clones were plated at a density of 20,000 cells/60-mm dish. A replicate plate of each clone was counted every 24 h using an automated cell counter (Nexelcom) as described previously (11). Data were plotted as change in cell number/day (n = 3–4).
Chromosomal Aberrations
Pol β+/+ MEFs expressing either WT or S229L Pol β were plated overnight in 10-cm dishes (106 cells/dish). Colcemid (Invitrogen) was added to the cells (0.1 μg/ml, 3 h) to arrest cells in mitosis. Cells were trypsinized, collected by centrifugation, washed once with PBS, and resuspended in a hypotonic solution (0.56% KCl) for 30 min at 37 °C to lyse the cells. The cells were fixed using Carnoy's fixative (75% methanol, 25% acetic acid) and dropped onto slides followed by staining with Giemsa, and well spread metaphases were identified under 100× objective (Zeiss). Images were taken using Spot Camera software (Diagnostic Instruments). Metaphase spreads were scored by eye for chromosomal fusions, breaks, and fragments. Scoring was performed blinded and validated by a second, independent person.
Statistics
Two-tailed t-tests and two-way analysis of variance were used as appropriate. Bonferroni's post hoc test was used to determine significant differences between the means of each group. All statistics were performed using GraphPad Prism version 5 (GraphPad Software, San Diego, CA), and data are represented as mean ± S.E. of all replicates.
RESULTS
S229L Pol β Is a Slow Polymerase
In an initial screen of Pol β variants identified in our original colon tumor study (6), S229L exhibited a decreased rate of catalysis compared with Pol β WT in a primer extension assay (6). Pre-steady-state burst kinetics, using a 1-base pair gapped DNA substrate (Table 1), revealed that S229L does not display a biphasic burst that is typical of Pol β, suggesting that the kinetic pathway of S229L differs from that of WT (6) (Fig. 1). The rate-limiting step of WT Pol β is product release, and the lack of burst kinetics by S229L indicates that the rate-limiting step of the variant is chemistry or a step preceding chemistry. The decreased rate was not due to general changes in the structure or stability of the protein as both the global folding and melting temperature of the proteins were similar (Fig. 2).
FIGURE 1.

S229L is a slow polymerase. Pre-steady-state kinetics experiments were performed by preincubating 300 nm 3C2M45 DNA substrate (Table 1) with 100 nm purified Pol β protein followed by the addition of 100 μm dCTP for various times. A, WT (filled circles) reactions were run on a time course ranging from 0.02 to 3 s. B, S229L (open triangles) pre-steady-state kinetic reactions were run on a time course ranging from 0.25 to 15 s. Products were resolved on 20% denaturing gels, imaged using a PhosphorImager, and quantified. Data were fit to the full burst equation (WT, kobs = 13 ± 2 s−1). S229L does not display biphasic burst kinetics, and a kobs could not be determined. Error bars, ±S.E.
FIGURE 2.
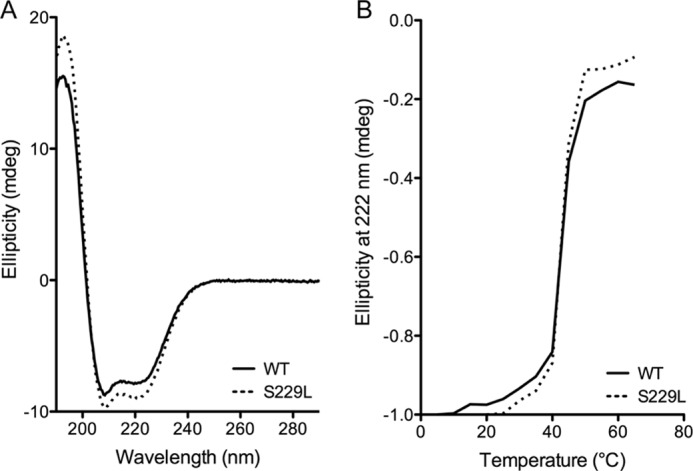
S229L Pol β has the same overall structure and thermal stability as WT Pol β. A, circular dichroism spectra of 1 μm WT (solid line) or S229L (dashed line) Pol β in 10 mm K2PO4 at 22 °C. B, thermal denaturation of 1 μm WT (solid line) or S229L (dashed line) Pol β analyzed in 10 mm K2PO4 at 222 nm.
