

Background: IDH1 R132H, implicated in glioblastoma and AML, produces the oncometabolite 2-HG.
Results: A detailed binding mechanism of a small molecule inhibitor (ML309) is proposed.
Conclusion: ML309 competes with α-KG but is uncompetitive with NADPH and rapidly and reversibly affects cellular 2-HG levels.
Significance: Understanding IDH1 R132H inhibition sets the stage for targeting IDH1 R132H for the treatment of cancer.
Keywords: Dehydrogenase, Drug Discovery, Enzyme Inhibitors, Glioblastoma, Small Molecules, AML, IDH1, Isocitrate Dehydrogenase, Enzyme
Abstract
Two mutant forms (R132H and R132C) of isocitrate dehydrogenase 1 (IDH1) have been associated with a number of cancers including glioblastoma and acute myeloid leukemia. These mutations confer a neomorphic activity of 2-hydroxyglutarate (2-HG) production, and 2-HG has previously been implicated as an oncometabolite. Inhibitors of mutant IDH1 can potentially be used to treat these diseases. In this study, we investigated the mechanism of action of a newly discovered inhibitor, ML309, using biochemical, cellular, and biophysical approaches. Substrate binding and product inhibition studies helped to further elucidate the IDH1 R132H catalytic cycle. This rapidly equilibrating inhibitor is active in both biochemical and cellular assays. The (+) isomer is active (IC50 = 68 nm), whereas the (−) isomer is over 400-fold less active (IC50 = 29 μm) for IDH1 R132H inhibition. IDH1 R132C was similarly inhibited by (+)-ML309. WT IDH1 was largely unaffected by (+)-ML309 (IC50 >36 μm). Kinetic analyses combined with microscale thermophoresis and surface plasmon resonance indicate that this reversible inhibitor binds to IDH1 R132H competitively with respect to α-ketoglutarate and uncompetitively with respect to NADPH. A reaction scheme for IDH1 R132H inhibition by ML309 is proposed in which ML309 binds to IDH1 R132H after formation of the IDH1 R132H NADPH complex. ML309 was also able to inhibit 2-HG production in a glioblastoma cell line (IC50 = 250 nm) and had minimal cytotoxicity. In the presence of racemic ML309, 2-HG levels drop rapidly. This drop was sustained until 48 h, at which point the compound was washed out and 2-HG levels recovered.
Introduction
Recent advances in tumor genome analysis have suggested novel roles for previously unappreciated genes in establishment and maintenance of the oncogenic state (1–3). Mutations in isocitrate dehydrogenase 1 (IDH1),3 a metabolic enzyme responsible for the conversion of isocitrate to α-ketoglutarate (α-KG), were annotated at a high frequency in an analysis of 22 human glioblastoma multiforme tumor samples. Interestingly, all these mutations were G395A, resulting in the conversion of an arginine at position 132 to a histidine (4). Because proteins with the IDH1 R132H missense mutation display decreased efficiency in the conversion of isocitrate to α-KG in vitro because Arg-132 is one of the substrate-binding arginine triads in the enzyme active site, these were at first believed to be loss-of-function mutations (5). However, the discovery of gain of function where IDH1 R132H results in a neomorphic enzymatic activity (Fig. 1), namely the conversion of α-KG to 2-hydroxyglutarate (2-HG), has profound implications for the role of IDH1 and its close homologue IDH2 in the metabolic activities of the cancer cell (6). As a dead-end metabolite, 2-HG accumulates to millimolar levels in cells with neoactive IDH1 (i.e. R132H or R132C) and IDH2 mutations (i.e. R172K) (7), and acts as an inhibitor of the α-KG-dependent epigenetic machinery (8, 9), blocking differentiation and promoting the proliferation of undifferentiated tumorous cells. It has recently been shown that 2-HG alone can promote leukemogenesis (10). Additionally, 2-HG suppresses the tricarboxylic acid (TCA) cycle and results in enhanced lipid metabolism (11). Inhibitors of 2-HG production by mutant IDH1 and IDH2 could have important clinical applications in the treatment of IDH mutated glioblastoma and acute myeloid leukemia (4, 5, 12, 13). Moreover, such inhibitors could help elucidate mechanism by which these mutations function in the context of the cancer cell metabolome. Therefore, there is a need for the development of inhibitors for mutant IDH1 and to gain an understanding of their mechanisms of action.
FIGURE 1.
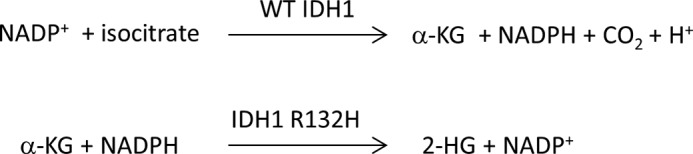
Enzyme reactions catalyzed by WT IDH1 and IDH1 R132H.
A previously reported high-throughput screen identified the first potent series of inhibitors of IDH1 R132H that were further optimized (14). The series consists of a phenyl-glycine scaffold with one stereocenter. One enantiomer was shown to be predominantly responsible for the activity of the racemic mixture. The inhibitor series was selective for mutant IDH1 over wild-type (WT) IDH1 and had excellent cell activity (IC50 = 70 nm), including the ability to lower 2-HG levels by ∼90% in an in vivo U87MG IDH1 R132H mouse tumor xenograft model (14). Recently, a member of this series was shown to delay growth and promote differentiation of glioma cells (15).
ML309, described herein, is a newly identified and characterized member of the phenyl-glycine series. ML309 is active in both biochemical and cell assays. The time dependence of the effect on 2-HG levels in cells was explored. To gain a deeper understanding of how the substrates, and phenyl-glycine scaffold inhibitors, such as ML309, interact with IDH1 R132H enzyme, a detailed characterization using kinetic and biophysical approaches was undertaken. Based on these results, a compound binding model was proposed that provides a plausible explanation of the inhibitory mechanism and that can be used for future structure and activity relationship studies.