S229L Pol β Binds DNA and dNTP with an Affinity Similar to WT, but Has a Defect in DNA Synthesis
To determine the biochemical mechanism underlying the slow rate of DNA synthesis by S229L Pol β, we first examined the ability of the protein to bind to a 1-base pair gapped DNA substrate (45AG) (Table 1) by EMSA. WT and S229L bound DNA with similar affinities (KD(DNA) = 4.1 ± 0.8 and 4.1 ± 0.3, respectively), indicating that DNA binding is not impaired and does not contribute to the DNA synthesis defect in S229L (Fig. 3 and Ref. 8). The rate of polymerization (kpol) and the apparent dNTP binding affinity (Kd(dNTP)) for both correct and incorrect nucleotides were determined using single turnover kinetics and the 3C2M45 DNA substrate (Fig. 4 and Table 1). These kinetic studies revealed that S229L binds both correct and incorrect dNTPs with affinity similar to that of WT (Table 2). However, there was a 4–5-fold reduction in the polymerization efficiency due to a decrease in the polymerization rate, kpol. This defect in polymerization accounts for the slowness of the S229L variant.
FIGURE 3.
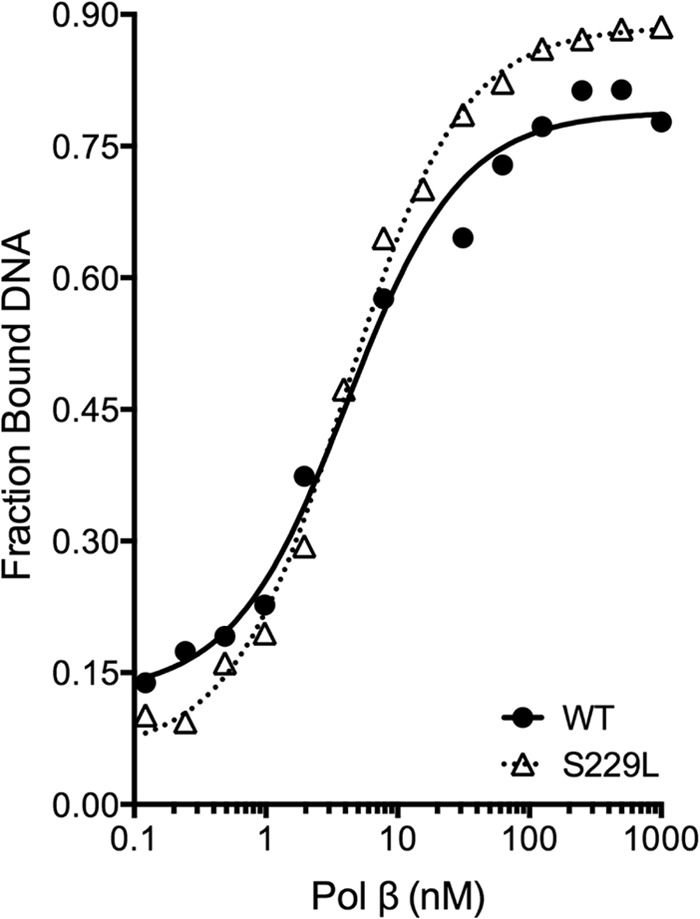
S229L has binding affinity for 1-bp gapped DNA similar to that of WT. Various concentrations of purified WT (filled circles) or S229L (open triangles) protein were incubated with 0.1 nm 45 AG DNA substrate (Table 1) for 15 min at room temperature and then resolved by native PAGE. (WT KD(DNA) = 4.1 ± 0.8 nm; S229L KD(DNA) = 4.1 ± 0.3 nm). In reference to the WT data: This research was originally published in Ref. 8.
FIGURE 4.