EXPERIMENTAL PROCEDURES
General Methods for Chemistry
All air- or moisture-sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents, such as dichloromethane, N,N-dimethylformamide, acetonitrile, methanol, and triethylamine, were purchased from Sigma-Aldrich. Analytical analysis was performed on an Agilent 1200 series LC/MS (Agilent Technologies, Santa Clara, CA). Sample dissolution in MeOH and analysis were performed at room temperature. The analytical column used was a Chiralcel OD column (4.6 × 150 mm, 5 μm). The mobile phase was 60:40 ethanol (absolute, 200 proof)/hexane (0.1% diethyl amine) at 1.0 ml/min. The sample was detected with a diode array detector at 220 and 254 nm. Optical rotation was determined with an in-line polarimeter (PDR-Chiral). Purity and enantiomeric excess were determined by this analytical method. All of the analogues tested in the biological assays have a purity greater than 95%. Preparative purification of racemic material was performed on an Agilent 1200 series Prep/LC (Agilent Technologies, Santa Clara, CA). The column used was a Chiralcel OD column (5 × 50 cm, 20 μm). The mobile phase was 60:40 ethanol (absolute, 200 proof)/hexane (0.1% diethyl amine) at 35 ml/min. Fraction collection was triggered by UV absorbance (220 nm). 1H NMR spectra were recorded on a Varian 400-MHz spectrometer. Chemical shifts are reported in ppm (CHD2OD at 3.31 ppm as internal standard) for MeOH-d4 solutions. High resolution mass spectrometry was recorded on Agilent 6210 time-of-flight LC/MS system. Confirmation of molecular formulae was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
Preparation of (−)- and (+)-2-(2-(1H-Benzo[d]imidazol-1-yl)-N-(3-fluorophenyl)acetamido)-N-cyclopentyl-2-o-tolylacetamide ((−)-ML309 and (+)-ML309)
A solution of isocyanocyclopentane (196 mg, 2.064 mmol), 3-fluoroaniline (229 mg, 2.064 mmol), and 2-(1H-benzo[d]imidazol-1-yl)acetic acid (364 mg, 2.064 mmol) in MeOH (6.9 ml) was treated with 2-methylbenzaldehyde (248 mg, 2.064 mmol). The reaction was warmed to 40 °C and stirred for 4 h and then concentrated under reduced pressure. The crude product was purified by silica chromatography (2:98 to 5:95 MeOH/dichloromethane), which afforded racemic ML309 (rac-ML309) (216 mg, 0.446 mmol) in 22% yield.
rac-ML309 (216 mg, 0.446 mmol) was separated by chiral chromatography to afford (−)-ML309 (73 mg, 0.151 mmol, >99% enantiomeric excess) and (+)-ML309 (66 mg, 0.136 mmol, >99% enantiomeric excess). (−)-ML309: LC-MS retention time: t1 (Chiralcel OD column; 60:40 EtOH/hexane isocratic) = 2.91 min; [α]D22 = −72.1° (c = 0.86, MeOH); 1H NMR (400 MHz, MeOH-d4) δ ppm 8.01 (s, 1 H), 7.89 (br s, 1 H), 7.65 (d, J = 7.8 Hz, 1 H), 7.41 (d, J = 7.8 Hz, 1 H), 7.32 (t, J = 6.4 Hz, 1 H), 7.27 (t, J = 6.8 Hz, 1 H), 7.14 (d, J = 6.8 Hz, 1 H), 7.07 (t, J = 7.2 Hz, 1 H), 6.99 (dt, J = 2.4, 8.0 Hz, 1 H), 6.88 (t, J = 7.2 Hz, 1 H), 6.81 (d, J = 7.2 Hz, 1 H), 6.56 (br s, 1H), 6.36 (s, 1H), 5.02 (d, J = 17.2 Hz, 1 H), 4.81 (d, J = 17.6 Hz, 1 H), 4.18 (tt, J = 6.8, 6.8 Hz, 1 H), 2.45 (s, 3 H), 1.94–1.83 (m, 2 H), 1.67–1.27 (m, 8 H); 19F NMR (282 MHz, MeOH-d4) δ, −113.03, −114.12 ppm; high resolution mass spectrometry (electrospray mass ionization) m/z (M+H)+ calculated for C29H29FN4O2, 485.2385; found 485.2335. (+)-ML309: LC-MS retention time: t1 (Chiralcel OD column; 60:40 EtOH/hexane isocratic) = 5.56 min; [α]D22 = + 75.3° (c = 0.76, MeOH); high resolution mass spectrometry (electrospray mass ionization) m/z (M+H)+ calculated for C29H29FN4O2, 485.2385; found 485.2367.
Absorption, Distribution, Metabolism, and Excretion, PK, Solubility, and Chemical Stability
rac-ML309 stability in aqueous solution, 5 mm glutathione, assay buffer, mouse and human microsomes, and human plasma and aqueous solubility were tested following methods described previously (16, 17). A 20 mm DMSO stock of rac-ML309 was dispensed in assay buffer (final concentrations of 2% DMSO, 15% MeCN, 83% assay buffer), 5 mm glutathione (final concentrations of 2% DMSO, 49% MeCN, 49% PBS), or Dulbecco's PBS buffer (2% DMSO, 25% MeCN, 73% Dulbecco's PBS), and the remaining compound was monitored by the area under the curve of the peak at 220 nm by LC/MS at 0, 0.5, 1, 2, 6, 24, and 48 h. The half-life of rac-ML309 was tested by in vivo mouse pharmacokinetics (PK) using wild-type BALB/c mice (18).
General Methods for Biology
Full-length IDH1 (EC 1.1.1.42 and UniProt accession number O75874), IDH1 R132H, and IDH1 R132C were expressed and purified as described previously (6, 14). Both IDH1 R132H and IDH1 R132C protein were expressed with N-terminal His6 tags and purified by standard metal-chelate affinity chromatography techniques; IDH1 R132H was expressed in Escherichia coli, and IDH1 R132C was expressed in insect cells using the BaculoDirect (Invitrogen) baculovirus expression system. Proteins were buffer-exchanged by G25 gel-filtration chromatography into 500 mm NaCl, 50 mm Tris, pH 8.0, 10% glycerol and stored at −80 °C. NADPH, α-KG, and buffer components were purchased from Sigma. Experiments were run at room temperature unless otherwise specified.
IDH1 R132H, IDH1 R132C, and IDH1 WT Biochemical Assays
The activity of IDH1 R132H and IDH1 R132C was measured in 384-well plates by coupling NADPH consumption to a diaphorase/resazurin-based detection system and measuring resorufin production (Ex544/Em590) as described previously (6, 14). For testing the potency of small molecule inhibitors, an end point-coupled assay system was used, with 2–4 nm enzyme. In this assay, the reactions were run in the α-KG to 2-HG direction with the concomitant oxidation of NADPH to NADP. For IDH1 R132H, a final concentration of 1 mm α-KG and 4 μm NADPH was used; for IDH1 R132C, a final concentration of 200 μm α-KG and 4 μm NADPH was used. The NADPH remaining at the end of a 1-h incubation reaction was measured by the addition of 20 μm resazurin and 10 μg/ml diaphorase, which facilitated the stoichiometric conversion of resazurin to the highly fluorescent resorufin. To test the reversibility of the binding of compound to enzyme, 10× IC50 of the compound and 100× enzyme were incubated for 1 h, at which point the sample was diluted 100-fold and the enzyme reaction was run in kinetic mode monitoring the consumption of NADPH at 340 nm. Data were normalized to control columns representing maximum signal (no enzyme) and minimum signal (all components). WT IDH1 activity was also measured with this coupled diaphorase/resazurin system, but in this case product (NADPH) could be measured directly. Readout interference was tested with a counterscreen assay that contained all components except the proteins. The data were fit in GraphPad Prism 5 (La Jolla, CA) using nonlinear least-squares curve fitting.
Stopped-Flow Kinetics and Assessment of Km(app), Ki, and Vmax(app)
For determination of the Km(app) of substrates, IDH1 R132H was assayed in 150 mm NaCl, 20 mm Tris-Cl, pH 7.5, 10 mm MgCl2, 0.03% BSA. Reactions where studied in an SFM-400 stop-flow spectrophotometer by monitoring the oxidation state of the co-factor at Ex340/Em450 and used 5–100 nm recombinant protein. Typical reactions were monitored for ∼5 s, and steady-state rates were extracted by linear regression with fluorescence converted to NADPH concentration by a standard curve of NADPH in assay buffer. The equations used to fit the substrate inhibition and product inhibition are described in detail in Ref. 19.
Microscale Thermophoresis Measurements of Binding Kinetics
IDH1 R132H was labeled with a fluorescent dye NT-647 using the manufacturer's protocol (NanoTemper Technologies, München, Germany). A 16-point titration series of rac-ML309 in DMSO, NADPH in water, α-KG in water, or combinations thereof was transferred to the protein (20 nm) in a buffer containing 0.05% BSA, 0.01% Tween 20, 2 mm β-mercaptoethanol, 20 mm Tris, pH 7.5, and 150 mm NaCl and equilibrated for 15 min. The final top concentrations were 25 μm for NADPH, 25 mm α-KG, or 500 μm rac-ML309 for the single agent tests. For the competition tests, the substrate was held constant and rac-ML309 was titrated, i.e. α-KG was fixed at 150 mm and rac-ML309 was titrated down from 500 μm or NADPH was fixed at 50 μm and rac-ML309 was titrated down from 500 μm. The final DMSO concentration for each protein-compound sample was 0.5% DMSO. Samples were loaded into Monolith NT hydrophilic treated capillaries (NanoTemper Technologies). A capillary scan was performed followed by the successive measurement of thermophoresis in each capillary on a NanoTemper Monolith NT.115 instrument at room temperature. The IR laser power and the LED power conditions are described in the figure legends. A laser on-time of 30 s and a laser-off time of 5 s were used. The experiment was performed in duplicate, and the fits are reported as mean ± S.D. Data normalization and curve fitting were performed using GraphPad Prism 5.
Surface Plasmon Resonance Binding Measurements
Binding experiments between IDH1 R132H and either NADPH or NADPH/rac-ML309 were performed using SPR measurements on a ProteOn XPR36 instrument (Bio-Rad Laboratories, Inc.) at 25 °C. IDH1 R132H (“ligand”) was immobilized at high surface densities (6000–7000 resonance units) on an activated ProteOn HTE nickel-nitrilotriacetic acid sensor chip (Bio-Rad Laboratories, Inc.) through the His tag on the IDH1 R132H protein according to the manufacturer's instructions. The ligand was injected five times at a concentration of 20 μg/ml at a flow rate of 30 μl/min for 1000 s across two of the six available channels. To perform the binding assays, the various compounds (“analytes”) were injected at different concentrations in running buffer at a flow rate of 100 μl/min for 120 s (NADPH or rac-ML309 alone) or 180 s (rac-ML309 with NADPH), and sensorgrams were recorded. Blank surfaces and interspots (a feature of the ProteOn interaction array system) were used for background corrections using the ProteOn software. The sensorgrams were fit using the ProteOn analysis software. The protein rapidly lost activity on the chip, and therefore the chip was regenerated and the ligand was reloaded for each enzyme-compound test. The analytes tested included NADPH (a five-point 1:3 dilution series starting at 1 μm) and NADPH/rac-ML309 (at 1 μm NADPH constant and a five-point 1:3 dilution of rac-ML309 starting at 3 μm). The running buffer used for ligand loading and testing of NADPH alone or rac-ML309 alone was TBS. For the tests with rac-ML309 and NADPH co-injections, the running buffer was TBS with 1 μm NADPH and 0.25% DMSO.