S229L Pol β has a lower polymerization rate than WT. A and C, representative plots show the incorporation of dCTP opposite G for WT (A) or S229L (C) at 37 °C. A preincubated solution containing 500 nm Pol β enzyme and 50 nm radiolabeled 3C2M45 DNA was combined with 10 mm MgCl2 and increasing concentrations of dCTP (0–1000 μm). Data were plotted as extended DNA formed versus time and fit to the single exponential equation to obtain the kobs for each dCTP concentration. B and D, secondary plots of the kobs versus dCTP concentration were fit to the hyperbolic equation to obtain kpol and Kd(dNTP) for WT (B) or S229L (D). Data are presented as mean ± S.E. (error bars; n = 2–4). Values are reported in Table 2.
TABLE 2.
Single-turnover misincorporation opposite template G
Kinetic constants obtained from single-turnover misincorporation experiments are listed for each enzyme (±S.E. of the fit). Efficiency was calculated by dividing kpol by Kd for each condition.
| dNTP | Enzyme | kpol | Kd | Efficiency | Fold changea | Fidelityb | Fold changec |
|---|---|---|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 | |||||
| dCTP | WT | 32.4 ± 0.4 | 5.7 ± 0.4 | 5.7 | 5 | ||
| S229L | 4.2 ± 0.1 | 3.7 ± 0.9 | 1.1 | ||||
| dATP | WT | 0.052 ± 0.002 | 170 ± 20 | 3.1 × 10−4 | 5 | 18,600 | 0.9 |
| S229L | 0.0098 ± 0.0003 | 170 ± 20 | 5.8 × 10−5 | 19,700 | |||
| dGTP | WT | 0.0113 ± 0.0009 | 310 ± 60 | 3.6 × 10−5 | 4 | 156,000 | 1 |
| S229L | 0.0025 ± 0.0001 | 250 ± 50 | 1.0 × 10−5 | 114,000 | |||
| dTTP | WT | 1.37 ± 0.05 | 1340 ± 90 | 1.0 × 10−3 | 4 | 5,560 | 1 |
| S229L | 0.46 ± 0.04 | 1900 ± 300 | 2.4 × 10−4 | 4,690 |
a Fold change in efficiency (WT/S229L).
b Fidelity + efficiencycorrect + efficiencyincorrect/efficiencyincorrect.
c Fold change in fidelity (WT/S229L).
BER Is Slow in the Presence of S229L Leading to the Accumulation of BER Intermediates
Next, we assessed the capacity of the S229L Pol β variant to participate in BER, given its slow catalytic rate in our biochemical experiments. The LPSD DNA substrate (Table 1) was incubated with UDG to excise the uracil, followed by incubation for 0–60 s with Pol β−/− MEF extract containing equally active amounts of purified WT or S229L Pol β enzyme. Although S229L was able to fill in the gap and remove the dRP group, forming an n+1 extended product, it did so at a slower rate than WT (Fig. 5, A and B). We subsequently performed clonogenic survival assays to assess whether S229L Pol β can participate in BER in vivo by expressing either WT or S229L in Pol β−/− MEFs. The data show that expression of S229L in Pol β−/− MEFs is able to rescue the cells from treatment with the alkylating agent MMS, but did not fully complement the BER defect of the Pol β-deficient MEFs compared with cells expressing WT Pol β (Fig. 5C). The lack of full complementation was not due to different levels of expression because Western blots showed that the two proteins are expressed at similar levels (Fig. 5D). To assess whether the slow rate of BER in S229L cells induced the accumulation of BER intermediates, we stably expressed either the S229L variant or WT in Pol β+/+ MEFs and performed an alkaline comet assay to quantify the amount of MMS-induced single-nucleotide gaps (SNGs) and single-strand breaks (SSBs). The Pol β+/+ MEF cell line was used because the S229L mutation is heterozygous and would normally be found with WT Pol β in a tumor. We used cells that expressed both WT and S229L Pol β at equivalent levels meaning that the cells exhibited 1:1 levels of exogenous to endogenous expression of Pol β (Fig. 5D). Following a 30-min treatment with MMS, S229L-expressing cells had a significant increase in SNGs/SSBs compared with WT cells. Moreover, even after allowing cells to recover for 30 min, cells expressing the S229L variant still had significant levels of SNGs/SSBs (Fig. 5E). These data suggest that cells expressing S229L do not efficiently fill in the SNG that is generated after removal of the alkylated bases that are induced by treatment with MMS. Interestingly, S229L-expressing cells had significantly more SNGs/SSBs than WT under basal conditions, indicating that the presence of the S229L variant leads to incomplete repair of endogenous lesions.
FIGURE 5.

S229L has impaired BER activity and induces the accumulation of BER intermediates. A, the LPSD DNA substrate was treated with UDG and incubated with Pol β−/− extract containing purified WT or S229L protein. B, quantification of n+1 product is shown as a function of total DNA. Data are presented as mean ± S.E. (error bars; n = 3). C, clonogenic survival assays were conducted with Pol β−/− MEFs expressing either WT or S229L Pol β or empty vector. Data are plotted as mean ± S.E. (n = 3–4). D, Western blotting was performed of Pol β in cell lines. Representative Western blots show expression of S229L Pol β in Pol β+/+ MEFs and Pol β−/− MEFs. Endogenous Pol β was used as a loading control in Pol β+/+ MEFs. Tubulin was used as a loading control in the Pol β−/− MEFs. The ratios of exogenously expressed Pol β protein to loading control are shown below the images of the blots. E, Pol β+/+ MEFs were left untreated or treated with 2 mm MMS for 30 min. Following MMS exposure, cells were allowed to recover for 0 or 30 min. Single-strand breaks were quantified by the alkaline comet assay. The percentage of tail DNA is plotted on the y axis. F, Pol β+/+ MEFs were left untreated or treated with 2 mm MMS for 2 h. Following MMS exposure, cells were allowed to recover for 0 or 2 h, stained with γH2AX antibody, and analyzed by flow cytometry to assess the levels of double-strand breaks. *, **, and *** denote p < 0.05, 0.01, and 0.001, respectively, comparing S229L versus WT within the same treatment group. ∧∧ and ∧∧∧ denote p < 0.01 and 0.001, respectively, comparing the treatment group versus untreated cells.
Deficient gap-filling by S229L Pol β could lead to the production of double-strand breaks (DSBs) if the replication fork collided with an unfilled gap. Therefore we estimated the levels of DSBs in the cells by quantifying γH2AX-positive cells by flow cytometry (Fig. 5F). After a 2-h treatment with MMS, cells expressing S229L had significantly more DSBs compared with WT cells. In addition, after a 2-h recovery period, there was no change in the amount of DSBs in the S229L-expressing cells that had delayed repair of the damage and/or continued to accumulate DSBs. Together, these data suggest that the slow catalytic rate of S229L impairs the ability of this variant to function in BER as efficiently as WT Pol β in vivo. The decrease in BER function induces the accumulation of BER intermediates, which could lead to genomic instability.
Expression of S299L Pol β Induces Chromosomal Aberrations
Pol β is the main polymerase involved in BER. S229L is a catalytically slow variant that performs BER, albeit with lower efficiency than WT Pol β. This defect in BER leads to the significant accumulation of BER intermediates (Fig. 5). We postulate that the inability of the slow S229L Pol β enzyme to fill the gaps in the DNA efficiently leads to chromosomal aberrations. To test whether this was the case, we analyzed metaphase spreads for chromosomal aberrations in Pol β+/+ MEFs expressing either WT or the S229L variant (Fig. 5D). Cells expressing S229L exhibited significantly increased levels of chromosomal fragments, fusions, and breaks compared with WT, as shown in Fig. 6. These data suggest that the loss of catalytic activity and decreased ability of the S229L Pol β variant to efficiently fill gaps during BER induce genomic instability in cells.
FIGURE 6.