Cell Line Culture (U87)
U87MG (human glioblastoma) cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA). U87MG is cultured in RPMI 1640, 2 mm l-Glutamine, 10 units/ml penicillin/streptomycin, and 10% FBS (Life Technologies). Cells were maintained in 5% CO2 at 37 °C.
Generation of IDH1 R132H-expressing Cell Lines
U87MG cells were infected with lentivirus containing full-length IDH1 R132H (U87MG pLVX-IDH1 R132H-neo or U87MG IDH1 R132H for short). For infection, cells were plated with the viral supernatant supplemented with 8 μg/ml Polybrene (5.0 × 104 cells/ml viral supernatant) and incubated in 5% CO2 at 32 °C for 24 h. After infection, transduced cells were selected using G418 (1 mg/ml) for 2 weeks to generate stable expression cells.
U87MG pLVX-IDH1 R132H-neo Cell-based Assays
U87MG pLVX-IDH1 R132H-neo cells were maintained in DMEM containing 10% FBS, 1× penicillin/streptomycin, and 500 μg/ml G418. Cells were seeded at a density of 5000 cells/well into 96-well white solid-bottom microtiter plates and incubated overnight at 37 °C and 5% CO2. The next day compounds were prepared in 100% DMSO and then diluted in medium for a final concentration of 0.2% DMSO. Medium was removed from the cell plates, and 200 μl of the compound dilutions were added to each well. After 48 h of incubation with compound at 37 °C, 100 μl of medium was removed from each well and analyzed by LC-MS for 2-HG concentrations as described in Ref. 6. The cell plates were then allowed to incubate another 24 h. At 72 h after compound addition, Promega CellTiter-Glo reagent (Madison, WI) was added to each well, and the plates were read for luminescence to determine any compound effects on growth inhibition (GI50) as described in Refs. 6 and 14).
For compound time course and washout studies, HT1080 cells (cultured as described in Refs. 6 and 14) that have endogenous expression of IDH1 R132C were incubated with compound similar to the cell-based assays with the following exceptions. Cells were plated into 6-well dishes. Upon each time point, cells were washed three times with PBS and harvested, and 2-HG was extracted as described previously (6).
RESULTS
ML309 Is a Selective Inhibitor of the IDH1 Mutants R132H and R132C in a Biochemical Assay in Comparison with Wild-type Enzyme
Containing a chiral center, rac-ML309 was separated into its (+) and (−) enantiomers (Fig. 2A). A coupled biochemical assay was used to determine the IC50 values for IDH1 R132H for the (+)-ML309 and (−)-ML309 enantiomers, which were 68 ± 1.2 and 29,000 ± 1.3 nm, respectively (Fig. 2B and Table 1), affirming the former as the eutomer and the latter as the distomer. Very similar activity was observed for the IDH1 R132C enzyme, giving IC50 values of 30 ± 1.4 and 15,000 ± 1.5 nm for (+)-ML309 and (−)-ML309, respectively (Fig. 2C and Table 1). rac-ML309 was weakly active in the IDH1 WT assay (IC50 >36 μm; Fig. 2D and Table 1). rac-ML309 showed no readout interference with the coupled diaphorase/resazurin detection system (data not shown). The binding of rac-ML309 to IDH1 R132H was found to be reversible by the method of compound/enzyme preincubation followed by rapid dilution and measurement of remaining enzyme activity (20) (Fig. 3).
FIGURE 2.

Biochemical and cellular IC50 values for ML309. A, chemical structure of ML309. B, IDH1 R132H biochemical assay for (+)-ML309 (triangles) and (−)-ML309 (open squares). C, IDH1 R132C biochemical assay for (+)-ML309 (triangles) and (−)-ML309 (open squares). D, IDH1 WT biochemical assay for rac-ML309. E, cytotoxicity of U87MG IDH1 R132H cells for (+)-ML309 (triangles) and (−)-ML309 (open squares). Error bars are mean ± S.D. from two duplicate experiments. F, inhibition of 2-HG in U87MG IDH1 R132H cells for (+)-ML309 (triangles) and (−)-ML309 (open squares). Error bars are mean ± S.D. from two replicates.
TABLE 1.
IC50 values (in nm) of rac-ML309, (+)-ML309, and (−)-ML309
Average values with S.E. are shown for two or three experimental replicates.
| Compound | IDH1 R132H | IDH1 R132C | IDH1 WT | Cell 2-HG levelsa | Cell cytotoxicitya |
|---|---|---|---|---|---|
| rac-ML309 | 96 ± 1.2 | 62 ± 1.2 | 36,000 ± 1.1 | 150 ± 1.7 | Noneb |
| (+)-ML309 | 68 ± 1.2 | 30 ± 1.4 | Noneb | 250 ± 1.7 | Noneb |
| (−)-ML309 | 29,000 ± 1.3 | 15,000 ± 1.5 | Noneb | Noneb | Noneb |
a U87MG IDH1 R132H cells were used. Cell 2-HG refers to an assay that measures 2-HG levels, and Cell pertains to results from cytotoxicity testing.
b None indicates that there was little to no effect over the concentration range tested, and no IC50 fit was obtained.
FIGURE 3.

Reversibility test of ML309. Activity values represent normalized (E = 100%, vehicle alone (Veh alone) = 0%) values. The error bars represent the error from the fit to the slope of enzyme activity versus time. E = IDH1 R132H, veh = DMSO, and I = ML309. pre-inc indicates that the compound (concentration = 10× IC50) was preincubated with 100×E for 30 min and then diluted 100× (achieving inhibitor concentration = IC10 and enzyme concentration = 1×) rather than the standard protocol of adding the inhibitor at the same final concentration and then directly measuring activity.