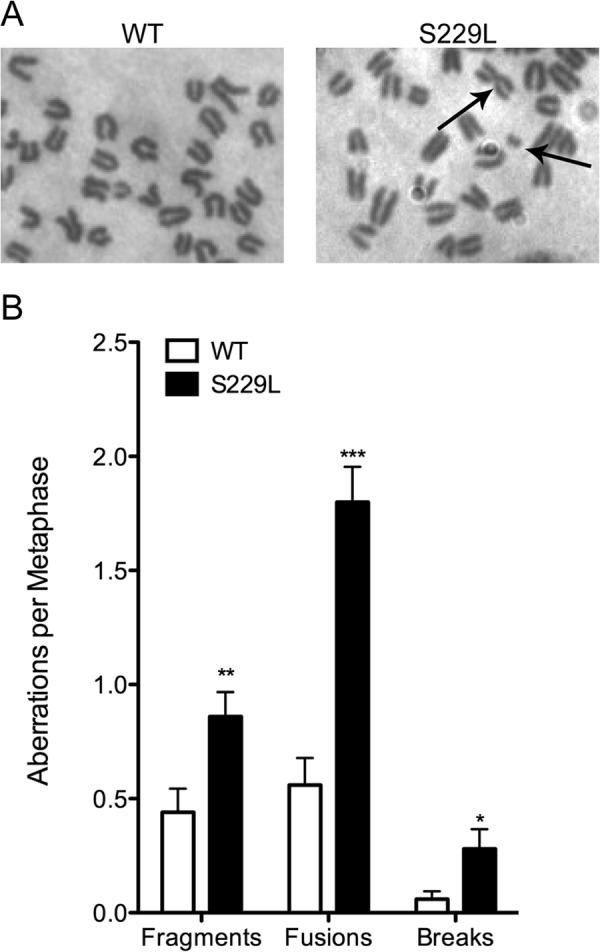
Expression of S229L Pol β induces chromosomal aberrations. Metaphase spreads of Pol β+/+ MEFs expressing either WT or S229L Pol β were analyzed for chromosomal aberrations. A, representative images of WT and S229L Pol β-expressing metaphase spreads. B, number of aberrations per metaphase. A total of at least 50 metaphases were scored for each cell line. *, **, and *** denote p < 0.05, 0.01, and 0.005, respectively.
S229L Induces Cellular Transformation
Given that genomic instability can lead to cellular transformation, we wished to determine whether expression of the S229L variant could induce cellular transformation. We expressed HA-tagged rat WT or S229L Pol β in C127 cells, a mouse mammary epithelial cell line, using a tetracycline (tet) off system. In this system, exogenous Pol β is expressed in the absence of tet and is shut off when tet is present in the medium. Six individual clones were selected that had 1:1 levels of expression of exogenous to endogenous Pol β (see example of one clone in Fig. 7A). Cells were serially passaged, and every fourth passage cells were set aside to assess focus formation, a hallmark of cellular transformation that occurs by the loss of contact-inhibited growth. Expression of S229L induced the formation of foci (Fig. 7B), and the transformation occurred at different rates indicating a stochastic mechanism. Note that previous work, performed at the same time as the S229L transformation experiments, shows that C127 cells expressing exogenous WT Pol β do not form foci (8). Another hallmark of cellular transformation is anchorage-independent growth. To determine whether expression of S229L induces anchorage-independent growth, we selected two clones (S229L25 and 31) and assayed for growth in soft agar. In both clones, we found that expression of S229L increased anchorage-independent growth (Fig. 7C). Under similar conditions, cells expressing exogenous WT Pol β do not induce anchorage-independent growth (8, 11). Cells expressing S229L also exhibit increased proliferation rates compared with their nonexpressing counterparts (Fig. 7D) and to cells expressing WT Pol β (11). To determine whether expression of S229L Pol β is required to maintain transformation, we turned off expression of exogenous Pol β by adding tet to the medium (Fig. 7A) and analyzed focus formation as described. Foci continued to form after expression was extinguished (Fig. 7E), indicating that cells are transformed through a heritable change that was induced by the expression of S229L.
FIGURE 7.

S229L Pol β induces cellular transformation. A, representative Western blot shows expression of S229L Pol β in C127 clones. C127 cells were grown in the presence of tetracycline (T) to turn off expression of the exogenous Pol β, tetracycline was removed to induce expression (NT), and expression was extinguished by adding tetracycline back to the media (NT→T). Endogenous Pol β was used as a loading control. B, C127 cells expressing (NT; dashed lines) or not expressing (T; solid lines) exogenous S229L Pol β were stained at every fourth passage and analyzed for foci formation, a loss of contact-inhibited growth. C, clones 25 and 31 were assayed for growth in soft agar when expressing (NT; open bars) or not expressing (T; filled bars) exogenous S229L Pol β. D, cells were plated at a density of 20,000 cells/60-mm dish and were counted every day for 5 days. Data were plotted as change in cell number per day. *, **, and *** denote p < 0.05, 0.01, and 0.005, respectively. E, expression of S229L Pol β was extinguished by adding tet back to the medium (NT to T) at passage 24 (indicated by arrow). Foci were counted every fourth passage.