Characterization of the Mechanism of Inhibition of rac-ML309
To better understand the effects of inhibitor binding on IDH1 R132H, we first determined the order of substrate binding. Steady-state enzymatic activity was measured while α-KG was held constant at 2 mm and NADPH was titrated from 64 to 5000 nm. Kinetic analysis of α-KG binding to IDH1 R132H was also undertaken, and our experiments revealed substrate inhibition at high concentrations of α-KG. This substrate inhibition was relieved at increasing concentrations of NADPH. Investigations of this phenomenon were performed at NADPH concentrations of 1, 5, 10, and 20 μm while α-KG was varied from 78 μm to 40 mm. Data for Km(app), Vmax(app), and h (Hill coefficient) were fit globally, whereas Ki was fit for each concentration of NADPH. These experiments are presented in Fig. 4, and the kinetic parameters obtained from these experiments are summarized in Table 2; results are suggestive of an obligate order of binding with NADPH binding preceding α-KG and confirm previously reported results (6, 21).
FIGURE 4.

Characterization of binding affinity of the substrate α-KG and co-factor NADPH. A, Km(app) determination for NADPH. [IDH1 R132H] = 5 nm; [α-KG] = 2 mm. [NADPH] as shown on x-axis of the graph. B, relief of α-KG substrate inhibition by increasing concentrations of the co-factor NADPH at 1 μm (circles), 5 μm (squares), 10 μm (triangles), and 20 μm (inverted triangles). [IDH1 R132H] = 5 nm; [α-KG] as shown on the x-axis of the graph. Data were fit to an allosteric sigmoidal binding model with substrate inhibition model. Error bars for A and B are mean ± S.D. from triplicate experiments. C, typical stopped-flow reaction trace. [IDH1 R123H] = 5 nm; [α-KG] = 2 mm and [NADPH] = 100 μm. Note that in this experiment, the NADPH and the enzyme were premixed for 60 min in one syringe and the α-KG was premixed in a second syringe. This pre-equilibration was done for all experiments as appropriate for the assay setup.
TABLE 2.
Kinetic parameters of substrate binding and inhibition of IDH1 R132H
| Fixed | Varied | Vmax(app)a | Km(app)a | ha,b | Kia |
|---|---|---|---|---|---|
| μm/min/mg | μm | mm | |||
| α-KG | NADPH | 2.2 ± 0.028 | 0.044 ± 0.011 | 1.6 ± 0.12 | (−) |
| NADPH (global fit) | α-KG | 1.8 ± 0.025 | 820 ± 11 | 1.1 ± 0.20 | |
| NADPH 1 μm | α-KG | 6.9 ± 0.43 | |||
| NADPH 5 μm | α-KG | 58 ± 5.7 | |||
| NADPH 10 μm | α-KG | 101 ± 11 | |||
| NADPH 20 μm | α-KG | 140 ± 17 |
a Average values with S.D. are shown for three experimental replicates.
b h + Hill coefficient.
Next, we determined the mechanism of inhibition of rac-ML309 with regard to NADPH and α-KG binding. Fig. 5 describes the effect on Vmax(app) and Km(app) as a function of inhibitor concentration. Increasing concentrations of rac-ML309 increased the apparent Km(app) of α-KG of the reaction while not affecting the apparent Vmax(app), indicating that the inhibitor is competitive with respect to substrate. Increasing concentrations of rac-ML309 decreased both the apparent Km(app) and the apparent Vmax(app) of the reaction with regard to NADPH, indicating that the mechanism of action is uncompetitive.
FIGURE 5.

Mechanism of action of ML309. A, stopped-flow kinetic investigation of the effect of rac-ML309 on the Vmax(app) and Km(app) of IDH1 R132H when NADPH is held constant at 100 μm and α-KG is varied as shown on the x-axis of the graph ([ML309]: 0 nm, circles; 50 nm, squares; 100 nm, triangles; 150 nm, inverse triangles; and 300 nm, diamonds). [IDH1 R132H] = 5 nm. Error bars represent the mean and S.D. of three replicates at each point. B, simultaneous titration of α-KG and rac-ML309 increases the Km(app) (squares) of the reaction, but not the Vmax(app) (circles), suggesting a competitive mode of action with regard to α-KG. Error bars represent the mean and S.D. of three replicates at each point. C, stopped-flow kinetic investigation of the effect of rac-ML309 on the Vmax(app) and Km(app) of IDH1 R132H when α-KG is held constant at 2 mm and NADPH is varied as shown on the x-axis of the graph ([ML309]: 0 nm, circles; 50 nm, squares; 100 nm, triangles; 150 nm, inverse triangles; and 300 nm, diamonds). [IDH1 R132H] = 5 nm. Error bars represent the mean and S.D. of three replicates at each point. D, simultaneous titration of NADPH and rac-ML309 at constant NADPH decreases both the Vmax(app) (squares) and the Km(app) (circles) indicative of an uncompetitive inhibitor mechanism of action with regard to NADPH. Error bars represent the mean and S.D. of three replicates at each point. E, microscale thermophoresis of α-KG shows the effect on the IC50 of rac-ML309 upon the addition of α-KG. Triangles, rac-ML309 with 150 mm α-KG; open squares, rac-ML309 without 150 mm α-KG. Laser power was 80%, and LED power was 50%. F, microscale thermophoresis of NADPH shows the effect on the rac-ML309 IC50 upon the addition of NADPH. Triangles, rac-ML309 with 50 μm NADPH; open squares, rac-ML309 without 50 μm NADPH. Laser power was 80%, and LED power was 50%.
To further characterize this effect, microscale thermophoresis methods were used to determine the apparent Kd of rac-ML309 binding in the absence of α-KG and in the presence of a saturating concentration of α-KG (Fig. 5E). There is a right shift in the Kd in the presence of 150 mm α-KG (from 20 ± 0.88 μm to >500 μM), indicating that α-KG and rac-ML309 compete for binding to IDH1 R132H.
Microscale thermophoresis was also used to measure the effect on the Kd of rac-ML309 binding in the presence and absence of saturating amounts of NADPH. As shown in Fig. 5F, the Kd decreases dramatically in the presence of NADPH (from 20 ± 0.88 μm to 410 ± 29 nm, consistent with the uncompetitive binding with respect to NADPH).
On/off Rate Analysis and Kd Determination from SPR
NADPH binding to IDH1 R132H was measured using SPR. The data were fit in both kinetic (Fig. 6A) and equilibrium modes (Fig. 6B). From a kinetic analysis, the Kd was determined to be 80 ± 0.94 nm with an on rate of ∼ 420,000 ± 8000 1/ms and an off rate of ∼0.034 ± 0.033 1/s. This agreed well with the equilibrium analysis, which produced a Kd of 88 ± 0.71 nm. These Kd values compare well with the recently reported Kd of 43 ± 9 nm (22). Additionally, the SPR data correlated well with the stopped-flow kinetic data described above, which yielded a Km(app) of 0.044 ± 0.011 μm. rac-ML309 alone has very little affinity for IDH1 R132H, and no binding was observed up to 3 μm (Fig. 6C). In the presence of saturating NADPH, the binding of rac-ML309 to IDH1 R132H increased. Fig. 6D shows the binding isotherms for rac-ML309 in the presence of NADPH. Fitting of the isotherms indicates that the on rate is ∼ 32,000 ± 3100 1/ms and the off rate is ∼ 0.0015 ± 0.000071 1/s. A binding constant of 48 ± 0.24 nm was determined, in good agreement with the IC50 observed in the enzymatic assay (see above).
FIGURE 6.

Surface plasmon resonance characterization of binding of NADPH and/or rac-ML309. A, NADPH alone in kinetic mode. Top concentration is 1 μm, and dilution is 1:3. Residuals to the fit are shown below the plot. RU, resonance units. B, NADPH alone in equilibrium mode. Top concentration is 1 μm, and dilution is 1:3. C, rac-ML309 alone in kinetic mode. Top concentration is 3 μm, and dilution is 1:3. D, NADPH and rac-ML309 together in kinetic mode. Top concentration of rac-ML309 is 3 μm, and dilution is 1:3. NADPH is held constant at 1 μm. Residuals to the fit are shown below the plot. For A, C, and D, one of the two replicates is shown with the fit. For B, the plot is of the averaged data and the corresponding fit, and the error bars were smaller than the symbol size. Injection start and stop times are indicated by vertical dashed lines.
ML309 Has Reasonable PK Properties
rac-ML309 has a kinetic aqueous solubility greater than 50 μm, making it useful for biochemical, biophysical, and cell-based studies. rac-ML309 was found to be highly stable in aqueous solution, 5 mm glutathione, assay buffer, and human plasma (98% remaining after 30 min in all instances). Despite the low stability in human and mouse microsomes, 6.2 and 7.7% remaining after 30 min, respectively, in mouse PK studies, the half-life of the compound was greater than 3 h. However, no brain exposure was observed.
ML309 Is Active in Cell Assays
In cells that contain the IDH1 R132H mutation, α-KG (the product of the WT IDH1 reaction) is converted into the oncometabolite 2-HG. The impact on 2-HG formation was determined in U87MG human glioblastoma cell line transfected with IDH1 R132H, and it was found that (+)-ML309 inhibited the production of 2-HG with an IC50 of 250 ± 1.7 nm, whereas (−)-ML309 showed no inhibition up to 5 μm (Fig. 2F). Notably, the shift between inhibitory potencies of the compound in the biochemical and cell assays is minimal, roughly 4-fold. As part of the above assay, the cytotoxicity was measured, and both the (+)-ML309 and the (−)-ML309 were nontoxic at the concentrations tested (Fig. 2E).
Time Dependence of 2-HG Reduction upon rac-ML309 Treatment and Recovery after Compound Washout
HT1080 cells, which harbor the IDH1 R132C mutation, were treated with 5 μm rac-ML309, and the 2-HG levels were monitored as described in Refs. 6 and 14. 2-HG levels rapidly decreased as shown in Fig. 7 (t½ ∼ 5 h) and remained low. The effect was reversed upon compound washout after 48 h.
FIGURE 7.
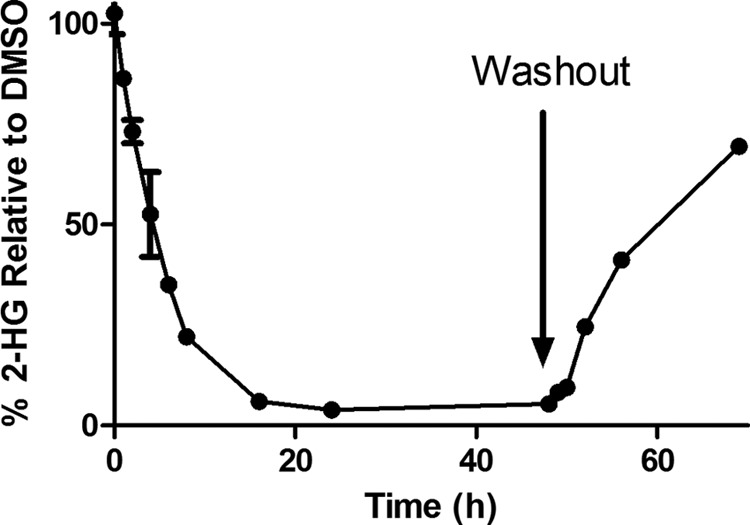
Time dependence of 2-HG levels following 5 μm rac-ML309 treatment and washout after 48 h in HT1080 cells, which harbor the IDH1 R132C mutation. Error bars are mean ± S.D. from three replicates.