DISCUSSION
The major cellular roles for Pol β include removal of the 5′-dRP group from the end of an SNG and filling in of the gap during BER. Mutations in the POLB gene that lead to a lack of or inefficient 5′-dRP removal or gap filling are likely to result in the accumulation of BER intermediates that lead to genomic instability and drive cancer. The S229L Pol β variant was identified in a stage 3 tumor in our screen of 134 human colorectal tumors (6). In total, 51 nonsynonymous mutations were identified in the POLB gene in that screen. Nonsynonymous mutations are predicted to alter the local structure and/or function of the protein, and the POLB mutations we identified are likely to have been subject to positive selection, as suggested by the ratio of nonsynonymous to synonymous substitutions. In addition, there were no corresponding mutations found in normal matched tissues, and a significantly higher proportion of the tumor mutations was identified in later stage tumors (p = 0.025), suggesting that some of these mutations drive cancer progression. In the present study, we characterized the S229L variant and found that it induces cellular transformation via genomic instability that likely arises as a result of its failure to fill gaps in DNA completely, which leads to an accumulation of BER intermediates. Together, the data presented herein support the role of the S229L Pol β variant as a driver of tumorigenesis.
The residue Ser-229 lies within the palm subdomain, which contains the active site that is responsible for catalysis. Although Ser-229 lies far from the active site, it is within close proximity (3.5 Å) to the template strand of DNA (templating base T-4). A nearby residue, Gly-231, lies within 2.8 Å of templating base T-5 and a previous study by our laboratory using molecular dynamics (8) suggested that when residue 231 was mutated from glycine to aspartic acid (G231D), there is a repulsion with the template strand of DNA that induces a distortion in the binding pocket of Pol β. This distortion in G231D prevents nucleotide binding and reduces the catalytic rate of the enzyme. Because the closely neighboring residues Ser-229 and Gly-231 are on a β-sheet adjacent to the template strand and both have decreased rates of catalysis, we hypothesized that the underlying biochemical mechanism would be similar. However, our data surprisingly revealed that S229L does not have a defect in binding of nucleotides and instead has a decreased polymerization rate that is independent of nucleotide binding. Thus, cellular transformation induced by the expression of these variants results from two entirely different biochemical mechanisms.
In vitro BER assays performed with cell extracts reveal that the gap-filling capability of S229L is disrupted. Expression of S229L in Pol β−/− MEFs is unable to fully complement the sensitivity to MMS treatment that is achieved by WT. The decreased observed catalytic rate for S229L most likely accounts for this phenotype. Patients who harbor this mutation in their tumor may be best treated with alkylating chemotherapies that are repaired by the BER pathway because the S229L variant is less capable of repairing the damage, leading to increased sensitivity of the cells in the tumor and increased cell death. This killing effect would be specifically potent for tumor cells and less harmful to normal cells, as S229L is not a known germ line variant of Pol β (17).
In the present study, we showed that expression of S229L induces cellular transformation and that this effect results from a heritable change because transformation continues even when S229L expression is extinguished. Cellular transformation is not due to overexpressing Pol β because cells expressing WT Pol β at similar levels did not exert this phenotype. In fact, because S229L exerts an effect in the presence of WT Pol β, it is likely that S229L acts in a dominant negative manner. S229L can bind DNA as tightly as WT and likely prevents WT Pol β from accessing gaps to which S229L is bound. This leads to the accumulation of BER intermediates which can be processed in an error-free manner by homology-directed repair, an error-prone manner, such as nonhomologous end joining, or can lead to cell death. We suggest that error-prone processing of the BER intermediates results in genomic instability that could confer a selective advantage to the cells. The cellular transformation we observe is likely not due to an increased frequency of point mutations, because the fidelity of the S229L is the same as for the WT polymerase. We point out that even though expression of S229L Pol β confers sensitivity to alkylating agents, treatment of cells harboring this variant with these types of drugs could drive carcinogenesis, especially if BER intermediates accumulate during the repair process.
In conclusion, we have shown that the human colorectal tumor variant S229L induces cellular transformation via genomic instability resulting from its slow polymerase activity. Pol β is mutated in 40% of human colorectal tumors and can drive tumorigenesis through genomic instability (6, 8). In combination with our previous work on the identification and characterization cancer-associated variants of Pol β, our work suggests that Pol β functions as a tumor suppressor.
This work was supported by National Institutes of Health Grants F32 CA156843 and T32 CA009259 from the NCI (to A. A. N.) and ES019179 from the NIEHS (to J. B. S.).
- BER
- base excision repair
- dRP
- deoxyribose phosphate
- DSB
- double-strand break
- MEF
- mouse embryonic fibroblast
- MMS
- methyl methanesulfonate
- Pol β
- polymerase β
- SNG
- single-nucleotide gap
- SSB
- single-strand break
- tet
- tetracycline.
REFERENCES
- 1. Loeb L. A., Springgate C. F., Battula N. (1974) Errors in DNA replication as a basis of malignant changes. Cancer Res. 34, 2311–2321 [PubMed] [Google Scholar]
- 2. Loeb L. A. (2011) Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer 11, 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes D. E., Lindahl T. (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 38, 445–476 [DOI] [PubMed] [Google Scholar]
- 4. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 5. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase beta deoxyribose phosphate lyase: substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 6. Donigan K. A., Sun K. W., Nemec A. A., Murphy D. L., Cong X., Northrup V., Zelterman D., Sweasy J. B. (2012) Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 287, 23830–23839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lang T., Dalal S., Chikova A., DiMaio D., Sweasy J. B. (2007) The E295K DNA polymerase β gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol. Cell. Biol. 27, 5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nemec A. A., Donigan K. A., Murphy D. L., Jaeger J., Sweasy J. B. (2012) Colon cancer-associated DNA polymerase β variant induces genomic instability and cellular transformation. J. Biol. Chem. 287, 23840–23849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murphy D. L., Jaeger J., Sweasy J. B. (2011) A triad interaction in the fingers subdomain of DNA polymerase β controls polymerase activity. J. Am. Chem. Soc. 133, 6279–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lowy D. R., Rands E., Scolnick E. M. (1978) Helper-independent transformation by unintegrated Harvey sarcoma virus DNA. J. Virol. 26, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donigan K. A., Hile S. E., Eckert K. A., Sweasy J. B. (2012) The human gastric cancer-associated DNA polymerase β variant D160N is a mutator that induces cellular transformation. DNA Repair 11, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sweasy J. B., Lang T., Starcevic D., Sun K. W., Lai C. C., Dimaio D., Dalal S. (2005) Expression of DNA polymerase β cancer-associated variants in mouse cells results in cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 102, 14350–14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamtich J., Starcevic D., Lauper J., Smith E., Shi I., Rangarajan S., Jaeger J., Sweasy J. B. (2010) Hinge residue I174 is critical for proper dNTP selection by DNA polymerase β. Biochemistry 49, 2326–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamtich J., Nemec A. A., Keh A., Sweasy J. B. (2012) A germline polymorphism of DNA polymerase β induces genomic instability and cellular transformation. PLoS Genet. 8, e1003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murphy D. L., Kosa J., Jaeger J., Sweasy J. B. (2008) The Asp285 variant of DNA polymerase β extends mispaired primer termini via increased nucleotide binding. Biochemistry 47, 8048–8057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dalal S., Chikova A., Jaeger J., Sweasy J. B. (2008) The Leu22Pro tumor-associated variant of DNA polymerase β is dRP lyase deficient. Nucleic Acids Res. 36, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamtich J., Speed W. C., Straka E., Kidd J. R., Sweasy J. B., Kidd K. K. (2009) Population-specific variation in haplotype composition and heterozygosity at the POLB locus. DNA Repair 8, 579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]