DISCUSSION
IDH1 R132H and IDH1 R132C have been identified as important targets for the development of therapeutics for glioblastoma and acute myeloid leukemia. Cells that harbor these mutations have significantly higher levels of 2-HG relative to WT cells (6). Decreasing the levels of 2-HG through inhibition of the 2-HG-producing IDH1 mutant enzymes is a potential avenue for therapeutic treatment.
Recent work at Agios Pharmaceuticals, Inc., resulted in the identification of a phenyl-glycine chemical series that showed potent inhibition in both biochemical and cell assays (14). A representative inhibitor from the phenyl-glycine series (ML309) was studied in detail here to provide a deeper understanding of the mechanism of inhibition. rac-ML309 was very stable in human plasma and had a half-life of over 3 h in a mouse PK study. The integrity of the compound is expected to be stable during the cell assays described herein. Although there was no observed penetration of the blood brain barrier in these normal healthy mice, there could still be penetration in a glioma setting in which the blood brain barrier may be compromised (23), and this compound could provide a good starting point for further optimization to improve drug-like properties and potency.
(+)-ML309 was potent against both IDH1 R132H and IDH1 R132C in a biochemical assay, indicating that it could be useful for both glioblastoma and acute myeloid leukemia treatment. It is exquisitely selective for the mutant forms of IDH1 over the WT form. Although a crystal structure is not currently available for ML309 bound to IDH1 R132H to definitively show the binding mode, one possibility for the preference of binding to IDH1 R132H over WT IDH1 is that IDH1 R132H has a more open conformation than WT IDH1 (22). The Kd measured for rac-ML309 binding to IDH1 R132H in the presence of NADPH (48 ± 0.24 nm) by SPR was very similar to the IC50 measured in the IDH1 R132H biochemical assay (68 ± 1.2 nm).
The substrate titration experiments allow us to propose a kinetic mechanism for the inhibitor. Our work suggests that NADPH is the first to bind the enzyme followed by α-KG. A previous study on IDH1 R132H using a combination of dead-end inhibition studies and primary kinetic isotope effects also concluded that NADPH is likely to bind first followed by α-KG (22). Given the uncompetitive nature of rac-ML309 with regard to NADPH and competitive nature with regard to α-KG, we propose that rac-ML309 binds after NADPH and prevents the binding of α-KG, stopping the catalytic cycle, which produces the oncometabolite 2-HG.
An understanding of the mechanism of action can provide assistance in series selection and lead optimization. NADPH binding may orient IDH1 R132H for productive inhibitor binding. It is possible that the (+)-ML309-binding site overlaps with that of α-KG and that the competitive inhibition is due to direct competition for the same binding site. Alternatively, (+)-ML309 could bind remotely and thus allosterically alter the α-KG-binding pocket. These two possibilities cannot be distinguished here, but hopefully a crystal structure with a member of the phenyl-glycine series will be forthcoming. Currently, a crystal structure is available for IDH1 with NADPH (6), and a structure with a small molecule (1-hydroxypyridine 2-one scaffold) inhibitor was published (24).
Understanding the inhibition modality is important when comparing different compound series, which may have different mechanisms of action. It is also important for properly setting up SPR and other biophysical measurements. For an uncompetitive inhibitor (or a mixed inhibitor in which the affinity is much greater for the ES form), a build-up of substrate leads to enhanced inhibitor affinity in contrast to competitive inhibitors, which can be overcome by high concentrations of substrate The cell activity was determined in both a cytotoxicity assay and an assay measuring product 2-HG formation. (+)-ML309 is not very toxic during the course of the in vitro cytotoxicity assay, consistent with previous data on a member of the phenyl-glycine series that indicated that the compound did not induce apoptotic cell death (15) but that it does have a profound effect on 2-HG formation. 2-HG is thought to behave as an oncometabolite by inhibiting multiple α-KG-dependent dioxygenases, which are important in a variety of biological processes, such as histone and DNA demethylation (8, 13). Indeed, it may be that epigenetic changes due to the presence of 2-HG contribute to tumorigenesis. A recent study showed that the effect of 2-HG on promoting leukemogenesis (i.e. growth factor independence and impaired differentiation) was reversible (10). The authors tested AGI-5198, a previously published member of the phenyl glycine series (14), and showed that this inhibitor reversed the impaired differentiation and growth factor independence of IDH1 R132H-expressing TF-1 cells. Inhibition of the formation of 2-HG should help mitigate the deleterious effect of the R132C and R132H mutations of IDH1.
To further understand the effect of ML309 on 2-HG levels, a time course was performed. Upon treatment of HT1080 cells, which harbor the IDH1 R132C mutation, with 5 μm rac-ML309, 2-HG levels rapidly dropped with a t½ ∼ 5 h and remained low until 48 h, at which time the compound was washed out and 2-HG levels recovered. The rapid recovery is consistent with reversible binding of rac-ML309. This rapid recovery of 2-HG levels also has implications for dosing regimens and indicates that continual coverage of the target may be necessary to keep 2-HG levels below deleterious levels, although this remains to be tested.
The two enantiomers of ML309 had vastly differing activities in the enzymatic and cellular assays, with (+)-ML309 emerging as the active enantiomer. Enantiopure (+)-ML309 was prepared by synthesizing rac-ML309 and isolating the enantiopure isomer by chiral HPLC. The absolute stereochemistry of (+)-ML309 could not be verified by an enantioselective synthesis because racemization occurs in the Ugi reaction used to prepare rac-ML309. However, it is likely that the (+)-ML309 enantiomer is (S)-ML309 and the (−)-ML309 enantiomer is (R)-ML309 by comparison with the previously characterized chiral phenyl-glycine series in which the activity was predominantly contained within the (S)-configured analogues (14).
In summary, the detailed mechanism of action of an inhibitor of IDH1 R132H was determined and validated using a battery of biochemical, cellular, and biophysical methods. The inhibitor binds to IDH1 R132H predominantly in the presence of NADPH and is competitive with respect to α-KG binding. The compound binds reversibly to the enzyme with modest on and off rates. rac-ML309 rapidly affected 2-HG levels in cells, and this effect could be reversed by compound washout. The detailed mechanistic evidence and proposed reaction diagram presented here will hopefully aid in the development of future mutant IDH1 inhibitors and in their evaluation as treatments for diseases, such as glioblastoma and acute myeloid leukemia.
Acknowledgments
We are grateful for the technical support teams from Bio-Rad-ProteOn and NanoTemper-MST (microscale thermophoresis) for helpful discussions. We acknowledge Ed Kerns, Amy Wang, and Kimloan Nguyen for the absorption, distribution, metabolism, and excretion and in vivo PK data and helpful discussions as well as William Leister and Jim Bougie for the help with chiral chromatography.
This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research, National Institutes of Health (U54 MH084681).
- IDH1
- isocitrate dehydrogenase 1
- α-KG
- α-ketoglutarate
- PK
- pharmacokinetics
- 2-HG
- 2-hydroxyglutarate
- rac-ML309
- racemic ML309
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Mardis E. R., Wilson R. K. (2009) Cancer genome sequencing: a review. Hum. Mol. Genet. 18, R163–R168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson C. B. (2009) Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 360, 813–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yates L. R., Campbell P. J. (2012) Evolution of the cancer genome. Nat. Rev. Genet. 13, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., Nikolsky Y., Busam D. A., Tekleab H., Diaz L. A., Jr., Hartigan J., Smith D. R., Strausberg R. L., Marie S. K., Shinjo S. M., Yan H., Riggins G. J., Bigner D. D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., Kinzler K. W. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., Velculescu V. E., Vogelstein B., Bigner D. D. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., Rabinowitz J. D., Cantley L. C., Thompson C. B., Vander Heiden M. G., Su S. M. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gross S., Cairns R. A., Minden M. D., Driggers E. M., Bittinger M. A., Jang H. G., Sasaki M., Jin S., Schenkein D. P., Su S. M., Dang L., Fantin V. R., Mak T. W. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., Ratcliffe P. J., Schofield C. J., Kawamura A. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G., Yan H., Kaelin W. G., Jr. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Losman J. A., Looper R. E., Koivunen P., Lee S., Schneider R. K., McMahon C., Cowley G. S., Root D. E., Ebert B. L., Kaelin W. G. (2013) (R)-2-Hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyata S., Urabe M., Gomi A., Nagai M., Yamaguchi T., Tsukahara T., Mizukami H., Kume A., Ozawa K., Watanabe E. (2013) An R132H mutation in isocitrate dehydrogenase 1 enhances p21 expression and inhibits phosphorylation of retinoblastoma protein in glioma cells. Neurol. Med. Chir. (Tokyo) 53, 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paschka P., Schlenk R. F., Gaidzik V. I., Habdank M., Krönke J., Bullinger L., Späth D., Kayser S., Zucknick M., Götze K., Horst H. A., Germing U., Döhner H., Döhner K. (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 28, 3636–3643 [DOI] [PubMed] [Google Scholar]
- 13. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Wang P., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., Frye S., Zhang Y., Xu Y. H., Lei Q. Y., Guan K. L., Zhao S. M., Xiong Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Popovici-Muller J., Saunders J. O., Salituro F. G., Travins J. M., Yan S., Zhao F., Gross S., Dang L., Yen K. E., Yang H., Straley K. S., Jin S., Kunii K., Fantin V. R., Zhang S., Pan Q., Shi D., Biller S. A., Su S. M. (2012) Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 3, 850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rohle D., Popovici-Muller J., Palaskas N., Turcan S., Grommes C., Campos C., Tsoi J., Clark O., Oldrini B., Komisopoulou E., Kunii K., Pedraza A., Schalm S., Silverman L., Miller A., Wang F., Yang H., Chen Y., Kernytsky A., Rosenblum M. K., Liu W., Biller S. A., Su S. M., Brennan C. W., Chan T. A., Graeber T. G., Yen K. E., Mellinghoff I. K. (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Avdeef A. (2007) High-throughput measurements of solubility profiles. in Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies, Verlag Helvetica Chimica Acta, pp. 305–325, Zurich [Google Scholar]
- 17. Di L., Kerns E. H., Li S. Q., Petusky S. L. (2006) High throughput microsomal stability assay for insoluble compounds. Int. J. Pharm. 317, 54–60 [DOI] [PubMed] [Google Scholar]
- 18. Korfmacher W. A. (2009) Advances in the integration of drug metabolism into the lead optimization paradigm. Mini Rev. Med. Chem. 9, 703–716 [DOI] [PubMed] [Google Scholar]
- 19. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, Wiley-VCH, New York [Google Scholar]
- 20. Copeland R. A. (2005) Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, pp. 125–128, John Wiley & Sons, Inc., Hoboken, NJ: [PubMed] [Google Scholar]
- 21. Pietrak B., Zhao H., Qi H., Quinn C., Gao E., Boyer J. G., Concha N., Brown K., Duraiswami C., Wooster R., Sweitzer S., Schwartz B. (2011) A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry 50, 4804–4812 [DOI] [PubMed] [Google Scholar]
- 22. Rendina A. R., Pietrak B., Smallwood A., Zhao H., Qi H., Quinn C., Adams N. D., Concha N., Duraiswami C., Thrall S. H., Sweitzer S., Schwartz B. (2013) Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 52, 4563–4577 [DOI] [PubMed] [Google Scholar]
- 23. Schneider S. W., Ludwig T., Tatenhorst L., Braune S., Oberleithner H., Senner V., Paulus W. (2004) Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol 107, 272–276 [DOI] [PubMed] [Google Scholar]
- 24. Zheng B., Yao Y., Liu Z., Deng L., Anglin J. L., Jiang H., Prasad B. V. V., Song Y. (2013) Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 4, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]